Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(5):485-496. doi:10.7150/ijbs.24681 This issue Cite
Research Paper
The Suppression of Medium Acidosis Improves the Maintenance and Differentiation of Human Pluripotent Stem Cells at High Density in Defined Cell Culture Medium
Weiwei Liu1, Zhili Ren1, Kai Lu1, Chengcheng Song1, Edwin Chong Wing Cheung1, Zhou Zhou2, Guokai Chen1 ![]()
1. University of Macau, Faculty of Health Sciences, Taipa, Macau
2. State Key Laboratory of Cardiovascular Disease, Beijing Key Laboratory for Molecular Diagnostics of Cardiovascular Diseases, Diagnostic Laboratory Service, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100037, China
Received 2018-1-1; Accepted 2018-2-26; Published 2018-4-5
Abstract

Cell density has profound impacts on the cell culture practices of human pluripotent stem cells. The regulation of cell growth, cell death, pluripotency and differentiation converge at high density, but it is largely unknown how different regulatory mechanisms act at this stage. We use a chemically defined medium to systemically examine cellular activities and the impact of medium components in high-density culture. We show that medium acidosis is the main factor that alters cell cycle, gene expression and cellular metabolism at high cell density. The low medium pH leads to inhibition of glucose consumption, cell cycle arrest, and subsequent cell death. At high cell density, the suppression of medium acidosis with sodium bicarbonate (NaHCO3) significantly increases culture capacity for stem cell survival, derivation, maintenance and differentiation. Our study provides a simple and effective tool to improve stem cell maintenance and applications.
Keywords: Human pluripotent stem cells, acidosis, glycolysis, cell death, differentiation
Introduction
Cell culture is a dynamic system that includes the cultured cells and their surrounding environment - culture medium. The nutrients and growth factors in the medium help maintain cell survival and their essential functions, while cellular metabolism could lead to changes in medium composition. For stem cells, the interaction and balance between cells and their culture media ultimately determine the quality of stem cells and their differentiation potential. A robust and consistent cell culture condition is the foundation to fully realize the potentials of human pluripotent stem cells (hPSCs) [1, 2]. In this study, we systematically studied the interactions between the culture medium and hPSCs at high density, and demonstrated the important roles of medium buffer capacity in stem cell derivation, maintenance and differentiation.
Cell density is an important environmental cue in stem cell culture[1]. In stem cell culture practices, different cell densities are required for specific applications [3-7]. For example, gene targeting and clonal selection experiments usually require individualized cells and subsequent culture at clonal density [8, 9]. On the other hand, regular maintenance and expansion use low- to medium-density cultures ranging from 20-70% confluency to maintain pluripotency and self-renewal ability [2, 10-12]. At the same time, hPSCs are often grown to higher confluency in order to obtain more cells for specific applications, such as large-scale production, cryopreservation and teratoma formation[13]. Furthermore, many differentiation procedures are conducted at confluent density to generate cardiac[14-16], hepatic[17], pancreatic[18, 19] and neural lineage cells[20].
Although cell density plays profound roles in stem cell practices, protocols are usually established empirically in terms of plating density with few mechanistic bases. Compared to clonal and low-density culture, a high-density culture has more cells in each unit of medium, which requires more growth factor stimulation and nutritional support, while releasing more cellular products into the medium. It was recently reported that high-density hPSC culture leads to decreased S phase in albumin-containing media[21]. In a separate study, people found that high-density culture leads to medium acidosis with DNA damage phenotypes in feeder culture, yet without obvious impact on S phase [4].
In this report, we used albumin-free E8 medium to understand whether common medium components could affect hPSC maintenance and differentiation at high density. The chemically defined composition of E8 medium allows us to study the contributions from individual medium components on hPSCs, and uncovers phenotypes previously masked in serum- or albumin-containing systems. We showed that medium acidosis led to hPSC cell cycle arrest and cell death at high density, which was not shown in previous reports[4, 21]. A screen of medium components identified medium buffer reagent as the key player that rescued cell survival phenotypes at high density. Based on these discoveries, we demonstrated that the suppression of medium acidosis is an effective way to improve the maintenance, derivation and differentiation of hPSCs in defined culture conditions.
Materials and Methods
hPSC culture maintenance and passaging
Human embryonic stem cell (hESC) H9 and H1 lines were used in this study. H9 was the main cell line used in mechanistic studies and the results were confirmed in H1. An iPSC line, ND1, was also used in differentiation studies.
Human ESCs (H1 and H9) and iPSCs (ND1) were maintained in E8 medium on matrigel-coated plates (Corning 354230 ) with daily medium change as previously described[22]. Because the cells were cultured in the incubator with 5% CO2, no additional NaHCO3 was added into E8 medium unless otherwise stated[22]. Cells were passaged using the EDTA method[23] in the presence of 5µM rock inhibitor Y-27632. Rock inhibitor was removed the next day and various treatments were started the day after. For conditioned medium collection, hESCs were grown to >90% confluency, and the culture medium was left in the well and collected after 24 hours. For cell counting, ESCs were harvested using TrypLE Select enzyme (Thermo Fisher Scientific) and the count of live cells were determined using a BD Accuri C6 flow cytometer (BD Biosciences) or a hemocytometer. For cell survival curve analysis, cells were plated on day -1 and treatment with NaHCO3 started on day 0. Culture medium was changed daily. The concentration of NaHCO3 added into the medium was 20mM unless specified otherwise. Other medium components were supplemented into the conditioned medium (Figure 1F) at the concentration used in E8[22].
Medium acidosis causes hESC death in high-density culture. (A) Representative growth curve of H9 culture. Cell count was determined by flow cytometry (n=3 for each time point). (B) Cell cycle analysis using Edu and propidium iodide (PI). Data representative of three independent experiments. (C) Caspase activation in high-density (HD) and low-density (LD) H9 culture, analyzed using CellEvent caspase 3/7 green detection reagent. Left, fluorescent and phase contrast images (scale bar: 400 µm). Middle and right, flow cytometry analysis (n=3). (D) Mitochondrial membrane potential of high-density and low-density cells measured using JC-1. (E) Low-density cells were cultured in fresh E8 or conditioned medium collected from high-density culture, and cell count determined after 24 hours (n=3). (F) 24-hr cell count of low-density cells cultured in high-density-conditioned medium (CM) with supplementation of various medium components (n=3). *, p < 0.05. ***, p < 0.001. (G) 24-hr cell count of low-density cells cultured in fresh E8 medium at different pH (n=3).
