Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(5):542-548. doi:10.7150/ijbs.24303 This issue Cite
Short Research Communication
Generation of Smurf2 Conditional Knockout Mice
Yi Tang1, Liu-Ya Tang1, Xuan Xu1, Cuiling Li2, Chuxia Deng2,3, Ying E. Zhang1 ![]()
1. Laboratory of Cellular and Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
2. Genetics of Development and Disease Branch, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, USA.
3. Present address: Faculty of Health Sciences, University of Macau, Avenida da Universidade, Taipa, Macau, China.
Received 2017-12-11; Accepted 2018-2-27; Published 2018-4-5
Abstract

Smad ubiquitin regulatory factor 2 (Smurf2) is a HECT domain-containing E3 ubiquitin ligase. Together with its closely related homolog Smurf1, Smurf2 was initially recognized as a negative regulator of transforming growth factor-β (TGF-β) and bone morphogenetic protein (BMP) signaling, but subsequent studies have expanded its function to regulate many different signaling pathways and play important roles in genomic stability, cell polarity, tissue homeostasis and carcinogenesis. Mice with conventional knockout of Smurf1 or Smurf2 alleles are viable, but conventional Smurf1 and Smurf2 double knockout mice were early embryonic lethal. In order to study the physiological function of Smurfs during late stage of embryonic development or in adult animals, we generated Smurf2flox/flox mice carrying a targeted mutation for conditional Smurf2 gene inactivation. We demonstrated that Cre-mediated recombination using Alb-Cre, a Cre line expressed in hepatocyte, results in specific deletion of the gene in liver tissue. We also showed that Cre-mediated recombination in mouse embryonic fibroblasts (MEFs) with Smurf2flox/flox genotype resulted in generation of Smurf2 knockout MEFs, and Smurf2 deficiency affects multiple signaling pathways. Therefore, this animal model will be useful to study the distinct roles of Smurf2 in different tissues at different ages.
Keywords: Smurf2, Liver, Mice, LoxP, Knockout
Introduction
Ubiquitination is a major form of post-translational modification that regulates essentially all aspects of cellular functions [1]. Conjugation of ubiquitin to a protein is carried out by sequential actions of three enzymes: an ubiquitin activating enzyme (E1), an ubiquitin conjugase (E2), and an ubiquitin ligase (E3). Because of diverse functions and physical contact with substrates, E3 ubiquitin ligases are of the most importance. Smurf1 and Smurf2 are two closely related members of the HECT family of ubiquitin E3 ligases, sharing a high degree of amino acid sequence homology and distinctive structural features of the HECT family of E3 ubiquitin ligases. These features include an amino-terminal phospholipid/calcium-binding C2 domain, two to four copies of WW repeats that mediate protein-protein interactions by binding to a PPXY (PY) motif in target proteins, and a carboxyl-terminal HECT domain that catalyzes the ubiquitination reaction [2]. Smurfs were initially recognized as a negative regulator of TGF-β/BMP signaling through interacting with PY motifs in most R-Smads and inhibitor Smads, thereby ubiquitinating Smads, or TGF-β/BMP or targeting other downstream components of the pathway [3-7]. Subsequent biochemical studies broadened the repertoire of Smurf substrates to include Axin, Ezh2, ID1/ID3, KLF5, MEKK1, Ptch1, Prickle1, RhoA, RNF20, Runx2/3, Talin, Traf4, etc., and extended their function to controlling cell polarity, differentiation, genomic stability and tumorigenesis [8-11].
To investigate the functions of Smurfs in vivo under physiological settings, mouse models with genetic modification of Smurf1 and Smurf2 have been generated [9, 12, 13]. In these conventional knock-out mice, Smurf1 or Smurf2 is disrupted in all cells, which prevents the effort for further studying of the role of Smurfs in specific types of cells and their corresponding cellular and molecular mechanisms. Furthermore, although mice that lack either Smurf1 or Smurf2 are viable, loss of both Smurfs leads to early embryonic lethality [9, 12, 13], which prevents us from studying the role of Smurfs in later embryonic development and diseases in adult stages such as aging related diseases. To overcome these disadvantages, we have generated a Smurf2 conditional null allele, the Smurf2flox allele, using the Cre/LoxP system.
