Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Author contributions
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(5):557-564. doi:10.7150/ijbs.24546 This issue Cite
Research Paper
PM2.5 induces autophagy-mediated cell death via NOS2 signaling in human bronchial epithelium cells
Xiao-Ming Zhu1, Qin Wang2, Wei-Wei Xing1, Min-Hui Long1, Wen-Liang Fu1, Wen-Rong Xia1, Chen Jin1, Ning Guo1, Dong-Qun Xu2 ![]() , Dong-Gang Xu1
, Dong-Gang Xu1 ![]()
1. Institute of Military Cognitive and Brain Sciences, Academy of Military Medical Sciences, Beijing, 100850, China
2. Institute for Environmental Health and Related Product Safety, China, CDC, Beijing, 100021, China
Received 2017-12-25; Accepted 2018-2-16; Published 2018-4-5
Abstract

The biggest victim of ambient air pollution is the respiratory system. Mainly because of the harmful components, especially the particulate matters with an aerodynamic diameter of ≤ 2.5µm (PM2.5), can be directly inhaled and deeply penetrate into the lung alveoli, thus causing severe lung dysfunction, including chronic cough, bronchitis and asthma, even lung cancer. Unfortunately, the toxicological mechanisms of PM2.5 associations with these adverse respiratory outcomes have still not been clearly unveiled. Here, we found that PM2.5 rapidly induced inflammatory responses, oxidative injure and cell death in human bronchial epithelium cells through upregulation of IL-6 expression, ROS production and apoptosis. Furthermore, PM2.5 specifically induced nitric oxide synthase 2 (NOS2) expression and NO generation to elevate excessive autophagy. Finally, disruption of NOS2 signaling effectively blocked autophayosome formation and the subsequent cell death. Our novel findings systemically reveled the role of autophagy-mediated cell death in PM2.5-treated human bronchial epithelium cells and provided potential strategy for future clinic intervention.
Keywords: Particulate matters two point five (PM2.5), Human bronchial epithelium cells, Autophagy-mediated cell death, NOS2 signaling, Clinic intervention
Introduction
Overexposed to the urban air pollution causes severe respiratory health problem of the susceptible population, especially the and the aged people [1-7]. The major component of urban air pollution is the impalpable particulate matter (PM) [8, 9]. Physically, those suspending particulate with aerodynamic diameter ≤ 2.5 mm (PM2.5) can be directly inhaled into the bronchus and deposited in the epithelium cells of the deepest pulmonary alveoli to cause primary or secondary disease of human body. In the past decade, numerous studies had exerted the detrimental effects of PM2.5 on respiratory system, including the bronchitis, chronic cough, asthma and even lung cancer [10-12]. However, the underlying toxicological mechanisms of PM2.5 associated with these lung dysfunction is still not thoroughly elucidated, which makes subsequent prevention and clinical treatment fewer effective.
Preliminarily studies reveal that many pro-inflammatory cytokines, such as interleukin 1 (IL1), interleukin 6 (IL6), interleukin 8 (IL8) and tumor necrosis factor (TNFα) are transiently or persistently secreted when exposure to PM2.5 in different types of cells [13-15]. Other cytological effects, like oxidative injury, aging and immoderately apoptosis are also reported [16-21]. These adverse factors may finally integrate to result in respiratory diseases. Interestingly, one recent study surprisingly shows that massive autophagy is induced in cardiovascular cells upon PM2.5 treatments. Besides, recent study further defines a form of autophagy-mediated cell death in chronic lung diseases, which showing an elevated autophagy level with progressing programmed cell death (PCD) [22, 23]. However, the exactly role of autophagy and the underlying mechanism of autophagy-mediated cell death in PM2.5-induced diseases is still unknown.