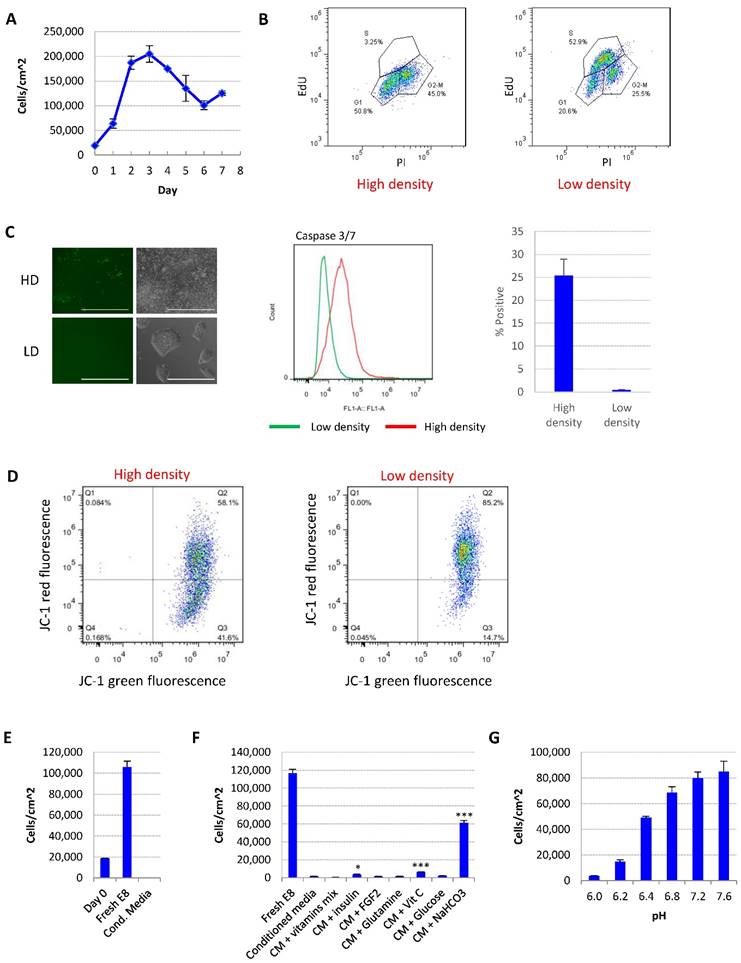
For the ease of discussion, in this report we define individualized cells <200,000 cells/cm2 or <70% confluence as low density, and >90% confluence as high density. Representative images of each condition are shown in Fig. 1C.
Apoptosis and cell cycle assays
For each assay, high-density and low-density ESC cultures were plated on day 0. The media was changed on day 1 (with 20mM NaHCO3 added if applicable) and assays were carried out on day 2. Caspase 3/7 activation were measured using CellEvent Caspase-3/7 Green Flow Cytometry Assay Kit (Molecular Probes) following manufacturer instructions. Mitochondrial membrane potential was measured using JC-1 dye (Molecular Probes) following manufacturer instructions. Cell cycle status was analyzed using Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit (Molecular Probes) following manufacturer instructions.
Cell-cycle reporter cell line
H1 hESCs were transduced with lentivirus to constitutively express mKO2-hCdt1(30/120)[24]. mKO2-positive populations were sorted with a BD FACSAria III cell sorter and plated as single cells in 48-well dishes. Following colony picking and further expansion, a second lentivirus transduction was performed to express mAG-hGeminin (1/110). Next, mAG-positive populations were FACS sorted and plated as single cells in 48-well dishes followed by colony picking and expansion of the FUCCI hESCs.
FUCCI plasmids mKO2-hCdt1(30/120) and mAG-hGeminin (1/110) were obtained from Dr. Atsushi Miyawaki (RIKEN, Japan). Lentiviruses were packaged in 293FT by transfection with polyethylenimine using the packaging plasmid psPAX2 and the envelope plasmid pMD2.G.
Medium component and pH analysis
Cell culture medium was collected from cell culture wells and centrifuged to remove debris. Content of glucose, glutamine and lactate were analyzed using Bioprofile FLEX Analyzer from Nova Biomedical. For medium pH measurement, the medium was equilibrated in cell culture incubators (37ºC, 5% CO2) for 30 minutes and the pH was determined using pH meter (Mettler Toledo).
Mito stress test
Oxygen consumption rates (OCR) were measured using the XF-96 Extracellular Flux Analyzer (Seahorse Biosciences). For Mito Stress Test, H1 cells (2 x 104/well) were seeded in E8 medium into XF96 cell culture microplates. The next day, cells were pre-incubated in XF assay media (XF base media supplemented with 25mM D-glucose, 2mM L-glutamine, and 1mM sodium pyruvate, with or without NaHCO3 or HCl treatment) for one hour before the Mito Stress Test were performed following manufacturer's protocol. After the assay, cells were lysed (10mM Tris/HCl pH7.5, 0.1% Triton X-100) and the protein content was determined using Bradford reagent for normalization.
Intracellular ATP content assay
Intracellular ATP content was measured using the ATP Determination Kit (Molecular Probes A22066). Briefly, cells were harvested, resuspended in water and then heated in a boiling water bath to lyse the cells. After centrifugation, the cell lysate was mixed with the luciferin-luciferase reagent from the assay kit and bioluminescence measured using a plate reader.