Materials and Methods
Generation of the Smurf2floxneo targeting construct
The targeting vector was constructed using Smurf2 genomic DNA isolated previously [12] and ploxPneo vector that contains a PGKneo cassette with two loxps at its both ends [14]. Briefly, a loxp site was first inserted into Smurf2 genomic DNA before the exon9, then a 7.96-kb Xba I fragment including exon 8-10 and flanking introns was inserted into the ploxPneo vector to generate the 5' homology recombination arm. The 3' homology recombination arm in the targeting vector was generated by inserting the 3.92-kb Xba I-BamH I fragment including exon 11 and flanking introns. The finished targeting construct, pLoxP-Smurf2floxneo, is shown in Figure 1A.
Generation of the Smurf2floxneo allele. (A) Strategy for generating the Smurf2 targeting vector and Smurf2floxneo (targeted) allele. Blue boxes represent exons. B, BamHI; X, XbaI; H, HpaI; N, NotI; p, primer. (B) PCR analysis of genomic ES cell DNAs for targeted events at 3' arm using primer p3 and p4. (C) PCR analysis of recombination events at 5' arm using primer p1 and p2.
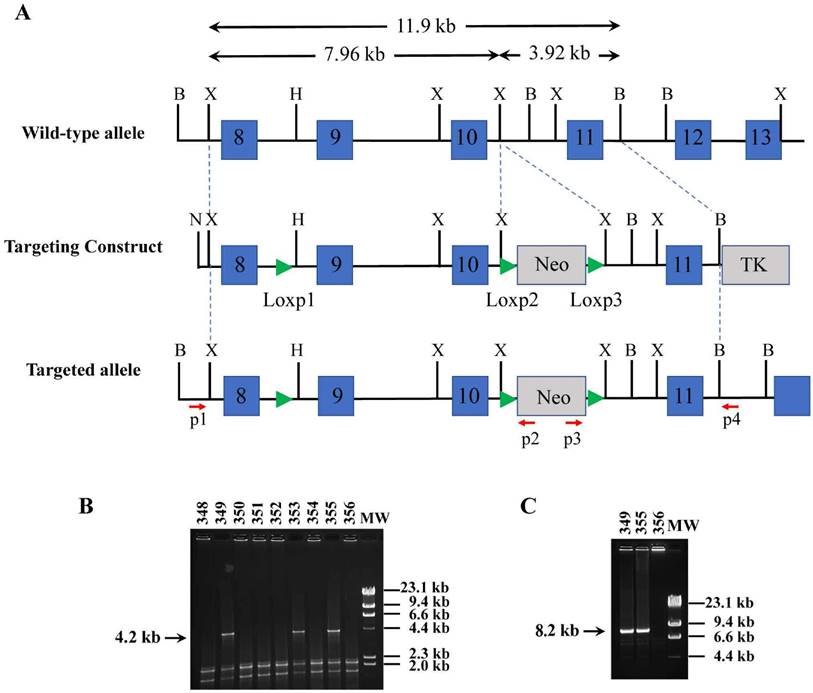
Transfection of ES cells and generation of germline chimeras
TC1 embryonic stem (ES) cells [15] were transfected with NotI-digested pLoxP-Smurf2floxneo by electroporation and selected using G418 and -(2-deoxy-2-fluoro-β-D-avabinofuranosyl-S-iodouracil (FIAU). G418- and FIAU- double resistant ES clones were screened for homologous recombination events within the Smurf2 locus by PCR. The 5' recombination was identified by PCR using the primer p1:( 5'- GCTTAGTGACCAAGTAGCAC -3') and p2:( 5'- CCAGACTGCCTTGGGAAAAGC-3'), which generated an 8.2-kb product. The 3' recombination was identified by PCR using the primer p3: (5'- CAGCTCATTCCTCCCACTC ATG -3') and p4: (5'- TCCTTTGCCTCCCAAGACTG -3'), which generated a 4.2-kb product. The targeted sites and integration of three Loxp sites were also confirmed by sequencing the above PCR products. Recombinant ES cells heterozygous for the targeted mutation were microinjected into blastocysts from C57/B6 mice to obtain germ-line transmission following standard procedures. The F1 agouti offspring were generated from breeding chimeric male with C57BL/6 female, and the presence of the Smurf2floxneo allele were analyzed by PCR. All mice were maintained and handled according to protocols approved by the Animal Care and Use Committee of the National Cancer Institute and National Institute of Diabetes, Digestive and Kidney Diseases, NIH.