In the normal physiological condition, autophagy orderly degrades impaired cytoplasmic components to maintain intercellular homeostasis through lysosome pathway [24]. But under various pathophysiological conditions, improper autophagy leads to unpredictable and finally promotes cellular senescence and morbidity [25]. Many signaling, such as AMPK and mTOR, are widely considered to precisely regulate autophagy upon nutrient starvation or growth factors stimulation [26]. While recently, the nitric oxide synthases (NOS) is also found to participate in regulation of autophagy [27]. In mammals, NOS family encodes three distinct isozymes: neuronal (NOS1), cytokine-inducible (NOS2) and endothelial (NOS3) [28]. It has been suggested that increased NOS2 production contributes to cell toxicity under many harmful stimuli. But the role of NOS2 in regulation of PM2.5-induced autophagy and cell death is still unrevealed.
In this study, we demonstrated that PM2.5 rapidly induced inflammatory responses, oxidative injure and cell death in human bronchial epithelium cells. Moreover, we found that PM2.5 specifically induced NOS2 expression and NO generation to elevate excessive autophagy. Disruption of NOS2 signaling effectively reduced autophagy level and subsequent cell death. Thus, our novel findings clearly reveled the mechanism of autophagy-mediated cell death in PM2.5-treated human bronchial epithelium cells and provided potential strategy for future clinic application.
Materials and Methods
Cell Culture
Human bronchial epithelial cells (BEAS-2B) were kindly provided by Dr. Chuanshu Huang as previously reported [29]. Briefly, the cells were cultured in DMEM (high glucose, Gibco, 12800-017) with 10% fetal bovine serum (Life Technologies, 16010-109) supplemented with antibiotic/antimycotic (Life Technologies, 15240-062) and incubated at 37℃ in 5% CO2 incubator.
Antibody and reagents
Anti-NOS1(YT3168), anti-NOS2 (YT3169) and anti-NOS3 (YT3171) were purchased from Immunoway; anti-LC3B (#3868) was purchased from Cell signaling technology; Anti-p-mTOR (YP0176) and anti-Caspase3 (YC0006), anti-p-PI3K (YP0765) were purchased from Immunoway. Anti-AMPK (YM3520), anti-mTOR (YT6120), anti-AMPK (YM3520), and anti-ERK (YM3516) were purchased from Immunoway. Anti-p-AMPK (YP0010) and Anti-p-ERK (YM1464) were also purchased from Immunoway. The PM2.5 samples were collected in Beijing city (China) as previously reported [29, 30]. Briefly, ambient PM2.5 was sampled between the 2nd and 3rd ring road of Beijing city, from October to December 2016. Then the filter was cut into 1 cm2 area of pieces, immersed in sterilized water and sonicated three times to extract the water-soluble components, followed by lyophilization and storage at -80℃. A certain quantity of PM2.5 was suspended and homogenized in DMEM supplemented with 2% FBS for using. The NOS2 siRNA (target sequence: 5'-GCAGAAUCCUUCAUGAAGUTT-3'), and control siRNA (target sequence: 5'-UGACCUCAACUACAUGGUUTT-3') were from GenePharma.
IL-6, NO and ROS detection
Cells were seeded into 96-well plates and cultured to 70% to 80% confluence. Then the cells were treated with PM2.5 at different doses or times. After treated for 24 hours, the supernatants were collected and analyzed the release of cytokines by using Human IL-6 ELISA Kit (Immunoway). For Nitric Oxide (NO) detection, the supernatants were analyzed by using Nitric Oxide (NO) assay kit (Nitrate reductase method, A012-1, Nanjing JianCheng Bioengineering Institute). For Reactive oxygen species (ROS) detection (E004, Nanjing JianCheng Bioengineering Institute), cells were trypsinized and stained with Reactive oxygen species Assay Kit, followed by Flow cytometry (FACS) analysis.
Western blot assay
After PM2.5 treatments, BEAS-2B cells were lysed with ice-cold RIPA buffer. The whole cell lysis was collected and separated by SDS-PAGE. Then the blots were incubated with the appropriate antibodies and were detected as indicated.