Microarray analysis
Total RNA was extracted with RNAiso Plus reagent (Takara #9109) and purified using RNAeasy mini kit (QIAGEN). Purified total RNA was then converted to cRNA using the TargetAmp™-Nano Labeling Kit for Illumina Expression BeadChip (Epibio) according to the manufacturer's instructions. cRNA samples were hybridized onto microarrays using the HumanHT-12 v4 Expression BeadChip Kit (Illumina) and the arrays were scanned on an iScanner (Illumina).
The microarray data was processed through the arrayanalysis.org portal (www.arrayanalysis.org). Data quality was inspected and assured via box plot and PCA plot. Background correction and quantile normalization were applied to the raw data. Then the variance stabilizing transformation (log2) was performed. Heatmap was generated with the {pheatmap} package in R to show the expression patterns. Hierarchical clustering was applied to both axes using Pearson correlation metric for similarity and complete linkage clustering. Analysis of functional enrichment on selected genes was performed using DAVID (https://david-d.ncifcrf.gov/). GEO accession number is GSE113016.
Reprogramming and iPSC generation
Reprogramming of human fibroblasts (CCD-1139Sk, ATCC® CRL 2708™) into iPSCs was carried out following published protocols[25, 26]. Following transduction on day 0, reprogramming cells were passaged on day 5 using TryPLE into E8-based reprogramming medium with butyrate. Medium was changed every other day. For NaHCO3 treatment, 20mM NaHCO3 was supplemented into the medium starting from day 15. iPSC colonies were stained using alkaline phosphatase substrate kit (Vector Laboratories) on day 25.
Real-time PCR
Total RNA was extracted using RNAiso Plus reagent (Takara #9109) and cDNA generated using High-Capacity cDNA Reverse Transcription Kit from Applied Biosystems (4368814). Real-time PCR of cDNA was performed using FastStart Universal SYBR Green Master Rox (Roche) on the Applied Biosystems QuantStudio 7 Flex Real-time PCR System. Primer sequences are provided in Supplemental Table 2.
Cardiac differentiation
The cardiac differentiation procedure was adapted from published protocols [15, 27]. Briefly, H9, H1 or ND1 cells maintained in E8 were passaged 1:3 onto matrigel-coated plates in E8 medium two days before differentiation. Medium was changed daily. The day before differentiation, 1mg matrigel / plate was mixed with cold E8 medium, overlaid onto cells and incubated overnight. Differentiation medium is prepared by supplementing E8 basal medium (DMEM/F12, L-ascorbic acid, selenium, transferrin) with 1x chemically defined lipid concentrate (Thermo Fisher 11905-031) and 1x penicillin/streptomycin. To induce differentiation, cells are switched to differentiation medium with the following supplementations: BMP4 (25ng/ml), bFGF (100ng/ml), and insulin (10µg/ml) from day 0 to day 2; IWR-1 (1µM) and heparin (1µg/ml) from day 4 to day 6; heparin (1µg/ml) and insulin (10µg/ml) from day 7 and after. Medium was changed daily.
FACS analysis
Cells were harvested using TrypLE Select enzyme (Thermo Fisher Scientific). Single cells were washed in PBS, fixed in 1% paraformaldehyde/PBS for 10 minutes at 37ºC, washed three times with PBS, and permeabilized in 90% methanol on ice for 30 minutes. The cells were then blocked with 1% BSA/PBS to prevent nonspecific binding, incubated with primary antibodies (diluted in 1% BSA) for one hour, washed three times, incubated with secondary antibodies (diluted in 1% BSA) for one hour, washed again, and finally resuspended in PBS for flow cytometry analysis using BD Accuri™ C6 Cytometer (FL1: FITC; FL2: PE). The following primary antibodies were used: Anti-brachyury (D2Z3J) antibody (Cell Signaling Technology 81694); Anti-cTnT Antibody (Developmental Studies Hybridoma Bank CT3).
Immunofluorescence and imaging
Cells were fixed in 4% paraformaldehyde for 20 minutes at room temperature, and rinsed three times with PBS. Permeabilization was performed with 0.2% Triton X-100 for 20 minutes at room temperature. Primary antibody was diluted with 2% BSA and incubated with cells overnight at 4ºC. After three washes with PBS for 5 minutes, cells were incubated with Alexa Fluor 488 secondary antibody (Jackson ImmunoResearch Laboratories) diluted in 2% BSA at 1:1000 ratio for 1 hour at room temperature. Finally, cells were washed with PBS for 5 minutes before Hoechst 33258 (Molecular Probes H1398) counter staining and slide mounting. Primary antibodies used: Anti-phosphorylated γH2A.X (S139) antibody (Abcam, ab2893); Anti-cTnT antibody (Developmental Studies Hybridoma Bank CT3).
Mounted slides were imaged with a Zeiss Axio Observer inverted microscope under 40x objective. Image Z-stacks were captured to cover the entire depth of the nuclei. Widefield images were deconvolved and maximum intensity projections were created from each deconvolved Z-stack.
Phase contrast images were captured using Invitrogen EVOS FL Auto Cell Imaging System.
Statistical analysis
Data are expressed as mean ± SD and statistical significance was determined by two-tailed Student's t-test. P < 0.05 was considered statistically significant.
Results
Medium acidosis in high-density culture leads to growth arrest and cell death in hESCs
We studied the effect of culture density on hESCs in E8 medium. After splitting at low density, the cell number first increased, and then significantly decreased after the cells reach confluence (Fig. 1A and S1A). The percentage of S phase cells was dramatically decreased at high density (Fig. 1B). Higher percentage of apoptotic cells (Fig. 1C) and loss of mitochondrial membrane potential (Fig. 1D) were observed in high-density culture, suggesting that more cells went through growth arrest and apoptosis at high density compared to lower density.
We hypothesized that the change of medium composition at high density led to the cell death. Conditioned medium collected from high-density hESC culture killed up to 90% of low-density cells in 24 hours (Fig. 1E). Through complementing experiments, it was found that the addition of vitamin C, insulin or NaHCO3 improved cell survival significantly, while NaHCO3 showed the most significant improvement (Fig. 1F). At the same time, decreasing the pH of fresh E8 medium with hydrochloric acid (HCl) to a pH level comparable to that of high-density culture significantly reduced hESC survival (Fig. 1G, Supplementary Table 1). Taken together, these evidences suggest that a decrease in medium pH is the major factor causing hESC cell death in medium from high-density cells.
