Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(5):577-585. doi:10.7150/ijbs.22220 This issue Cite
Research Paper
Chemical and genetic inhibition of STAT3 sensitizes hepatocellular carcinoma cells to sorafenib induced cell death
Linna Xie1,2*, Yanhua Zeng1*, Zichan Dai3, Wensheng He2, Huozhao Ke2, Qiaofa Lin1, Yan Chen4, Jingjing Bu1, Dexin Lin1 ![]() , Min Zheng 1,3
, Min Zheng 1,3 ![]()
1. Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
2. Department of Food and Bioengineering, Fujian Vocational College of Bioengineering, Fuzhou, Fujian, 350007, China.
3. Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, Fujian Medical University, Fuzhou, Fujian, 350108, China.
4. Key Laboratory of Ministry of Education for Gastrointestinal Cancer, Fujian Medical University, Fuzhou, Fujian, 350108, China.
*These authors contribute equally to this work.
Received 2017-8-3; Accepted 2018-3-1; Published 2018-4-25
Abstract

Hepatocellular carcinoma (HCC) has become the second leading cause of cancer related death, with an increasing death rate in recent years. For advanced HCC, sorafenib is the first-line FDA approved drug, with no more than 3 months' overall survival advantage. Recently, a novel strategy has been proposed to improve sorafenib efficacy through enhancing the ability of sorafenib to induce cell death. STAT3 plays a key role in cancer development and recurrence by promoting cell proliferation, survival and immune evasion through its well-established function as a transcription factor in cancer. Notably, STAT3 transcription activity, indicated by its phosphorylation on Y705 is heterogeneous in different liver cancer cell lines. And sorafenib attenuates STAT3 phosphorylation on Y705. However, the role of STAT3 in sorafenib induced cell death is still largely unknown. Here, we show that liver cancer cells also exhibit heterogeneous sensitivities to sorafenib induced cell death, which co-relates with the STAT3-Y705 phosphorylation levels and JAK1/2 expression levels in Hep3B, Huh7 and HepG2 cells. Furthermore, overexpression or knockdown of STAT3 could switch HCC cells between resistant and sensitive to sorafenib induced cell death, which could be partially due to its regulation on Mcl-1, an anti-apoptotic protein. Finally, both inhibitors of STAT3 SH2 domain (S3i-201) or STAT3 upstream kinases JAKs (JAK inhibitor I) could synergistically enhance sorafenib induced cell death. Taken together, these data strongly suggest that STAT3 is not only a downstream effector of sorafenib, but also a key regulator of cellular sensitivity to sorafenib induced cell death, which provide support for the notion to develop STAT3-targeting drugs to improve clinical efficacy of sorafenib in liver cancer.
Keywords: STAT3, HCC, Sorafenib, Cell death
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common cancer worldwide, often accompanied by chronic liver disease due to viral hepatitis, alcohol abuse or non-alcoholic steatohepatitis [1]. Because liver cancer is an extraordinary heterogeneous malignant disease [2] and could be induced by multiple etiologies[3], treatment of liver cancer is usually unsatisfied and the five-year survival rate is only reaching 26% [4]. Until 2012, liver cancer has become the second leading cause of cancer related death [5].
The first line FDA-approved targeted drug for advanced hepatocellular carcinoma (HCC) is sorafenib, which was originally developed as an inhibitor targeting on Raf kinase, a key regulator of cell proliferation [6]. Pharmacological profile showed that sorafenib could also block multiple kinases including VEGFRs, PDGFRβ, c-Kit and RET in addition to Raf family members [7]. This property confers sorafenib as a powerful drug simultaneously targeting on multiple oncogenic kinases. As expected, preclinical data showed its competent inhibitory effect on tumor formation and progression both ex-vivo andin vivo, and FDA approved sorafenib as a targeted drug for advanced HCC in 2007 [8]. However, the clinical benefit of sorafenib treatment is still limited with no more than 3 months overall survival advantage [9].
The molecular mechanisms underlying sorafenib resistance in HCC are still largely unknown. Notably, sorafenib treated cancers rarely completely regressed and that sorafenib alone did not induce massive cell death [9]. Indeed, as a single agent, sorafenib weakly induced cell death, either apoptosis [10] or ferroptosis [11] in cell-type and drug-dose dependent manner. However, in combination with chemical inhibitors or shRNAs targeting on key regulators, sorafenib induces strong cell death in multiple HCC cell lines [12-15], indicating a potential to improve sorafenib efficacy against HCC through enhancing its ability to induce cell death. Therefore, identifying the key regulators and strategies to enhance sorafenib induced cell death might be the key to meet the challenges of advanced HCC treatment.