Flow cytometry analysis
Cells were fixed by 4% (vol/vol) paraformaldehyde or 70% ethanol (vol/vol). Then the ratio of apoptotic cells was detected by FITC Annexin V Apoptosis Detection Kit (BD pharmingenTM) according to the instruction manual.
Autophagy assay
For Immunofluorescence staining, BEAS-2B cells were fixed by 4% (vol/vol) paraformaldehyde. After washing, cells were stained with LC3B antibody and FITC-conjugated secondary antibody. Then cells were counted under the confocal microscope (ZEISS, LSM510 META) and scored for positive LC3B staining. And the presence of autophayosome in bronchial epithelial cells was directly assessed by transmission electron microscopy (TEM)-based analysis (TEM, Hitachi, H7650).
Statistical Analysis
All experiments were performed at least three times. All data were presented as the mean standard deviation (SD), and analyzed using Student's t test and one-way analysis of variance. p < 0.05 was considered statistically significant.
Results
PM2.5 induced cell death, ROS generation and IL6 expression in BEASE-2B cells
To determine the adverse role of PM2.5 on human bronchial epithelial cells, we first treated BEASE-2B epithelial cell line with different concentration of PM2.5, which was collected from the main streets of Beijing city, China (from October to December 2016). As shown in Fig. 1A, morphological characteristic of cell death was clearly emerged as the gradually increasing concentration of PM2.5 treatments. While the MTT proliferation assay also showed that the cell reproductive capacity was significantly decreased after a series of PM2.5 treatments (Fig.1B). Using Annexin-V/PI double staining assay, we further demonstrated that PM2.5 remarkably but moderately induced apoptosis of BEASE-2B cells (Fig.1C and D). These results confirmed that PM2.5 generally triggered cell apoptosis as previous reported. Most interestingly, we surprisingly found that the cellular level of reactive oxygen species (ROS) and interleukin 6 (IL6) were dramatically increased after high concentration of PM2.5 treatments in a short period (Fig.1E and F). Therefore, we speculated that the rapidly induction of oxidative injure and extensive inflammatory reaction in human bronchial epithelial cells might eventually contribute to the majority of cell death [31].
PM2.5 induced cell death, ROS generation and IL6 expression in BEASE-2B cells. (A) Representative pictures showed the morphological cell death after a different concentration of PM2.5 treatments. (B) The MTT assay showed the decreased proliferation ability after a different concentration of PM2.5 treatments. Data was calculated from 3 independent experiments (Error bar, mean ± s.d.). *p<0.05, **p<0.01, ***p<0.001. (C) Representative FACS results showed the increased apoptosis rate of BEASE-2B cells after a different concentration of PM2.5 treatments. (D) The apoptosis ratio was counted in C. Data was calculated from 3 independent experiments (Error bar, mean ± s.d.). *p<0.05, **p<0.01, ***p<0.001. (E) The level of ROS was detected at indicated times after 100μg/mL PM2.5 treatments. Data was calculated from 3 independent experiments (Error bar, mean ± s.d.). *p<0.05, **p<0.01, ***p<0.001. (F) The level of IL6 was detected at indicated times after 100μg/mL PM2.5 treatments. Data was calculated from 3 independent experiments (Error bar, mean ± s.d.). *p<0.05, **p<0.01, ***p<0.001.