Establish a conditional allele for Smurf2 and genotype analysis
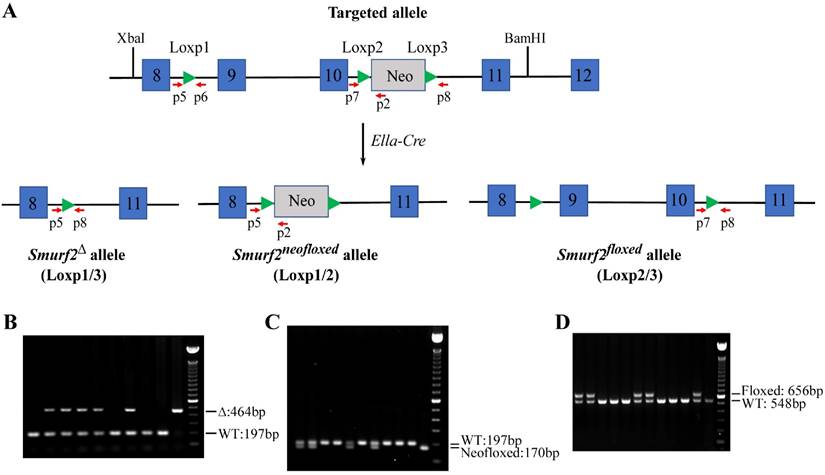
To establish exon 9 and exon 10 floxed Smurf2 allele, Neo cassette were removed by crossing mice with targeted allele with Ella-Cre transgenic mice [16]. Genotypes of the mice bearing the Smurf2∆, Smurf2neofloxed, Smurf2flox and wild-type allele were determined by PCR analysis as illustrated in Figure 2. The primer sequences are: p5: 5'- GATTGCAGGTTTGTGACACC-3', p6: 5'- AGACAACTCAGAGGAAGAGG, p7: 5'- GAGGTCAGGGATGATTTT GG, p8: 5'- ACAAGGGTTTACCAGTGCTC. Genomic DNA from Smurf2flox/flox mice were sequenced to confirm correct targeted sites and two LoxP sites after removing of the Neo cassette. Mice carrying Alb-Cre were purchased from Jackson Labs (www.jax.org/strain/003574). The Cre transgene was detected using primer set of 5'- CCTGTTTTGCACGTTCACCG-3' and 5'- ATGCTTCTGTCCGTTTGCC G -3'.
Validation of exons 9-10 and neo gene in Smurf2floxneo targeted alleles deleted by Cre recombinase. (A) Map of Smurf2floxneo allele. The position of primers was marked in the map. (B-D) Mice containing Smurf2floxneo alleles were crossed with EIIa-Cre transgenic mice resulting in three kinds of deletion between three Loxp sites. Position of primers was also marked. (B) Primer p5, p6 and p8 amplify fragments of 456 bp from Smurf2∆ allele with deletion exons 9-10 and neo cassette between Loxp1 and Loxp3 (Loxp1/3), and a 197 bp fragment from the wild type (WT) allele. (C) Primer p2, p5 and p6 amplify fragments of 170 bp from Smurf2neoflox allele with deletion exons 9-10 between Loxp1 and 2 (2/3), and a 197 bp fragment from the WT allele. (D) Primer p7 and p8 amplified a 656 bp fragment from Smurf2flox allele with deletion neo between Loxp2 and 3 (Loxp2/3), and a 548 bp fragment from the WT allele.