STAT3 (Signal Transducer and Activator of Transcription 3) is a pivotal transcriptional factor of multiple tumor promoting genes and anti-apoptotic genes, like Mcl-1 (myeloid cell leukemia sequence 1) [16-18]. In response to growth factors and inflammatory factors, STAT3 is activated through phosphorylation on 705th tyrosine and 727th serine, leading to its trans-localization from cytoplasm to nucleus [19]. In liver cancer cells, Sorafenib dephosphorylates and inhibits STAT3 activity through activating phosphatase SHP2 [13, 20], leading to down-regulation of Mcl-1 [21]. RFX-1/SHP-1 activation could also overcome sorafenib resistance by dephosphorylating STAT3 [22]. However, the role of STAT3 in determining cellular sensitivity to sorafenib induced cell death in HCC is still largely unknown.
Here, we show that HCC cell lines exhibit heterogeneous STAT3-Y705 phosphorylation and cellular sensitivities to sorafenib induced cell death. STAT3-Y705 is constitutively phosphorylated in sorafenib resistant Huh7 and HepG2 cells, but not in sorafenib sensitive Hep3B cells. Over-expression of STAT3 confers Hep3B cells with resistance to sorafenib induced cell death. Reversely, genetic knockdown of STAT3 could sensitize Huh7 cells to sorafenib induced cell death. Supportively, anti-apoptotic Mcl-1 was down regulated in STAT3 knocked down Huh7 and HepG2 cells and up-regulated in STAT3 overexpressed Hep3B cells. Importantly, inhibitors of STAT3 itself by S3i-201 or its upstream kinase JAKs by JAK inhibitor I (JAKi) could synergistically enhance sorafenib induced cell death in both Huh7 and HepG2 cells. Taken together, our data suggest that STAT3 contribute to sorafenib resistance, and inhibition of STAT3 could improve sorafenib efficacy by enhancing sorafenib induced cell death in liver cancer.
Results
STAT3 contributes to cellular resistance to sorafenib induced cell death in HCC cells
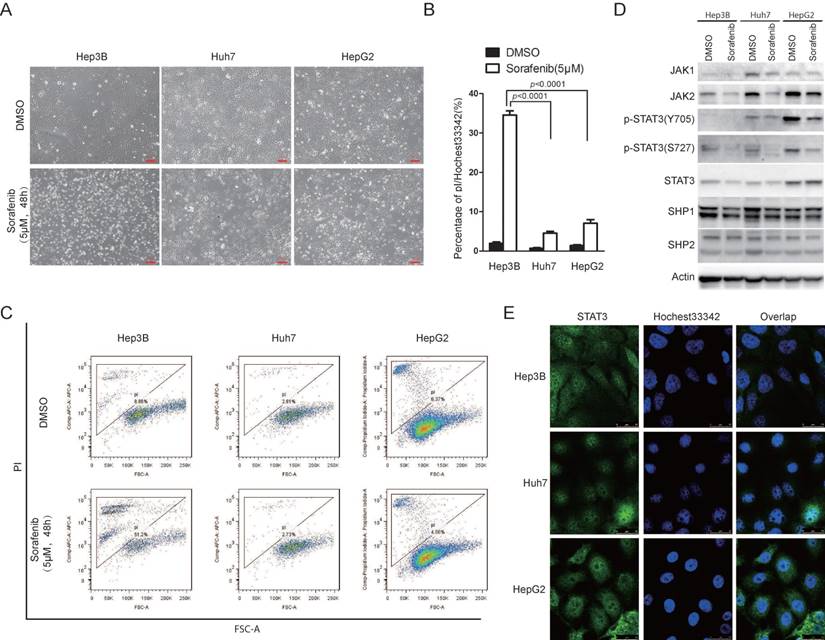
Sorafenib is the first FDA approved targeted drug for advanced hepatocellular carcinoma [9]. However, the efficacy of sorafenib in HCC is still limited [9]. Recently, accumulating evidence showed that sorafenib could not only inhibit cell growth, but also be able to induce cell death [23]. In the light of heterogeneity of HCC cells, we examined the effect of sorafenib to induce cell death in different HCC cell lines. We found that different HCC cell lines exhibited heterogeneous sensitivities to sorafenib induced cell death. Hep3B cells were more sensitive to sorafenib induced cell death compared to Huh7 and HepG2 cells, indicated by microscopy observation (Figure 1A), pI/Hoechst staining assay (Figure 1B) and pI staining followed by FACS analysis (Figure 1C). Interestingly, STAT3 Y705 phosphorylation was much higher in Huh7 and HepG2 cells than that in Hep3B cells (Figure 1D). Supportively, STAT3 nuclear localization was much more intensive in Huh7 and HepG2 cells (Figure 1E). Since tyrosine kinase JAK and phosphatase SHP family are well-known regulators of STAT3 Y705 phosphorylation, we also examined the expression of JAK1/2 and SHP1/2. As shown in figure 1D, JAK1/2 expression was also lower in Hep3B cells than that in Huh7 and HepG2 cells. These data indicate that STAT3 activities might play a key role in cellular sensitivity to sorafenib induced cell death in HCC cells.