Autophagy level was significant elevated in PM2.5-treated BEASE-2B cells
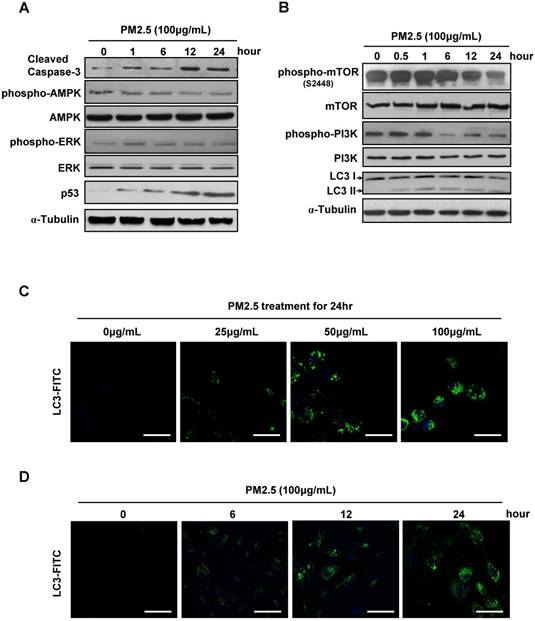
Except for massive ROS generation and sharply immunoreaction, we next investigated the more accurate mechanism underlying the PM2.5-induced cell death. Consistently, the caspase-3 and p53 expression level were gradually increased after high concentration of PM2.5 treatments, reflecting the upregulation of apoptosis in BEASE-2B cells. Meanwhile, the phosphorylation levels of AMPK and REK were slightly changed, suggesting not the stress signaling pathway but more specific signaling pathways might be activated to possibly initiate cell apoptosis (Fig.2A). In contrast, we notably found that the phosphorylation levels of mTOR and PI3K were significantly decreased, while the autophagosome marker LC3-II was continuous accumulated after high concentration of PM2.5 treatments (Fig.2B). These results suggested that PM2.5 severely suppressed the metabolism pathways and subsequently induced autophagy formation in BEASE-2B cells. Immunofluorescence staining of endogenous LC3-II further clearly showed the elevated autophagy level in BEASE-2B cells after different concentration of PM2.5 treatments (Fig.2C). Moreover, the autophagy was quickly emerged after a high concentration of PM2.5 treatments, suggesting autophagy might act as a rapid response mechanism in BEASE-2B cells when exposed to PM2.5 (Fig.2D).
Autophagy level was significant elevated in PM2.5-treated BEASE-2B cells. (A and B) Immunoblot analysis of BEASE-2B cells at indicated times after 100μg/mL PM2.5 treatments. (C) Representative immunofluorescence images showed the increased autophagy level after a different concentration of PM2.5 treatments. (D) Representative immunofluorescence images showed the increased autophagy level at indicated times after 100μg/mL PM2.5 treatments.
PM2.5 induced NO production and NOS2 expression in BEASE-2B cells
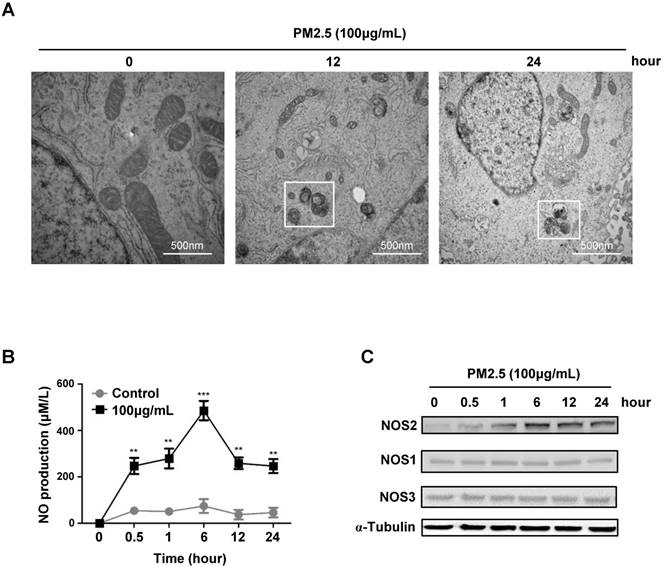
To further investigate whether the elevated autophagy level was related to PM2.5-induced epithelial cells death. We performed the high resolution transmission electron microscopy scaning, and immediately found the apparently formation of autophagosome after a high concentration of PM2.5 treatments, while the global programmed cell death phenotype was simultaneously appeared in the cellular content, like blebbing, cell shrinkage, karyopyknosis, mitochondrial atrophy (Fig.3A). These data further proved the possibility that PM2.5-induced autophagy might lead to cell death and urged us to explore the underlying mechanisms that regulate of autophagy-mediated cell death. Previous study showed that the nitrogen signaling could active autophagy in certain cells, we then detected and indeed found that the production of nitric oxide (NO) level was dramatically increased after a high concentration of PM2.5 treatments (Fig.3B). Most interestingly, we further found the induction of NO was specifically mediated by the upregulation of NOS2 expression (Fig.3C). Collectively, these results suggested that PM2.5-induced autophagy-mediated cell death might mainly though the activation of NOS2-NO signaling axis.