Isolation of mouse embryonic fibroblasts (MEFs)
Embryonic fibroblasts were generated by standard techniques. Briefly, E14.5 embryos were dissected individually, all inner organs, head, limb and tail removed and remaining tissues minced by cutting. After incubation in 2.5 ml trypsin at 37°C for 5-10 minutes, cells were resuspended and plated on 10 cm plates in DMEM supplemented with 10% FBS. After three passages, homogenous fibroblast cells were apparent and used for experiments.
Adenovirus infection of MEFs
Recombinant adenoviruses, Ad-GFP and Ad-Cre were obtained from Vector BioLabs (Malvern, PA). Infection of MEFs was conducted in 6-well plates in DMEM supplemented with 5% FBS at a multiplicity of infection (MOI) of 100. After 24 hours of infection, cells were recovered in the regular culture medium for additional 24 hours. Cells were then treated with TGF-β (4 ng/ml) or BMP2 (100 ng/ml), BMP4 (50 ng/ml) for 1 hour after being starved for 2 hours in DMEM containing 0.2% FBS.
Western blot
MEFs grown on six wells were harvested in lysis buffer containing 25 mM Tris-HCl, pH 7.5, 300 mM NaCl and 1% Triton with protease and phosphatase inhibitors. Tissues were minced by a Dounce homogenizer using 1-2 ml RIPA buffer. A total of 30 µg of protein were separated on 10% SDS-PAGE and transferred to PVDF membranes. After blocking of the membranes by 5% dry milk/TTBS, membranes were incubated in following primary antibody solutions: anti-Smurf1 (Novus, 1D7); anti-Smurf2 (Abcam, EP629Y3); anti-Smad1 (Cell Signaling, 9743); anti-Smad2 (Abcam, EP784Y); anti-Smad3 (Abcam, ab28379), anti-Smad5 (Abcam, EP619Y), anti-phospho-Smad1/5 (Cell Signaling, 41D10), anti-phospho-Smad2 (Cell Signaling, 138D4); anti-phospho-Smad3 (Rockland, 600-401-919), GAPDH (Santa Cruz, 0411), HSC70 (Santa Cruz, B-6). Protein detection was carried out using HRP-coupled species-specific secondary antibodies and ECL solution, exposed to Hyperfilm ECL.
Luciferase reporter assay
TGF-β (CAGA12-Luc), BMP4 (BRE-Luc), Sonic hedgehog (Shh) (8XGli-Luc) and Wnt-3a (TOPflash) reporter assays were carried out in MEFs. After transfected with the firefly luciferase reporters and the Renilla luciferase control reporter and incubated for 24 hours, Smurf2flox/flox or Smurf1-/-;Smurf2flox/flox MEFs were infected with either control adenovirus expressing LacZ or adenovirus expressing Cre for 2 days, and treated with different ligands for 16 hours. The signal of firefly luciferase was normalized to that of Renilla luciferase. The fold activation was calculated with dividing the relative luciferase unit (RLU) of the treated group by that of the untreated group. Results are shown as relative fold change ± S.D. (error bars) (n = 3 or 4).