JAK1/2-STAT3 pathway is associated with cellular sensitivity to sorafenib induced cell death. (A, B, C) Hep3B, HepG2 and Huh7 cells were treated with 5uM Sorafenib for 48hours. Cell death was monitored under microscopy (A), and cell death assay was performed by staining with pI and Hoechst 33342 followed by photograph under microscopy and analyzed by Image J (B) or staining with pI and analyzed by flow cytometry (C). (D) The expressing levels of JAK1, JAK2, STAT3, SHP1, SHP2, actin and phosphorylation levels of STAT3 were determined by western blot using the antibodies, respectively. (E) STAT3 localization was examined in Hep3B, Huh7 and HepG2 cells.
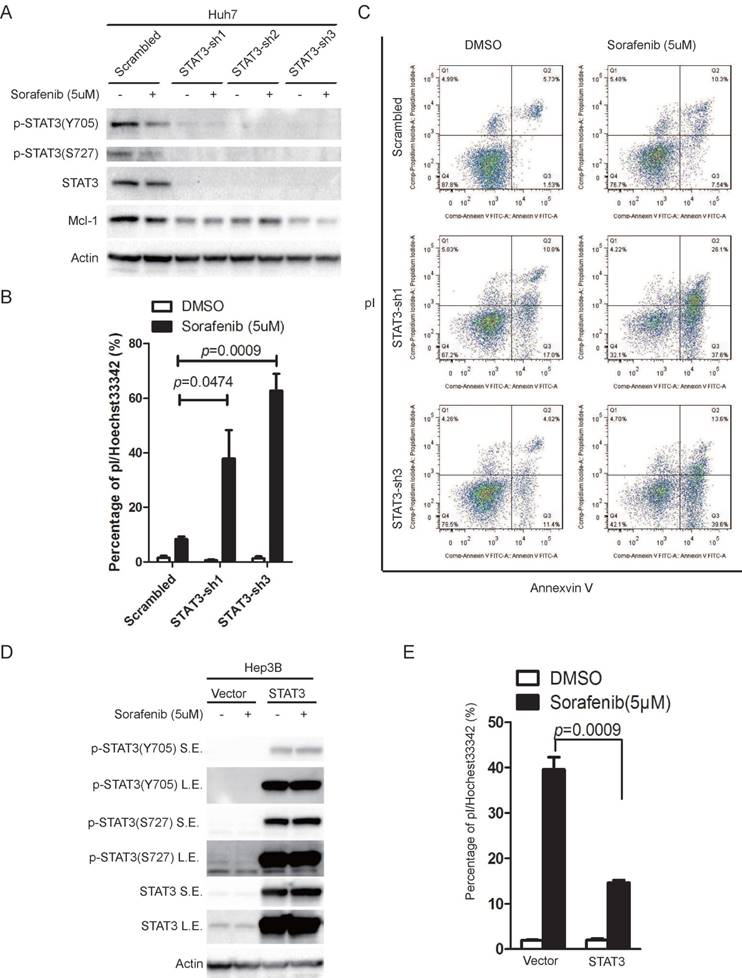
In order to determine the role of STAT3 in sorafenib induced cell death in HCC cells, we knocked down STAT3 in Huh7 cells and over-expressed STAT3 in Hep3B cells, using a lentivirus based system. The ability of sorafenib to induce cell death was monitored. Knockdown of STAT3 reduced STAT3 protein levels and phosphorylation on Y705 or S727 (Figure 2A), while sorafenib induced cell death was significantly increased, indicated by pI/Hoechst staining assay (Figure 2B), and double confirmed by pI/Annexin V staining followed by FACS analysis (Figure 2C). Inversely, over expression of STAT3 in Hep3B increased STAT3 protein levels and phosphorylation on Y705 or S727 (Figure 2D), while sorafenib induced cell death was dramatically attenuated (Figure 2E).
STAT3 contributes to cellular resistance to sorafenib induced cell death. (A) STAT3 was knocked down in Huh7 cells using lentivirus expressing STAT3 shRNAs. Cells were treated with or without 5uM sorafenib for 48 hours. The expression levels of STAT3 and its phosphorylation levels were determined. (B) Huh7 cells expressing scramble shRNA or STAT3 shRNAs were treated with 5uM sorafenib for 72 hours, and then stained with pI and Hoechst, photographed under microscopy and analyzed by Image J. (C) Cells were treated as in (B), and then stained with pI/Annexin V and analyzed by flow cytometry. (D) STAT3 was over-expressed in Hep3B cells using lentivirus expressing STAT3. Cells were treated with or without 5uM sorafenib for 24 hours. The expression levels of STAT3 and its phosphorylation levels were determined. S.E. stands for short exposure, while L.E. represents long exposure. (E) Hep3B cells expressing STAT3 and its parallel control were treated with 5uM sorafenib for 48hours. Cell death assay was performed as in (B). Data of three independent replicates are presented as the mean+/-s.e.m., n=3.