PM2.5 induced NO production and NOS2 expression in BEASE-2B cells. (A) Representative TEM images showed a high-magnification of double-membrane autophagic vesicles in BEASE-2B cells after a different concentration of PM2.5 treatments. Double-membrane autophagic vesicles were indicated in white squares. (B) The level of NO was detected at indicated times after 100μg/mL PM2.5 treatments. Data was calculated from 3 independent experiments (Error bar, mean ± s.d.). *p<0.05, **p<0.01, ***p<0.001. (C) Immunoblot analysis of NOS expression in BEASE-2B cells at indicated times after 100μg/mL PM2.5 treatments.
PM2.5 induced autophagy-mediated cell death through NOS2 signaling
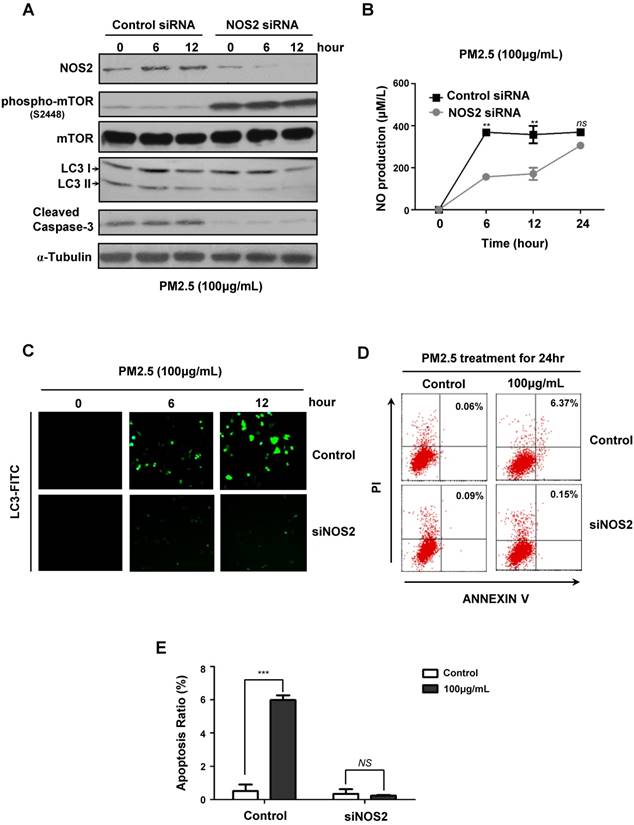
To further verify the critical role of NOS2-NO signaling in PM2.5-induced autophagy and subsequent cell death, we next knocked-down NOS2 expression by using siRNA in BEASE-2B cells. When compared to control, the NOS2 protein level was efficiently reduced in NOS2 knocked-down cells, whereas the phosphorylation levels of mTOR was increased, suggesting the pro-survival signaling was reactivated (Fig.4A). Most importantly, after NOS2 knocked-down, the expression level of autophagyosome marker LC3-II was significantly eliminated, following the cell apoptosis marker caspase-3 was immediately diminished when compared to control (Fig.4A). These data firstly and comprehensively revealed the unexpected role of NOS2 in promoting PM2.5-induced autophagy and autophagy-mediated cell death in bronchial epithelial cells. Meanwhile, the NO level was also significantly reduced in NOS2 knocked-down cells, suggesting NO might act as a direct downstream effector molecule to trigger PM2.5-induced cell death (Fig.4B). Consistently, immunofluorescence staining further demonstrated that autophagy level was clearly decreased in NOS2 knocked-down cells, suggesting the specific role of autophagy in PM2.5-induced cell death (Fig.4C). Furthermore, FACS analysis clearly showed the dramatically decreased level of apoptosis in NOS2 knocked-down cells when exposed to PM2.5 (Fig.4D and E). Taken together, our study in the first time reveled the detailed mechanism of autophagy-mediated cell death in PM2.5-treated human bronchial epithelium cells.