Increased buffer capacity rescues high-density cell death
We further showed that the addition of buffer reagent NaHCO3 into E8 medium improved hESC maintenance at high-density, as it increased cell survival (Fig. 2A), promoted more cells into S phase (Fig. 2B and S1B), decreased the percentage of apoptosis cells (Fig. 2C), restored mitochondrial membrane potential (Fig. 2D) and decreased DNA damage at high density (Fig. S1C). Similar protective effect was observed with other buffering agents including HEPES and sodium phosphate (Fig. S1D). Cells from high-density culture had poor viability after passaging, which was significantly improved by NaHCO3 treatment (Fig. 2E). The pluripotency markers are maintained in NaHCO3-treated cells after multiple passages (Figure S1E). These results confirmed that the increased buffer capacity by NaHCO3 improved the medium's ability to sustain cell functions at high density.
pH modulation reverses cell death phenotypes. (A) 48-hr cell count of high-density culture in the presence of different concentrations of NaHCO3 (n=3). Medium was changed once at 24 hours. (B) Cell cycle analysis by Edu-PI in high-density and low-density culture with or without NaHCO3 (20mM) supplementation. (C) Caspase activation in high-density and low-density culture with or without NaHCO3 (20mM) treatment, analyzed by flow cytometry using CellEvent caspase 3/7 green detection reagent. (D) Mitochondrial membrane potential measured using JC-1. (E) Passaging efficiency of high-density cells maintained with or without NaHCO3 (20mM) supplement (n=3). Ri, rock inhibitor (Y27632 10µM). (F) Medium pH of high-density H9 culture with or without 1% (w/v) bovine serum albumin (BSA) (n=3). (G) 48-hr cell count of H9 high-density culture with or without BSA (n=3). HD, high-density. LD, low-density. Ctrl, control.
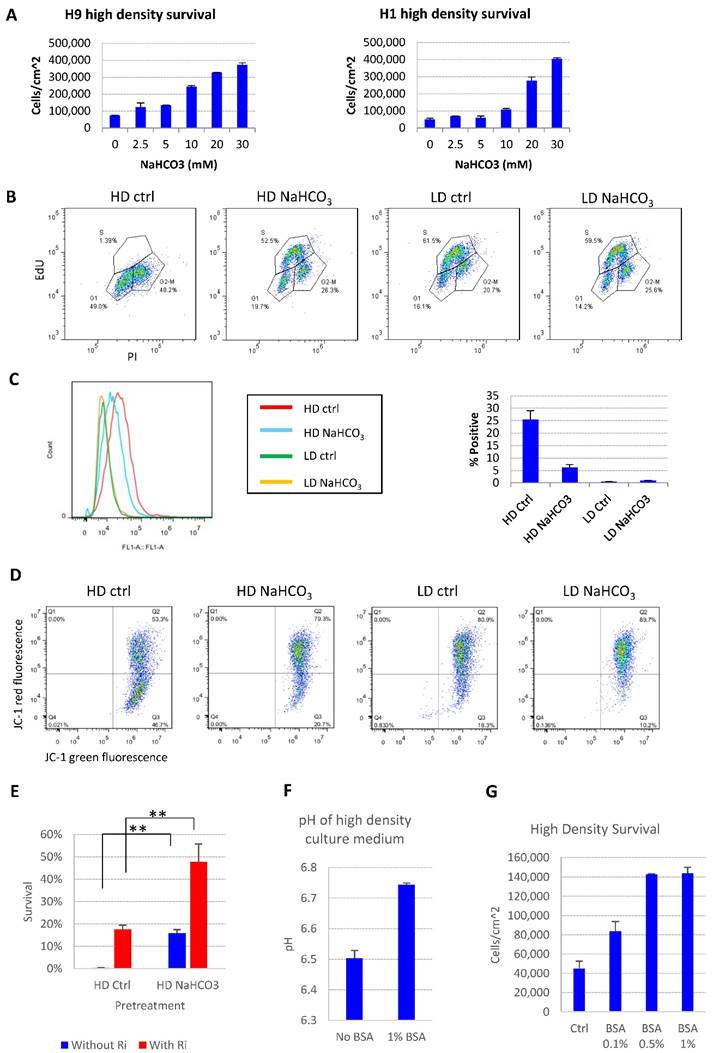
The cell death phenotype in high-density hESC culture in E8 medium was not reported under similar conditions with other albumin-containing media[4, 21], so we tried to explore possible mechanisms. The E8 medium does not include albumin, while traditional media, such as mTeSR, contain 0.5% to 1% albumin[11]. We demonstrated that the presence of albumin suppressed the pH decrease by high density culture (Fig. 2F), and deterred the pH decrease by the addition of lactic acid in vitro (Fig. S1F-G). As expected, the presence of albumin suppressed cell death at high density (Fig. 2G), although the beneficial effect of albumin is not as strong as direct NaHCO3 supplementation (Fig. 2A). These data indicate that traditional culture conditions might obtain extra buffer capacity through albumin compared to albumin-free media, which may have masked the cell death phenotype at high density in traditional media.