Results and Discussion
To construct the Smurf2floxneo allele, we have made a targeting vector that contains three Loxp sites, the first two Loxp sites flanking the entire exons 9 and 10 (Figure 1A). In this way, existence of Cre recombinase would lead to deletion of exons 9 and 10, and create a premature stop codon that terminates protein translation prior to the last two highly conserved WW domains and the HECT ubiquitin ligase domain. This targeting strategy is the same as the conventional Smurf2 knockout mice that we generated previously, in which the expression of the truncated peptide is very low and does not have a dominant function [12]. The neo cassette was flanked by the last two Loxp sites, which enable us to remove the neo cassette by Cre recombinase as well. After transfecting the Not I-linearized targeting vector pLoxP-Smurf2floxneo into TC1 ES cells, four targeted ES clones were detected by PCR (Figure 1B and 1C) and confirmed by sequencing. Two positive ES clones were microinjected into blastocysts harvested from C57/BL6 mice to generate male chimeric mice, which were bred with female C57BL/6 mice to produce F1 agouti offsprings carrying one copy of the targeted Smurf2floxneo allele.
To test whether neo gene and exons 9-10 in Smurf2floxneo allele can be deleted by the Cre recombinase and to generate a neo-less conditional knockout allele, heterozygous (Smurf2floxneo/+) mice were crossed with EIIa-Cre transgenic mice that express Cre recombinase in germline [17]. The offspring were identified by PCR with primers shown in Figure 2A. As expected, we found that this crossing breeding generated three types of alleles due to Cre-mediated recombination among three Loxp sites. A 464 bp fragment was amplified from Smurf2Δ allele with deletion of neo gene and exons 9-10 between Loxp1 and Loxp3 (Figure 2B). Smurf2neofloxed allele with deletion of exons 9-10 was detected by producing an amplified 170 bp fragment (Figure 2C). The PCR-amplified 656 bp fragment indicated the excision of the neo gene, which produced Smurf2flox allele (conditional knockout allele) in Smurf2flox/+ heterozygous mice (Figure 2D).
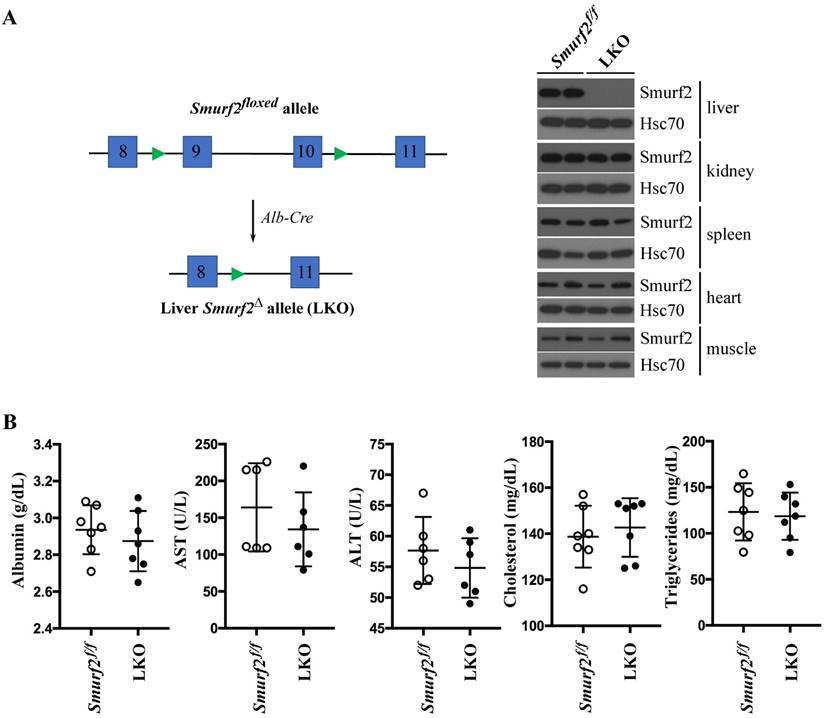
To verify gene inactivation in tissue specific manner in mice, we generated homozygous Smurf2flox/flox mice, then crossed them with mice that carry an Alb-Cre transgene [18] to assess the activity of Smurf2 conditional knockout allele in liver tissues (Figure 3A). Western blot analysis indicated that the Cre-mediated deletion occurs only in liver tissue, but not in other tissues where the Alb-Cre transgene is not expressed, such as the kidney, spleen, heart and muscle (Figure 3A). These data indicate that the Smurf2flox allele can be recombined to delete exons 9-10 in a tissue-specific fashion, thus the Smurf2 conditional mouse model should be very useful for studying Smurf2 functions during later stages of embryonic development. Like the Smurf2 knockout mice [19], histological analysis of H&E stained liver sections also did not reveal any apparent phenotype up to 12 months of age (data not shown). In addition, the liver function and lipid metabolism in these hepatocyte-specific Smurf2 knockout mice, (Alb-Cre;Smurf2flox/flox or LKO), were normal (Figure 3B).