Taken together, these data demonstrate that STAT3 plays a key role in determining the cell sensitivity to sorafenib induced cell death in HCC cells.
STAT3 protects HCC cells from sorafenib induced cell death through regulating Mcl-1 expression
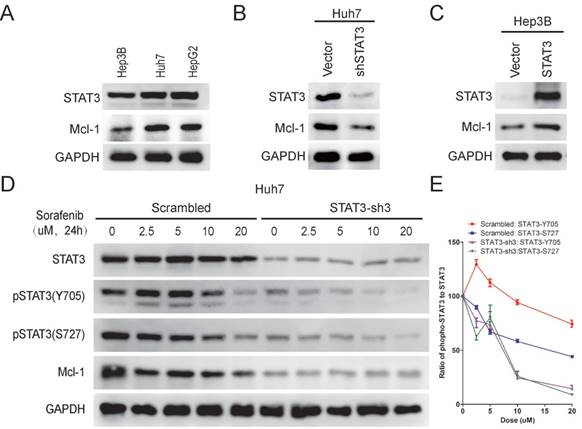
STAT3 has been well known to regulate the expression of Mcl-1 [20, 24], a member of Bcl-2 family, which plays a key role in anti-apoptosis [25]. It has also been shown that sorafenib treatment leads to down-regulation of Mcl-1[20]. Therefore, we examined Mcl-1 expression in Hep3B, Huh7 and HepG2 cells. As shown in Figure 3A, Mcl-1 expression was higher in Huh7/HepG2 cells than Hep3B cells, which correlated with their STAT3-Y705 phosphorylation levels and sensitivities to sorafenib induced cell death. Moreover, knockdown of STAT3 reduced Mcl-1 expression in Huh7 cells (Figure 3B), while over expression of STAT3 increased Mcl-1 expression in Hep3B cells (Figure 3C). Furthermore, sorafenib induced down-regulation of Mcl-1 in Huh7 cells, but not in STAT3 knocked-down Huh7 cells, indicating the essential role of STAT3 in sorafenib induced down regulation of Mcl-1 (Figure 3D). Taken together, these data indicates that STAT3 might protect HCC cells from sorafenib induced cell death through regulating Mcl-1 expression.
STAT3 protects HCC cells from sorafenib induced cell death through regulating Mcl-1 expression. (A) The expression levels of STAT3, Mcl-1 and GAPDH in Hep3B, Huh7 and HepG2 were determined by Western Blot using anti-STAT3, anti-Mcl-1 and anti-GAPDH antibodies, respectively. (B) STAT3 was knocked down in Huh7 cells. The expression levels of Mcl-1 and STAT3 levels were determined. (C) STAT3 was over-expressed in Hep3B cells. The expression levels of Mcl-1 and STAT3 were determined. (D, E) STAT3 was knocked down in Huh7 cells. Cells were then treated with different doses of sorafenib for 24hours. The expression levels of Mcl-1, STAT3 and its phosphorylation levels were determined (D). The ratio of pSTAT3 to STAT3 was quantified using Image J (E).
Inhibition of STAT3 enhances sorafenib induced cell death in HCC cells
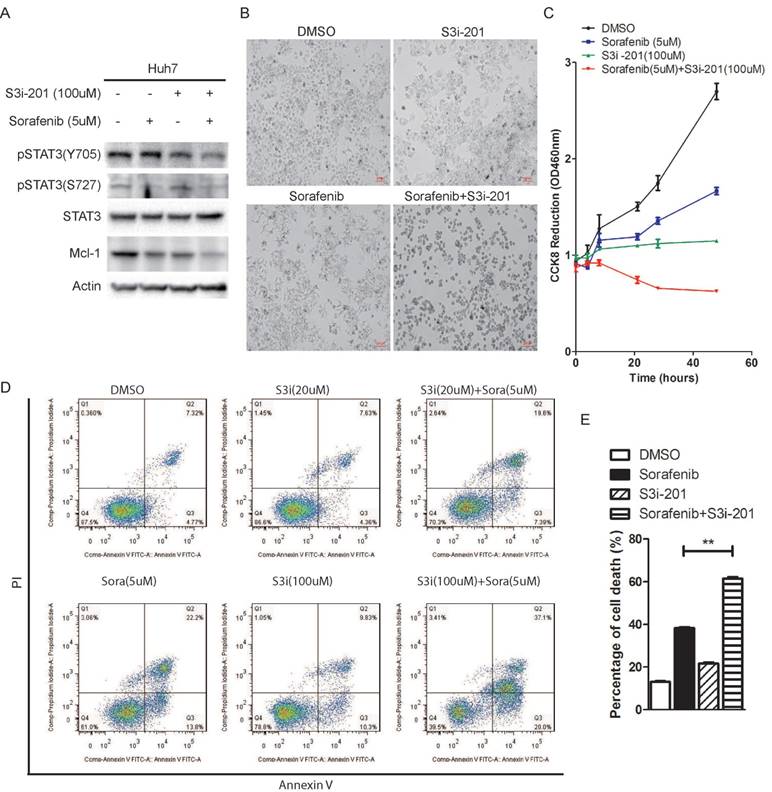
In order to investigate the effect of STAT3 inhibition on sorafenib induced cell death, we used a STAT3 inhibitor, S3i-201, which was identified through a structure-based virtual screening from the NCI chemical libraries [26]. Huh7 cells were treated with Sorafenib in the absence or presence of S3i-201. As shown in figure 4A and 4B, combination of S3i-201 with sorafenib lead to down-regulation of STAT3 phosphorylation and Mcl-1 expression, and shrinkage of Huh7 cells, while sorafenib or S3i-201 alone lead to reduction of cell density. Consistently, tetrazolium based cell growth assay showed that sorafenib or S3i-201 alone lead to inhibition of cell growth, while combination of both drugs completely blocked cell growth, as shown by CCK8 assay (Figure 4C). Annexin V/pI staining also showed that combination of S3i-201 could dramatically enhance the ability of sorafenib to induce cell death (Figure 4D and 4E).