The increased buffer capacity rescues metabolic and gene expression changes in high-density culture
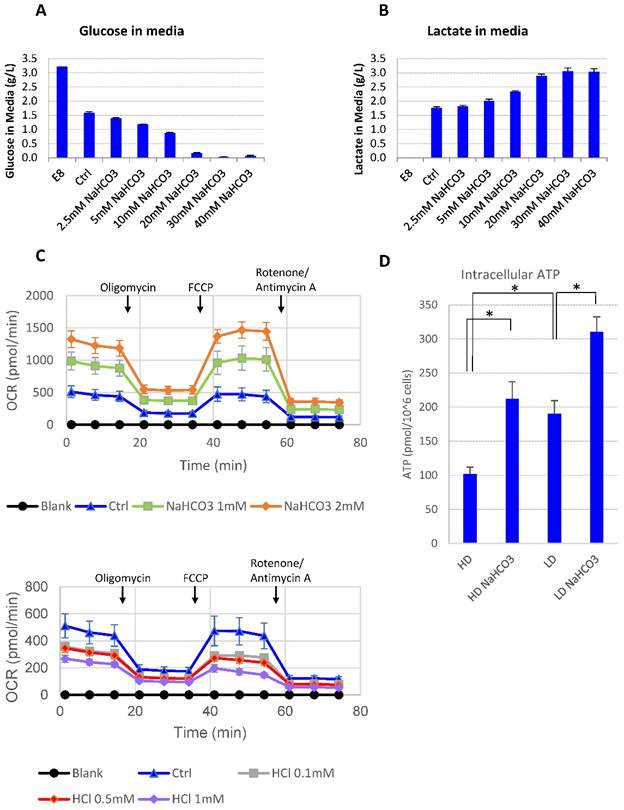
It was reported that lactic acid produced through glycolysis in hPSCs could lead to medium acidosis [28]. In order to understand the impact of high culture density on growth arrest and cell death, we looked into cellular metabolic changes at high density. At high cell density, more glucose was consumed along with more lactic acid release compared to low-density culture (Supplemental Table 1). However, there was always a significant amount of glucose left unconsumed (Supplemental Table 1). It suggests that glucose consumption may be inhibited at high density. Indeed, when the buffer capacity was elevated with NaHCO3, the high-density hESCs resumed to consume the remaining glucose in the culture media until the concentration approached zero (Fig. 3A), while the production of lactate increased simultaneously (Fig. 3B). It suggests that high-density culture caused pH decrease and reduced glucose consumption through glycolysis, either directly or through cell cycle arrest. In addition, glucose consumption through oxidative phosphorylation was also enhanced in the presence of additional NaHCO3. Both the basal oxygen consumption rate (OCR) and the spare respiratory capacity (SRC) of H1 cells increased as NaHCO3 was supplemented into the assay system, while addition of HCl decreased OCR and SRC values (Fig. 3C). Consistent with increased glycolysis activity and increased oxygen consumption, the intracellular ATP content in high-density culture was significantly upregulated by NaHCO3 treatment (Fig. 3D).
Medium pH controls glucose consumption and metabolism. (A-B) Culture media were collected from high-density culture supplemented with various concentrations of NaHCO3. Glucose (panel A) and lactate (panel B) concentrations were analyzed using Bioprofile FLEX Analyzer (n=3). (C) Oxygen consumption rate (OCR) measured by mito stress test with or without NaHCO3 or HCl in the assay medium (n=3). (D) Intracellular ATP content of H9 cell lysate from high-density and low-density culture (n=3). *, p < 0.05.
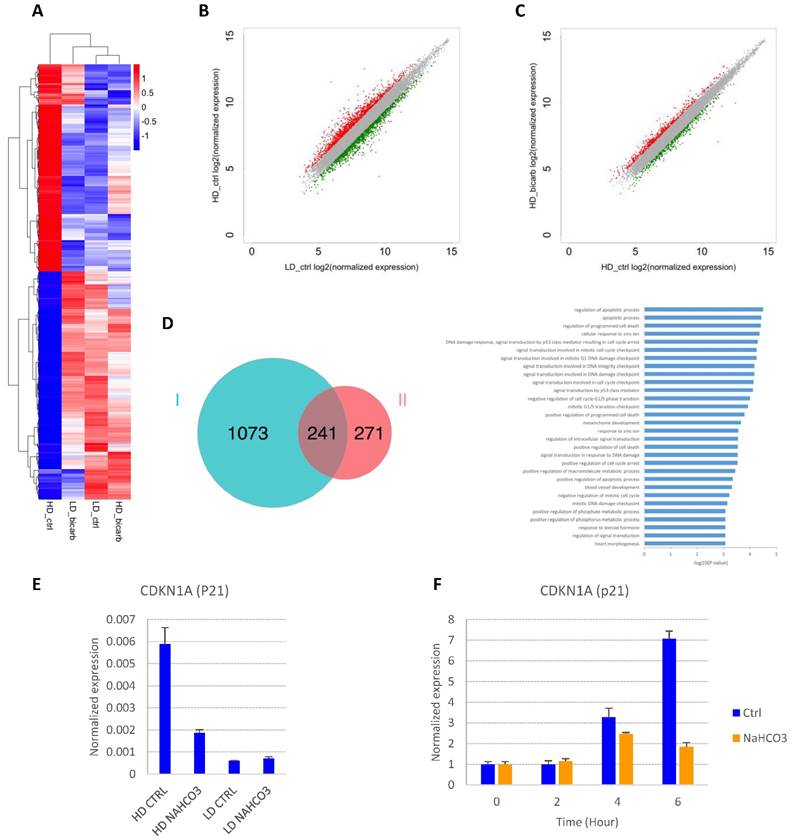
In order to explore the impact of cell density and buffer capacity on gene expression, we analyzed the global gene expression profiles in E8 at different cell density with or without additional NaHCO3 (Fig. 4A-D and S2A-B). There is a significant overlap between genes up-regulated in high-density culture and genes down-regulated by NaHCO3 treatment (Fig. 4D). Of note, p53-related apoptosis pathway was enriched in this gene pool, including cyclin-dependent kinase inhibitor p21 (CDKN1A). Consistent with previous report[21], the transcription of p21 was found to be significantly up-regulated in high-density cells compared to low-density culture, and down-regulated by NaHCO3 treatment (GEO accession No. GSEXXX). The density-dependent expression of p21 was confirmed by real-time PCR (Fig. 4E). We also found that p21 could be directly induced in cells exposed to high-density-conditioned medium within a few hours, which can be rescued by NaHCO3 (Fig. 4F). These findings are consistent with the cell cycle phenotypes observed and indicate that p21 might be a critical factor in the cell growth arrest and cell death at high density.