PM2.5 induced autophagy-mediated cell death through iNOS signaling. (A) Immunoblot analysis of NOS2 knockdown and control BEASE-2B cells at indicated times after 100μg/mL PM2.5 treatments. (B) The level of NO was detected in NOS2 knockdown and control BEASE-2B cells at indicated times after 100μg/mL PM2.5 treatments. Data was calculated from 3 independent experiments (Error bar, mean ± s.d.). *p<0.05, **p<0.01, ***p<0.001. (C) Representative immunofluorescence images showed the decreased autophagy level in NOS2 knockdown cells when compared to control at indicated times after 100μg/mL PM2.5 treatments. (D) Representative FACS results showed the decreased apoptosis rate in NOS2 knockdown BEASE-2B cells. (E) The apoptosis ratio was counted in D. Data was calculated from 3 independent experiments (Error bar, mean ± s.d.). *p<0.05, **p<0.01, ***p<0.001.
Discussion
With the development of the heavy industries and less control of the sources of pollution, the urban air is becoming continually over-contaminated. The growing morbidity of respiratory system caused by the inhalant particulate matters (PMs) in the urban air pollution is widely recognized, but the toxicological mechanism underlying these lung dysfunction is still not systemically revealed. Hugh efforts has been made to uncover the substantial function of PM2.5 on respiratory system, and several regulation mechanisms are constantly discovered, including sharply oxidative injure and excessive immune response, which finally lead to programmed cell death. Traditionally, autophagy is deemed to orderly degrade impaired cytoplasmic components and reuse these metabolites to maintain intercellular homeostasis and subsistence. Interestingly, recent studies indicate that excessive autophagy may lead to cell death. How autophagy triggers cell death and the underlying mechanism of autophagy-mediated cell death is still unknown. The upstream signal molecule triggering autophagy is also unknown. Although previously studies indicated the contradictory role of nitric oxide (NO) in autophagy formation [32, 33], the exactly role of NO in autophagy under PM2.5 exposure is largely unrevealed.
In our study, we demonstrated that PM2.5 rapidly induced autophagosome formation and subsequently cell death in human bronchial epithelium cells. We further found that PM2.5 specifically induced NOS2 expression and NO generation to elevate excessive autophagy. Most importantly, disruption of NOS2 signaling effectively blocked autophayosome formation and subsequently cell death. But how NO triggers autophagosome formation is still needed to explored. Moreover, due to complexity component of PM2.5, the exactly constituent caused cell death is also needed for further analysis. Except for PM2.5 induced cell death, another important aspect is that whether PM2.5 can also trigger human lung cell aging, as abnormal cell dysfunction always along with cell aging [34]. In sum, our novel findings partially reveled the mechanism of autophagy-mediated cell death that induced by PM2.5 in human bronchial epithelium cells and provided a potential target to remit the PM2.5-induced respiratory diseases.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (No.81170255) and Beijing Municipal Natural Science Foundation (BJNSF) (No.7164285).