STAT3 inhibitor S3i-201 synergistically enhances sorafenib induced cell death. (A) Huh7 cells were treated with S3i-201 (100uM), sorafenib (5uM) or its combination for 24 hours. The expression of STAT3 and Mcl-1, and the phosphorylation level of STAT3 were determined. (B, C) Huh7 cells were treated with 100uM S3i-201 (100uM), Sorafenib (5uM) or their combination for 48 hours. Cell death was monitored by microscopy (B). Cell metabolic activities were monitored by CCK8 assay (C). Data of three independent replicates are presented as the mean+/-s.e.m., n=3. (D, E) Huh7 cells were treated with 5uM sorafenib, 20uM or 100uM S3i-201, or their combination. Cell death assay was performed by pI/Annexin V staining and followed by flow cytometry analysis. Data of three independent replicates are presented as the mean+/-s.e.m., n=3.
Janus kinase (JAK) family members are well known as STAT3 upstream kinases responsible for STAT3 phosphorylation on tyrosine 705. Indeed, JAK inhibitor I (JAKi) alone lead to de-phosphorylation of STAT3 on Y705 [27], but not S727 (Figure 5A and 5C). Long term sorafenib treatment alone resulted in de-phosphorylation on both Y705 and S727 phosphorylation on STAT3 (Figure 5A and 5C), as shown previously [20]. Combinational treatment of sorafenib with JAKi could lead to synergistic de-phosphorylation of STAT3 and Mcl-1 down-regulation in both Huh7 and HepG2 cells (Figure 5A and 5C). Meanwhile, either sorafenib or JAKi alone had mild effect on cell death, while combination of sorafenib and JAKi dramatically increase the rate of cell death in both Huh7 and HepG2 cells (Figure 5B and 5D). By contrast, sorafenib alone could lead to drastic cell death in Hep3B cells where STAT3 phosphorylation on Y705 is barely detectable, while the effect of combinational treatment to induce cell death was much modest (Figure 5E and 5F). Furthermore, the synergistic effect of sorafenib and JAK inhibitor was also double confirmed using pI staining followed by FACS analysis (Figure 5G). Notably, although long-term sorafenib treatment alone could effectively down-regulate STAT3 phosphorylation levels, there are still residual p-STAT3 (Y705) and p-STAT3(S727) left. Only combination of sorafenib and STAT3 inhibitors could dramatically induce cell death, but not sorafenib alone, indicating that residual p-STAT3(Y705) and p-STAT3(S727) might still protect HCC cells from sorafenib induced cell death. In addition, STAT3 inhibitors might synergistically contribute to sorafenib sensitivities through abrogating the residual STAT3 activities.
JAK inhibitor I enhanced sorafenib induced cell death in STAT3 active HCC cells. (A, C, E) HCC cells were treated with 2uM JAK inhibitor I (JAKi), 5uM Sorafenib or their combination. The expression levels of STAT3, phospho-STAT3 (Y705) phospho-STAT3 (S727), Mcl-1 and GAPDH were determined by Western Blot. (B, D, F) Cell death assays were performed by pI staining and photographed under microscopy. Cell death rate was calculated by Image J. Data of three independent replicates are presented as the mean+/-s.e.m., n=3. (G) Huh7 cells were treated with JAKi (2uM), Sorafenib (5uM) and their combination for 60hours. Cell death assay was performed by pI staining and followed by flow cytometry analysis.