Gene expression changes in response to culture density and medium pH. (A) Heat map from hierarchical clustering of differentially expressed genes (DEGs) in high-density and low-density cells with or without NaHCO3 (20mM) treatment. (B) Scatter plot of gene expression in high-density versus low-density culture. Differentially expressed genes are marked in red (up-regulated in high-density culture) or green (down-regulated in high-density culture). (C) Scatter plot of gene expression in NaHCO3-treated versus untreated high-density cells. Differentially expressed genes are marked in red (up-regulated by NaHCO3 treatment) or green (down-regulated by NaHCO3 treatment). (D) Venn diagram (left) summarizing the overlap between genes up-regulated in high-density culture (Set I) and genes down-regulated by NaHCO3 treatment in high-density culture (Set II), and gene ontology (GO) biological process analysis (right) of the genes in the overlapping pool. (E) Verification of p21 gene expression changes by real-time PCR on high-density (HD) and low-density (LD) H9 culture (n=3). (F) Low-density H9 cells were exposed to high-density-conditioned medium for the specified time, and p21 expression examined by real-time PCR (n=3). Expression levels are normalized to time zero.
The increased buffer capacity improves hPSC maintenance and iPSC derivation
Based on the impact of additional buffer capacity on cell survival and metabolism, we explored its application in hPSC culture practices. We first evaluated whether buffer capacity could affect cell proliferation under different medium change schedules. Supplementation of NaHCO3 in culture medium significantly improved cell proliferation with daily medium change (Fig. 5A) and every other day medium change schedules (Fig. 5B). It suggests that increased buffer capacity has positive effects on hPSC maintenance, and allows less frequent medium changes.
Application of pH modulation in cell culture maintenance and somatic cell reprogramming. (A-B) Growth curves of H9 cells maintained with or without NaHCO3 (20mM) supplementation. H9 cells were maintained for four days with either daily (panel A) or alternate-day (panel B) medium change without passaging (n=3 for each time point). (C) Alkaline phosphatase staining of iPSC colonies on day 25 of human fibroblast reprogramming. (D) Phase contrast images of representative iPSC colonies on day 18 and day 24 of reprogramming, with or without additional NaHCO3.
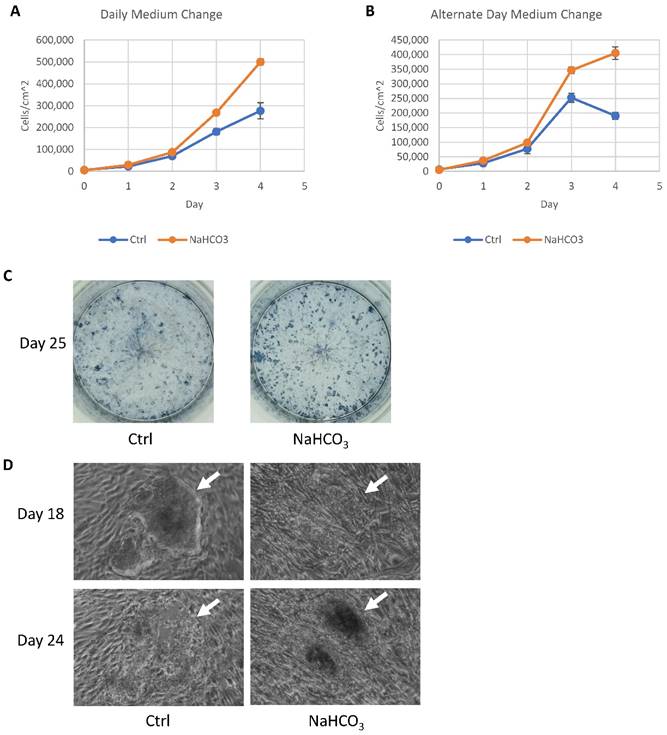
During reprogramming, an efficient iPSC derivation often generates too many colonies that lead to medium acidosis before the iPSC colonies could be effectively picked around 25 days after induction. We tested whether the increased buffer capacity could help sustain the iPSC derivation. When many iPSC colonies were present, increased buffer capacity by NaHCO3 helped maintain colony number (Fig. 5C). In regular medium, iPSC colonies lost their integrity from day 18 to day 25 (Fig. 5D, indicated by white arrow), while the colonies were maintained with increased buffer capacity. This result demonstrated that higher buffer capacity allows more consistent iPSC derivation and production.
The increased buffer capacity enhances BMP4-driven cardiac differentiation
Many differentiation protocols use high-density culture at 80-100% confluency as the starting culture [14, 15, 17, 18, 20]. Researchers sometimes encountered difficulties to adopt those existing differentiation protocols in defined, albumin-free culture, which is a vital step in the translation of research findings into clinical applications. Our findings on high-density hPSC culture offers a new strategy in optimization of differentiation protocols for albumin-free culture.