Generation of liver specific Smurf2 knockout mice. (A) Liver-specific inactivation of the Smurf2 conditional allele by Alb-Cre transgene was revealed by Western blot analysis of liver extracts from homozygous Smurf2flox/flox or LKO (Alb-Cre;Smurf2flox/flox) mice. (B) No significance difference was observed in liver function: albumin, alanine transaminase (ALT) and aspartate transaminase (AST), or serum lipid content: triglyceride and cholesterol, between male adult Smurf2flox/flox and LKO mice.
We also isolated MEFs from Smurf2flox/flox embryos. After infection of these MEFs with a control adenovirus expressing GFP (Ad-GFP) or adenovirus expressing Cre recombinase (Ad-Cre), we obtained the control wild type MEFs regarding to Smurf2 expression, Ad-GFP;Smurf2flox/flox, and MEFs with Smurf2 deficiency, Ad-Cre;Smurf2flox/flox (Figure 4A). We treated these cells with TGF-β, BMP2 or BMP4, and did not observe any significant difference in Smad protein level or p-Smad levels among these cells (Figure 4A and 4B). However, when subjected to reporter assays, we found that TGF-β-dependent (CAGA)12-Luc activity were much higher in Smurf2 deficiency MEFs (Figure 4C). These results were consistent with previous reports that Smurf2 does not directly target TGF-β family of receptors or Smads for polyubiquitination and degradation, instead it regulates the sensitivity of the TGF-β family signaling via monoubiquitination that disrupts formation of Smad complex [12, 20]. Smurfs were also reported to regulate many different pathways other than TGF-β and BMPs. For example, in sonic hedgehog (Shh) pathways, Smurfs promote Ptch1 to undergo polyubiquitination and degradation, thereby, activate Gli1-mediated transcription [10]; while in canonical Wnt signaling, Smurf1 and Smurf2 were shown to mediate ubiquitination of Axin, a negative regulator of Wnt/β-catenin signaling [21, 22]. We found that Smurf2 deficiency severely inhibited Shh-mediated Gli1-dependent 8xGli1-Luc activity and Wnt3-mediated β-catenin/Tcf reporter TOPFLASH activity (Figure 4D).
Multiple signaling pathways were affected by Smurf2 deficiency. MEFs from Smurf2flox/flox embryos were infected with adenovirus expressing Cre to remove Smurf2 alleles. Control cells were infected with adenovirus expressing GFP or LacZ. (A-B) Smurf2 deficiency did not affect Smad protein level or ligand-stimulated C-terminal Smad phosphorylation in MEFs. Cells were treated with (A) TGF-β or (B) BMP-2 or BMP-4 for 1 hour. Whole cell lysates were subjected to Western blot analysis using specific antibodies as indicated. (C) Smurf2 deficiency significantly increased TGF-β (CAGA12-Luc)-dependent transcription reporter activities, but not much on BMP (BRE-Luc)-dependent activities. (D) Smurf2 deficiency inhibited Shh-induced 8xGli1-Luc and Wnt3A-induced TOPFLASH reporter activities. Results are shown as relative fold change ± S.D. (error bars) (n = 3 or 4). Statistically significant difference (p < 0.01) after ligand treatment is indicated by *; statistically significant differences (p <0.01) between control and Smurf2 deficient cells after ligand treatment are indicated by #, respectively.