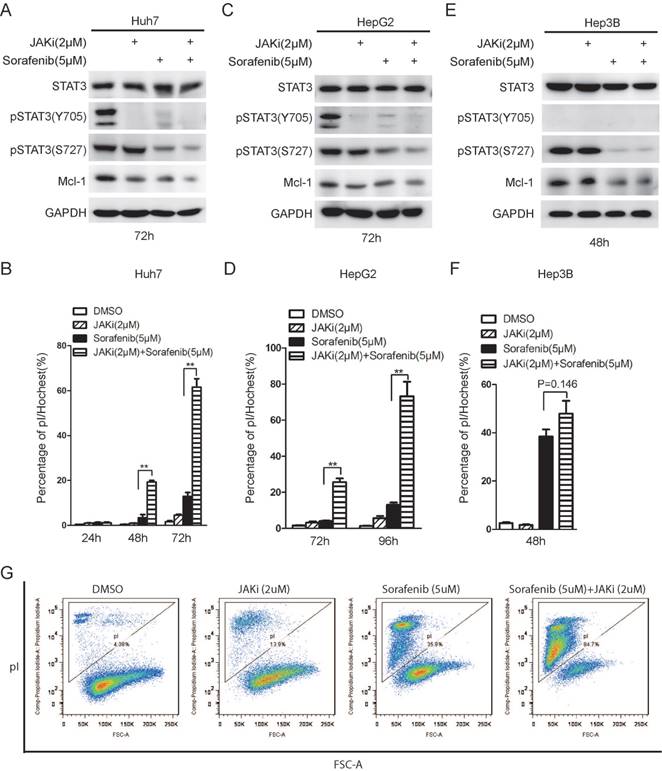
Taken together, these data suggest that STAT3 inhibition could synergistically improve efficacy by enhancing sorafenib induced cell death.
Discussion
In contrast to most cancers, the incidence and death rate of liver cancer is increasing, in part due to the lack of novel and potent drugs and treatments. The first-line targeted drug for advanced HCC is sorafenib, which was approved by FDA in 2007 [9]. However its overall clinical benefit is still limited [28]. Thus, one of the major challenges for biomedical studies of sorafenib is to improve its efficacy against HCC. Accumulating evidences show that sorafenib is capable to induce cell death in high dose [21]. Therefore, identification of biomarkers and regulators in determining HCC cell sensitivity to sorafenib induced cell death might contribute to clinical benefit from sorafenib.
In this study, we show that three HCC cell lines (Hep3B, Huh7 and HepG2) response to sorafenib induced cell death in a different way. Hep3B with low STAT3 Y705 phosphorylation is much more sensitive to sorafenib induced cell death than HepG2 and Huh7 cells, where STAT3 is highly phosphorylated on Y705. Importantly, up-regulation of STAT3 increases resistance to sorafenib induced cell death in Hep3B cells, while depletion of STAT3 reduces cellular resistance in Huh7 and HepG2 cells, indicating STAT3 plays a pivotal role in protecting HCC cells from sorafenib induced cell death.
STAT3 inactivation is accompanied with down-regulation of multiple anti-apoptotic proteins, such as Mcl-1, survivin and Bcl-XL [16]. These genes serve as protective shield against spear of sorafenib. Indeed, our study also shows that Mcl-1 is tightly controlled by STAT3 expression. In concert with previous works, our data also show that sorafenib attenuates STAT3 phosphorylation on both 727th serine and 705th tyrosine, indicating STAT3 activities could be suppressed by sorafenib [20]. Taken together, it suggests that STAT3 might serve as a “bumper” to protect cancer cells from sorafenib. Recent researches identified many key factors playing similar roles in sorafenib induced cell death, such as Erk, AIB1 and Pin1 [29-31]. The expression or activities of these genes are also down regulated by sorafenib, while genetic or chemical inhibition of these genes could enhance sorafenib induced cell death. Interestingly, STAT3 could be phosphorylated by Erks [32], while AIB1 and STAT3 are documented to be regulated by Pin1 [33, 34], indicating that a network of genes might play a role of “bumper” in sorafenib induced cell death.
One might easily propose that attenuating or eradicating the bumper might enhance sorafenib efficacy. Indeed, STAT3 inhibitor or STAT3 upstream kinase JAK inhibitor could synergistically enhance sorafenib induced cell death. Supportively, combinational treatment with other drugs, such as all-trans retinoic acid (ATRA) [15, 31] or bufalin [30] could dramatically enhance the ability of sorafenib to induce cell death in HCC cells. These findings strengthen the potential strategy to improve sorafenib efficacy by combinational treatment and enhancing sorafenib induced cell death in liver cancer cells.