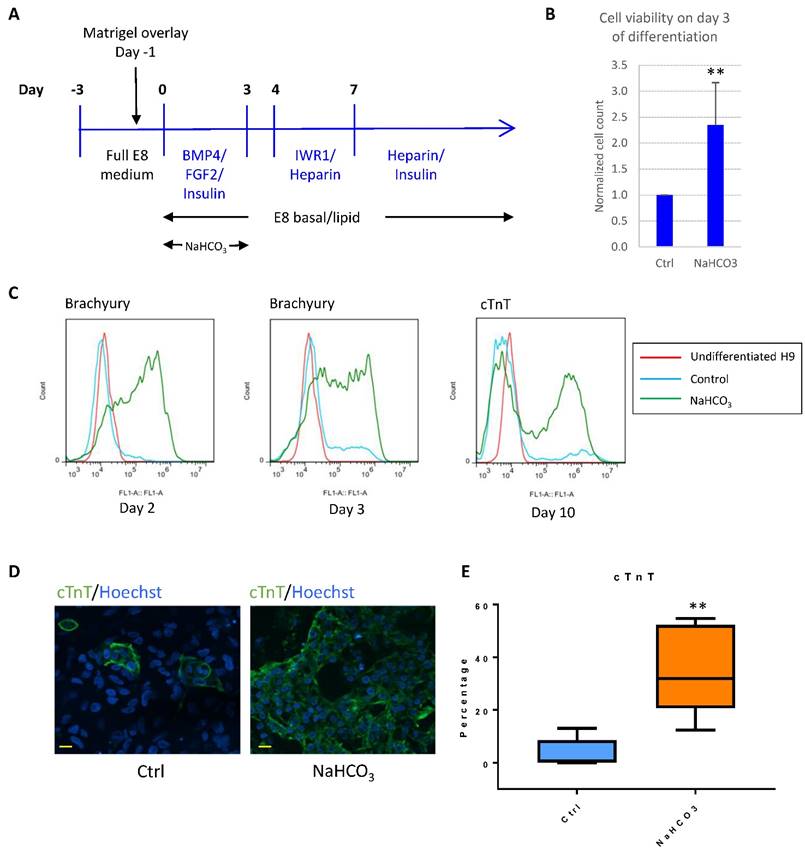
For example, BMP4 was successfully used to induce cardiomyocyte differentiation in albumin-containing media[15], and we found that BMP4 was generally unsuccessful as the initiator for cardiomyocyte differentiation in E8-based defined culture (Fig. 6A). We asked whether pH modulation could improve differentiation in defined culture. In the first 3-4 days after initiation of differentiation, we observed severe cell death, while additional NaHCO3 significantly improved cell survival (Fig. 6B). In addition, treatment with additional NaHCO3 promoted epithelial-mesenchymal transition (Fig. S3A), and early mesoderm markers Brachyury (encoded by the T gene) and MIXL1 were highly elevated (Fig. 6C and S3B). Finally, NaHCO3 treatment resulted in significant improvement in cardiomyocyte production, as shown by the elevated cardiomyocyte markers cTnT and NKX2.5 (Fig. 6C-E and S3C). The cardiac-promoting effect were reproducible in human ESC lines H9 (Fig. S3C), H1 (Fig. S3D) and iPSC line ND1 (Fig. S3E). These results suggest that NaHCO3 supplementation helped improve BMP4-initiated cardiac differentiation at high density, which could be a useful approach for other lineage-specific differentiation.
pH modulation improves cardiac differentiation in albumin-free system. (A) Schematic drawing of BMP4-initiated cardiac differentiation protocol in defined media. (B) Cell survival on day 3 of differentiation with or without NaHCO3 (20mM) treatment. Cell counts were normalized to that of the control wells (n=5). (C) FACS analysis of Brachyury on day 2 (left) and day 3 (middle) of differentiation, and cTnT on day 10 of differentiation (right). Data representative of three independent experiments. (D) Immunostaining of cTnT on day 14 of differentiation. Scale bar, 20µm. (E) Box and whiskers plot showing percentage of cTnT-positive cells on day 10 of differentiation in control and NaHCO3-treated cells (n=5 independent experiments). **, p < 0.01.
Discussion
Cell culture consistency is a central problem in stem cell applications, and great efforts have been taken to improve medium formula, extracellular matrix and handling methods [1, 7, 9]. The control of medium pH is a fundamental factor in cell culture[29]. Many key cellular processes are pH-dependent, such as glucose transportation[30], glycolysis[31], actin filament assembly[32] and integrin activation[33]. This report calls for renewed attention to pH and buffer capacity in stem cell applications.
The buffer capacity is often an afterthought nowadays, because almost all base media contain buffering reagents to sustain consistent neutral pH. People often assumed that the base medium has sufficient capacity to support various culture conditions. However, this report demonstrated that the hPSCs at high density could push the regular cell culture capacity over its limit. Compared to somatic cells such as fibroblasts, hPSCs could reach 10-fold more cells in each surface unit. Because of the large numbers of cells in the system, the hPSCs pose a unique challenge to medium capacity in both nutritional supply and pH control. In addition, hPSCs rely on glycolysis to generate energy and cell-building materials for cell proliferation[34], producing lactic acid during the process. The lactic acid then leads to medium acidosis with pH to as low as pH6.3 (Supplementary Table 1). The acidic environment leads to cell cycle arrest, stoppage of glucose consumption, inefficient passaging and cell death, and these phenomena are detrimental to stem cell maintenance and downstream applications. By increasing the medium buffer capacity, we were able to rescue the phenotypes essential for a successful stem cell culture. This simple maneuver could greatly improve the quality and consistency of stem cell culture. In this study, we also found other factors such as insulin and vitamin C that could rescue high-density cell survival. More study could be done to investigate additional mechanisms and further improve the medium consistency.
Our data also revealed the sensitive nature of defined albumin-free culture. We found that albumin probably served as a buffering agent[35] to delay medium acidosis to around pH6.4 when cell death is triggered (Fig. 1G and Supplemental Table 1). The buffer effect likely explains why the cell death phenotypes in hPSCs were not previously reported in albumin-containing culture systems[4, 21]. Albumin is a well-reported differentiation modulator in cardiac differentiation[36], and our study suggests that the buffer function of albumin might also play a role to improve differentiation, as increased buffer capacity improved cardiac differentiation in albumin-free media (Fig. 6).
In order to improve stem cell maintenance and derivation, methods such as thermo-stable FGF and controlled-release method have been developed to decrease the medium change frequency[37, 38]. This study showed that additional NaHCO3 could significantly improve the medium culture capacity. The addition of NaHCO3 helped support cell proliferation with less-frequent medium change, sustained iPSC derivation at high colony density, and promoted stem cell differentiation at high density.