Author contributions
M.Z. performed experiments and interpreted data. Q.W. collected the experiments materials. W.X., H. L., L.F., and R.X. participated in molecular biology experiments. C.L. performed and analyzed the FACS data; N.G. supervised the collaborations. Q.X. and G.X. conceived of and designed the studies, supervised the work, and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schikowski T, Sugiri D, Reimann V. et al. Contribution of smoking and air pollution exposure in urban areas to social differences in respiratory health. BMC public health. 2008;8:179
2. van der Zee S, Hoek G, Boezen HM. et al. Acute effects of urban air pollution on respiratory health of children with and without chronic respiratory symptoms. Occupational and environmental medicine. 1999;56:802-12
3. Calderon-Garciduenas L, Engle R, Mora-Tiscareno A. et al. Exposure to severe urban air pollution influences cognitive outcomes, brain volume and systemic inflammation in clinically healthy children. Brain and cognition. 2011;77:345-55
4. Calderon-Garciduenas L, Franco-Lira M, Henriquez-Roldan C. et al. Urban air pollution: influences on olfactory function and pathology in exposed children and young adults. Experimental and toxicologic pathology: official journal of the Gesellschaft fur Toxikologische Pathologie. 2010;62:91-102
5. Calderon-Garciduenas L, Mora-Tiscareno A, Franco-Lira M. et al. Decreases in Short Term Memory, IQ, and Altered Brain Metabolic Ratios in Urban Apolipoprotein epsilon4 Children Exposed to Air Pollution. Journal of Alzheimer's disease: JAD. 2015;45:757-70
6. Charpin D, Penard-Morand C, Raherison C. et al. [Long-term exposure to urban air pollution measured through a dispersion model and the risk of asthma and allergy in children]. Bulletin de l'Academie nationale de medecine. 2009;193:1317-28 discussion 28-9
7. Segala C. Health effects of urban outdoor air pollution in children. Current epidemiological data. Pediatric pulmonology Supplement. 1999;18:6-8
8. Li P, Xin J, Wang Y. et al. Association between particulate matter and its chemical constituents of urban air pollution and daily mortality or morbidity in Beijing City. Environmental science and pollution research international. 2015;22:358-68
9. Bauer M, Moebus S, Mohlenkamp S. et al. Urban particulate matter air pollution is associated with subclinical atherosclerosis: results from the HNR (Heinz Nixdorf Recall) study. Journal of the American College of Cardiology. 2010;56:1803-8
10. Badyda AJ, Grellier J, Dabrowiecki P. Ambient PM2.5 Exposure and Mortality Due to Lung Cancer and Cardiopulmonary Diseases in Polish Cities. Advances in experimental medicine and biology. 2017;944:9-17
11. Kam W, Delfino RJ, Schauer JJ, Sioutas C. A comparative assessment of PM2.5 exposures in light-rail, subway, freeway, and surface street environments in Los Angeles and estimated lung cancer risk. Environmental science Processes & impacts. 2013;15:234-43
12. Xing YF, Xu YH, Shi MH, Lian YX. The impact of PM2.5 on the human respiratory system. Journal of thoracic disease. 2016;8:E69-74
13. Yan Z, Wang J, Li J. et al. Oxidative stress and endocytosis are involved in upregulation of interleukin-8 expression in airway cells exposed to PM2.5. Environmental toxicology. 2016;31:1869-78
14. Wang YL, Gao W, Li Y, Wang YF. Concentration-dependent effects of PM2.5 mass on expressions of adhesion molecules and inflammatory cytokines in nasal mucosa of rats with allergic rhinitis. Eur Arch Otorhinolaryngol. 2017;274:3221-9
15. Monn C, Becker S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicology and applied pharmacology. 1999;155:245-52