Acknowledgements
Research in Y.E. Zhang's group is supported by the intramural research program of the US National Institutes of Health, US National Cancer Institute, Center for Cancer Research; Research in C. Deng's group was supported by the intramural program of US National Institutes of Health, US National Institute of Diabetes, Digestive and Kidney Diseases.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26:399-422
2. Scheffner M, Kumar S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochimica et biophysica acta. 2014;1843:61-74
3. Imamura T, Oshima Y, Hikita A. Regulation of TGF-beta family signalling by ubiquitination and deubiquitination. J Biochem. 2013;154:481-9
4. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH. et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365-75
5. Lin X, Liang M, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem. 2000;275:36818-22
6. Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci U S A. 2001;98:974-9
7. Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400:687-93
8. David D, Nair SA, Pillai MR. Smurf E3 ubiquitin ligases at the cross roads of oncogenesis and tumor suppression. Biochimica et biophysica acta. 2013;1835:119-28
9. Narimatsu M, Bose R, Pye M, Zhang L, Miller B, Ching P. et al. Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell. 2009;137:295-307
10. Yue S, Tang LY, Tang Y, Tang Y, Shen QH, Ding J. et al. Requirement of Smurf-mediated endocytosis of Patched1 in sonic hedgehog signal reception. eLife. 2014:3
11. Zou X, Levy-Cohen G, Blank M. Molecular functions of NEDD4 E3 ubiquitin ligases in cancer. Biochimica et biophysica acta. 2015;1856:91-106
12. Tang LY, Yamashita M, Coussens NP, Tang Y, Wang X, Li C. et al. Ablation of Smurf2 reveals an inhibition in TGF-beta signalling through multiple mono-ubiquitination of Smad3. Embo J. 2011;30:4777-89
13. Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX. et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell. 2005;121:101-13
14. Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998;95:3667-72
15. Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911-21
16. Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E. et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860-5
17. Xu X, Li C, Garrett-Beal L, Larson D, Wynshaw-Boris A, Deng CX. Direct removal in the mouse of a floxed neo gene from a three-loxP conditional knockout allele by two novel approaches. Genesis. 2001;30:1-6
18. Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM. et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305-15
19. Blank M, Tang Y, Yamashita M, Burkett SS, Cheng SY, Zhang YE. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nature medicine. 2012;18:227-34
20. Zhou F, Xie F, Jin K, Zhang Z, Clerici M, Gao R. et al. USP4 inhibits SMAD4 monoubiquitination and promotes activin and BMP signaling. EMBO J. 2017;36:1623-39
21. Fei C, Li Z, Li C, Chen Y, Chen Z, He X. et al. Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/beta-catenin signaling. Mol Cell Biol. 2013;33:4095-105
22. Kim S, Jho EH. The protein stability of Axin, a negative regulator of Wnt signaling, is regulated by Smad ubiquitination regulatory factor 2 (Smurf2). J Biol Chem. 2010;285:36420-6
Author contact
![]() Corresponding author: Ying E. Zhang, Laboratory of Cellular and Molecular Biology, Center for Cancer Research, National Cancer Institute, NIH, Building 37, RM 2056B, Bethesda, Maryland 20892-4256. Tel: (240) 760-6861; e-mail: zhangyinnih.gov
Corresponding author: Ying E. Zhang, Laboratory of Cellular and Molecular Biology, Center for Cancer Research, National Cancer Institute, NIH, Building 37, RM 2056B, Bethesda, Maryland 20892-4256. Tel: (240) 760-6861; e-mail: zhangyinnih.gov