Methods and Materials
Cell culture and Reagents
Huh7, HepG2 and Hep3B cells were bought from Cell Bank of Chinese Academy of Sciences. Cells were cultured in high glucose DMEM (#12800-017; GIBCO) with 10% FBS (#10437-028; GIBCO). All the cells were cultured in 37 degree centigrade with 5% CO2. GAPDH antibody (#HC301) and Cell Counting Kit-8 (CCK-8) (#FC101) were from Transgene (Beijing, China). Antibodies of anti STAT3 (#9139), anti phospho-STAT3 at Y705 (#9145) or anti phospho-STAT3 at S727 (#94994) were from Cell Signaling (Davers, MA). Mcl-1 (#16225-1-AP), JAK1 (#66466-1-Ig), JAK2 (17670-1-AP), SHP1 (24546-1-AP), SHP2 (20145-1-AP) antibodies was from Proteintech (Wuhan, China). TurboFect Transfection Reagent (#R0532) was from Life Technologies. Sorafenib (#sc-220125) was purchased from Santa Cruz (Santa Cruz, CA) and Medchemexpress (#HY-10201, Shanghai, China). JAK inhibitor I (#420099) was from Millipore (Billerica, MA). Polybrene (#107689) and STAT3 inhibitor S3i-201 (#SML0330) was from Sigma (St. Louis, MO).
RNA interference
STAT3 shRNA expressing lentivirus was bought from Genechem (Shanghai, China). The shRNA sequences are 5'-GCTGACCAACAATCCCAAGAA-3' for STAT3-sh1, 5'- GCACAATCTACGAAGAATCAA-3' for STAT3-sh2, and 5'-GCAAAGAATCACATGCCACTT-3' for STAT3-sh3. To establish STAT3 knocked down cells, Huh7 and HepG2 cells were infected with lentivirus expressing STAT3 shRNA and supplemented with 4ug/ml polybrene.
Cell viability assay
Cells were seeded in a 96-well plate and were treated as indicated. 10ul of CCK-8 was added into 90ul DMEM culture medium. Cells were then incubated at 37℃ and 5% CO2 for 1 hour. Measure the absorbance using Multi-scan GO (Thermo Scientific) at 450nm with 620nm as reference wavelength.
Cell death assay
pI/Hoechst33342 double staining was described previously [35, 36]. Briefly, HCC cells were stained with 5ug/mL pI and 5ug/mL Hoechst33342 after treatment as indicated. Cells were photographed under fluorescence microscopy (Zeiss Axio Observer A1). pI positive cells were considered as dead cells. Hoechst33342 positive cells were considered as total cells. The number of pI or Hoechst positive cells was quantified using Image J (Wayne Rasband, National Institutes of Health, Bethesda, MD, USA), respectively. The percentage of cell death was quantified by pI positive cells divided by Hoechst33342 positive cells. Alternatively, cells were stained with pI and analyzed by flow cytometry as previously described [31]. Briefly, pI incorporation and cell size were quantified by flow cytometry. pI negative cells with normal size were considered as live cells. pI positive cells with smaller size were considered as dead cells. Annexin V/pI staining was performed following manual instruction of Annexin-V-FLUOS Staining kit (#11988549001) from Roche (Mannheim, Germany).
Statistical analysis
Data were expressed as means±s.e.m. The two sample t-test was used to compare differences between treated groups and their paired controls.
Acknowledgements
This research was supported by the Natural Science Foundation of Fujian Province (No. 2015J01293), the Educational Scientific Research Project of Young Teachers in Fujian Province (No. JA14410), the Science and Technology Planning Project of Fujian Province (No. 2014Y2008), the Natural Science Fund for Distinguished Young Scientist of Fujian Province (No. 2015J06017), the Joint Science and Technology Innovation Fund Project of Fujian Province (No. 2016Y9043), the Young and Middle-aged Key Personnel Training Program from Fujian Provincial Health and Family Planning Commission (No. 2017-ZQN-58), Fujian Environmental Protection Science and Technology Project (No. 2016R015), the Collaborative Innovation Center for Stem Cells Translational Medicine (Fujian 2011 Program).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Casadei Gardini A, Tenti E, Masini C, Nanni O, Scarpi E, Valgiusti M. et al. Multicentric survey on dose reduction/interruption of cancer drug therapy in 12.472 patients: indicators of suspected adverse reactions. Oncotarget. 2016
2. Li L, Wang H. Heterogeneity of liver cancer and personalized therapy. Cancer letters. 2015
3. Dhanasekaran R, Bandoh S, Roberts LR. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Research. 2016;5:879
4. Simard EP, Ward EM, Siegel R, Jemal A. Cancers with increasing incidence trends in the United States: 1999 through 2008. CA: a cancer journal for clinicians. 2012;62:118-28
5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66:7-30
6. Kolch W, Kotwaliwale A, Vass K, Janosch P. The role of Raf kinases in malignant transformation. Expert reviews in molecular medicine. 2002;4:1-18
7. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA. et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nature reviews Drug discovery. 2006;5:835-44
8. Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M. et al. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. Journal of Clinical Oncology. 2005;23:965-72
9. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F. et al. Sorafenib in advanced hepatocellular carcinoma. The New England journal of medicine. 2008;359:378-90
10. Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D. et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer research. 2006;66:11851-8
11. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B. et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. International journal of cancer. 2013;133:1732-42
12. Sun X, Niu X, Chen R, He W, Chen D, Kang R. et al. Metallothionein-1G Facilitates Sorafenib Resistance through Inhibition of Ferroptosis. Hepatology (Baltimore, Md). 2016
13. Hung M-H, Tai W-T, Shiau C-W, Chen K-F. Downregulation of signal transducer and activator of transcription 3 by sorafenib: a novel mechanism for hepatocellular carcinoma therapy. World Journal of Gastroenterology. 2014;20:15269-74
14. Guan DX, Shi J, Zhang Y, Zhao JS, Long LY, Chen TW. et al. Sorafenib enriches epithelial cell adhesion molecule-positive tumor initiating cells and exacerbates a subtype of hepatocellular carcinoma through TSC2-AKT cascade. Hepatology. 2015;62:1791-803
15. Ishijima N, Kanki K, Shimizu H, Shiota G. Activation of AMP-activated protein kinase by retinoic acid sensitizes hepatocellular carcinoma cells to apoptosis induced by sorafenib. Cancer science. 2015;106:567-75
16. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nature reviews Cancer. 2009;9:798-809
17. Timofeeva OA, Tarasova NI, Zhang X, Chasovskikh S, Cheema AK, Wang H. et al. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc Natl Acad Sci U S A. 2013;110:1267-72
18. Tai WT, Cheng AL, Shiau CW, Liu CY, Ko CH, Lin MW. et al. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3. Molecular cancer therapeutics. 2012;11:452-63
19. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nature reviews Cancer. 2014;14:736-46
20. Blechacz BRA, Smoot RL, Bronk SF, Werneburg NW, Sirica AE, Gores GJ. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology (Baltimore, Md). 2009;50:1861-70
21. Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS, Cheng AL. et al. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell death & disease. 2013;4:e485
22. Su JC, Tseng PH, Wu SH, Hsu CY, Tai WT, Li YS. et al. SC-2001 overcomes STAT3-mediated sorafenib resistance through RFX-1/SHP-1 activation in hepatocellular carcinoma. Neoplasia. 2014;16:595-605
23. Louandre C, Ezzoukhry Z, Godin C, Barbare J-C, Mazière J-C, Chauffert B. et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. International journal of cancer. 2013;133:1732-42
24. Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA. et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42:1329-38
25. Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS. et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104-9
26. Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR. et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391-6
27. Pedranzini L, Dechow T, Berishaj M, Comenzo R, Zhou P, Azare J. et al. Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer research. 2006;66:9714-21
28. Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA: a cancer journal for clinicians. 2012;62:394-9
29. Yuen JSP, Sim MY, Sim HG, Chong TW, Lau WKO, Cheng CWS. et al. Combination of the ERK inhibitor AZD6244 and low-dose sorafenib in a xenograft model of human renal cell carcinoma. International Journal of Oncology. 2012;41:712-20
30. Li M, Wang W, Dan Y, Tong Z, Chen W, Qin L. et al. Downregulation of amplified in breast cancer 1 contributes to the anti-tumor effects of sorafenib on human hepatocellular carcinoma. Oncotarget. 2016
31. Zheng M, Xu H, Liao XH, Chen CP, Zhang AL, Lu W. et al. Inhibition of the prolyl isomerase Pin1 enhances the ability of sorafenib to induce cell death and inhibit tumor growth in hepatocellular carcinoma. Oncotarget. 2017;8:29771-84
32. Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Molecular and cellular biology. 1997;17:6508-16
33. Yi P, Wu RC, Sandquist J, Wong J, Tsai SY, Tsai MJ. et al. Peptidyl-prolyl isomerase 1 (Pin1) serves as a coactivator of steroid receptor by regulating the activity of phosphorylated steroid receptor coactivator 3 (SRC-3/AIB1). Molecular and cellular biology. 2005;25:9687-99
34. Lufei C, Koh TH, Uchida T, Cao X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene. 2007;26:7656-64
35. Belloc F, Dumain P, Boisseau MR, Jalloustre C, Reiffers J, Bernard P. et al. A flow cytometric method using Hoechst 33342 and propidium iodide for simultaneous cell cycle analysis and apoptosis determination in unfixed cells. Cytometry. 1994;17:59-65
36. Guaitolini CR, Taffarel MO, Teixeira NS, Sudano MJ, Freitas PM, Lopes MD. et al. Post-thaw viability ofin vivo-produced canine blastocysts cryopreserved by slow freezing. Theriogenology. 2012;78:576-82
Author contact
![]() Corresponding authors: Min Zheng at mzhengfjmu.edu.cn and Dexin Lin at dexin9997com.
Corresponding authors: Min Zheng at mzhengfjmu.edu.cn and Dexin Lin at dexin9997com.