16. Li R, Kou X, Xie L. et al. Effects of ambient PM2.5 on pathological injury, inflammation, oxidative stress, metabolic enzyme activity, and expression of c-fos and c-jun in lungs of rats. Environmental science and pollution research international. 2015;22:20167-76
17. Dong C, Song WM, Shi YW. [Study on the oxidative injury of the vascular endothelial cell affected by PM2.5]. Wei sheng yan jiu = Journal of hygiene research. 2005;34:169-71
18. Ding A, Yang Y, Zhao Z. et al. Indoor PM2.5 exposure affects skin aging manifestation in a Chinese population. Scientific reports. 2017;7:15329
19. Nwanaji-Enwerem JC, Dai L, Colicino E. et al. Associations between long-term exposure to PM2.5 component species and blood DNA methylation age in the elderly: The VA normative aging study. Environment international. 2017;102:57-65
20. Wang W, Deng Z, Feng Y. et al. PM2.5 induced apoptosis in endothelial cell through the activation of the p53-bax-caspase pathway. Chemosphere. 2017;177:135-43
21. Hu R, Xie XY, Xu SK. et al. PM2.5 Exposure Elicits Oxidative Stress Responses and Mitochondrial Apoptosis Pathway Activation in HaCaT Keratinocytes. Chinese medical journal. 2017;130:2205-14
22. Ropolo A, Bagnes CI, Molejon MI. et al. Chemotherapy and autophagy-mediated cell death in pancreatic cancer cells. Pancreatology. 2012;12:1-7
23. Lee TG, Jeong EH, Kim SY. et al. The combination of irreversible EGFR TKIs and SAHA induces apoptosis and autophagy-mediated cell death to overcome acquired resistance in EGFR T790M-mutated lung cancer. International journal of cancer. 2015;136:2717-29
24. Monastyrska I, Klionsky DJ. Autophagy in organelle homeostasis: peroxisome turnover. Molecular aspects of medicine. 2006;27:483-94
25. Ulasov IV, Shah N, Kaverina NV. et al. Tamoxifen improves cytopathic effect of oncolytic adenovirus in primary glioblastoma cells mediated through autophagy. Oncotarget. 2015;6:3977-87
26. Munson MJ, Ganley IG. MTOR, PIK3C3, and autophagy: Signaling the beginning from the end. Autophagy. 2015;11:2375-6
27. Ratovitski EA. DeltaNp63alpha/IRF6 interplay activates NOS2 transcription and induces autophagy upon tobacco exposure. Archives of biochemistry and biophysics. 2011;506:208-15
28. Yadav VP, Dangi SS, Chouhan VS. et al. Expression analysis of NOS family and HSP genes during thermal stress in goat (Capra hircus). International journal of biometeorology. 2016;60:381-9
29. Xu X, Wang H, Liu S. et al. TP53-dependent autophagy links the ATR-CHEK1 axis activation to proinflammatory VEGFA production in human bronchial epithelial cells exposed to fine particulate matter (PM2.5). Autophagy. 2016;12:1832-48
30. Xia WR, Fu W, Wang Q. et al. Autophagy Induced FHL2 Upregulation Promotes IL-6 Production by Activating the NF-kappaB Pathway in Mouse Aortic Endothelial Cells after Exposure to PM2.5. International journal of molecular sciences. 2017:18
31. Pinheiro da Silva F, Nizet V. Cell death during sepsis: integration of disintegration in the inflammatory response to overwhelming infection. Apoptosis: an international journal on programmed cell death. 2009;14:509-21
32. Sarkar S, Korolchuk VI, Renna M. et al. Complex inhibitory effects of nitric oxide on autophagy. Molecular cell. 2011;43:19-32
33. Xaus J, Comalada M, Valledor AF. et al. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood. 2000;95:3823-31
34. Qiao Y, Li Q, Du HY. et al. Airborne polycyclic aromatic hydrocarbons trigger human skin cells aging through aryl hydrocarbon receptor. Biochemical and biophysical research communications. 2017;488:445-52
Author contact
![]() Corresponding authors: Dong-Gang Xu, E-mail: xudgac.cn and Dong-Qun Xu, E-mail: dongqunxucom.
Corresponding authors: Dong-Gang Xu, E-mail: xudgac.cn and Dong-Qun Xu, E-mail: dongqunxucom.