In summary, we identified medium pH change as the major causative factor for the cell death and cell culture inconsistency in hPSCs. By suppressing medium acidosis, we significantly improved hPSC survival and differentiation at high density. This discovery provides pH modulation as a simple but useful tool to consider in stem cell applications in the future.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We would like to thank Dr. Mahendra Rao for discussion. This project was supported by University of Macau Multi-Year Research Grant MYRG2015-00228-FHS, MYRG2015-00229-FHS and Macao Science and Technology Development Fund FDCT/131/2014/A3. We would like to thank the Biological Imaging & Stem Cell Core Facility at the Faculty of Health Sciences, University of Macau.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen KG, Mallon BS, McKay RD, Robey PG. Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell. 2014;14:13-26
2. International Stem Cell Initiative C, Akopian V, Andrews PW, Beil S, Benvenisty N, Brehm J. et al. Comparison of defined culture systems for feeder cell free propagation of human embryonic stem cells. In Vitro Cell Dev Biol Anim. 2010;46:247-58
3. Hsiao C, Lampe M, Nillasithanukroh S, Han W, Lian X, Palecek SP. Human pluripotent stem cell culture density modulates YAP signaling. Biotechnol J. 2016;11:662-75
4. Jacobs K, Zambelli F, Mertzanidou A, Smolders I, Geens M, Nguyen HT. et al. Higher-Density Culture in Human Embryonic Stem Cells Results in DNA Damage and Genome Instability. Stem Cell Reports. 2016;6:330-41
5. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J. et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010;24:1106-18
6. Liu Y, Munoz N, Bunnell BA, Logan TM, Ma T. Density-Dependent Metabolic Heterogeneity in Human Mesenchymal Stem Cells. Stem Cells. 2015;33:3368-81
7. Chetty S, Pagliuca FW, Honore C, Kweudjeu A, Rezania A, Melton DA. A simple tool to improve pluripotent stem cell differentiation. Nat Methods. 2013;10:553-6
8. Chen G, Hou Z, Gulbranson DR, Thomson JA. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell. 2010;7:240-8
9. Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T. et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681-6
10. Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD. et al. Chemically defined conditions for human iPSC derivation and culture. Nat Methods. 2011;8:424-9
11. Ludwig TE, Levenstein ME, Jones JM, Berggren WT, Mitchen ER, Frane JL. et al. Derivation of human embryonic stem cells in defined conditions. Nat Biotechnol. 2006;24:185-7
12. Vallier L, Alexander M, Pedersen RA. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J Cell Sci. 2005;118:4495-509
13. Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, Dunn NR. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. 2009;2:198-210
14. Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD. et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855-60
15. Zhang J, Klos M, Wilson GF, Herman AM, Lian X, Raval KK. et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res. 2012;111:1125-36
16. Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661-80
17. Agarwal S, Holton KL, Lanza R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells. 2008;26:1117-27
18. Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S. et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26:443-52
19. Gage BK, Webber TD, Kieffer TJ. Initial cell seeding density influences pancreatic endocrine development during in vitro differentiation of human embryonic stem cells. PLoS One. 2013;8:e82076
20. Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275-80
21. Wu J, Fan Y, Tzanakakis ES. Increased culture density is linked to decelerated proliferation, prolonged G1 phase, and enhanced propensity for differentiation of self-renewing human pluripotent stem cells. Stem Cells Dev. 2015;24:892-903
22. Beers J, Gulbranson DR, George N, Siniscalchi LI, Jones J, Thomson JA. et al. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat Protoc. 2012;7:2029-40
23. Liu W, Chen G. Cryopreservation of human pluripotent stem cells in defined medium. Curr Protoc Stem Cell Biol. 2014;31(1C):171-189
24. Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H. et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487-98
25. Sommer CA, Stadtfeld M, Murphy GJ, Hochedlinger K, Kotton DN, Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27:543-9
26. Beers J, Linask KL, Chen JA, Siniscalchi LI, Lin Y, Zheng W. et al. A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Sci Rep. 2015;5:11319
27. Lin Y, Linask KL, Mallon B, Johnson K, Klein M, Beers J. et al. Heparin Promotes Cardiac Differentiation of Human Pluripotent Stem Cells in Chemically Defined Albumin-Free Medium, Enabling Consistent Manufacture of Cardiomyocytes. Stem Cells Transl Med. 2016
28. Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell. 2012;11:589-95
29. Ceccarini C, Eagle H. pH as a determinant of cellular growth and contact inhibition. Proc Natl Acad Sci U S A. 1971;68:229-33
30. Kaminskas E. The pH-dependence of sugar-transport and glycolysis in cultured Ehrlich ascites-tumour cells. Biochem J. 1978;174:453-9
31. Halperin ML, Connors HP, Relman AS, Karnovsky ML. Factors that control the effect of pH on glycolysis in leukocytes. J Biol Chem. 1969;244:384-90
32. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11:671-7
33. Paradise RK, Lauffenburger DA, Van Vliet KJ. Acidic extracellular pH promotes activation of integrin alpha(v)beta(3). PLoS One. 2011;6:e15746
34. Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596-606
35. Caironi P, Gattinoni L. The clinical use of albumin: the point of view of a specialist in intensive care. Blood Transfus. 2009;7:259-67
36. Burridge PW, Thompson S, Millrod MA, Weinberg S, Yuan X, Peters A. et al. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS One. 2011;6:e18293
37. Lotz S, Goderie S, Tokas N, Hirsch SE, Ahmad F, Corneo B. et al. Sustained levels of FGF2 maintain undifferentiated stem cell cultures with biweekly feeding. PLoS One. 2013;8:e56289
38. Chen G, Gulbranson DR, Yu P, Hou Z, Thomson JA. Thermal stability of fibroblast growth factor protein is a determinant factor in regulating self-renewal, differentiation, and reprogramming in human pluripotent stem cells. Stem Cells. 2012;30:623-30
Author contact
![]() Corresponding author: Guokai Chen, Ph.D., Faculty of Health Sciences, University of Macau, E12-4013, Taipa, Macau SAR, 999078. Phone: (853)-8822 4985; Fax: (853)-8822 2314; Email: guokaichenmo.
Corresponding author: Guokai Chen, Ph.D., Faculty of Health Sciences, University of Macau, E12-4013, Taipa, Macau SAR, 999078. Phone: (853)-8822 4985; Fax: (853)-8822 2314; Email: guokaichenmo.