Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(6):586-598. doi:10.7150/ijbs.23256 This issue Cite
Research Paper
FGF23 and Fetuin-A Interaction in the Liver and in the Circulation
Deborah Mattinzoli1, Masami Ikehata1, Koji Tsugawa1, Carlo M Alfieri1,2, Paola Dongiovanni3, Elena Trombetta4, Luca Valenti5,6, Aldamaria Puliti7,8, Lorenza Lazzari9, Piergiorgio Messa1,2,6 ![]()
1. Renal Research Laboratory Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, 20122 Milan, Italy
2. Unit of Nephrology, Dialysis and Renal transplant Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
3. Internal Medicine, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
4. Flow Cytometry and Experimental Hepatology Service, Clinical Chemistry and Microbiology Laboratory, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
5. Department of pathophysiology and transplantation, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
6. Università degli Studi di Milano, Italy
7. DiNOGMI, University of Genoa, Italy
8. Medical Genetics Unit, Istituto Giannina Gaslini, Italy
9. Cell Factory Unit of Cell Therapy and Cryobiology, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
Received 2017-10-10; Accepted 2018-2-2; Published 2018-4-25
Abstract

Recently it has been demonstrated that Fetuin-A, an anti-inflammatory protein synthesized by the liver, is produced also in bone by an FGF23-regulated pathway. FGF23 has been also demonstrated to induce inflammatory cytokine production in the liver. This study aimed to explore if FGF23 plays a role in the Fetuin-A production in the liver cells too and the possible relationships with FGF23 pro-inflammatory effects.
FGF23 and Fetuin-A were studied in liver, kidney and in plasma with immunochemistry, immunoprecipitation, western blot, chromatin immunoprecipitation, duolink, ELISA, qrtPCR methodology.
FGF23 is produced, but not secreted by the liver cells. In hepatocytes and circulation, FGF23 was present only strictly linked to Fetuin-A, while Fetuin-A was found also in unbounded form. No link was observed in the kidney. FGF23 up to 600 pg/ml stimulates, while, at higher concentrations, reduces Fetuin-A expression. Notably, overall the range of concentrations, FGF23 stimulates Fetuin-A promoter, TNFα and IL6 expression. In the nucleus, FGF23 seems to act as a direct transcription factor of Fetuin-A promoter.
These results suggest that FGF23 played a direct regulatory role in Fetuin-A expression in liver cells with a biphasic effect: Fetuin-A progressively increases when FGF23 increases up to 400-600 pg/mL, and declines at higher FGF23 concentrations. These results lead us to hypothesize: a) a possible epigenetic post-transcriptional regulation; b) a possible counter-regulatory effect of FGF23 induced inflammatory cytokines (TNFα/ NF-κB mechanism). This study could add an additional key for the interpretation of the possible mechanisms linking FGF23, Fetuin-A and inflammation in CKD patients and suggests a role for FGF23 as transcription factor.
Keywords: Chronic kidney disease, FGF23, Fetuin-A, Liver, Circulation, Inflammation
Introduction
Bone, kidney and intestine are the organs involved in maintaining the balance of phosphate and calcium both in normal subjects and in chronic kidney disease (CKD) patients.
FGF23, an osteocyte (OS) derived hormone, plays a key role in this network [1-5]. It has been widely reported that FGF23 levels can increase from the beginning of kidney diseases, achieving very high blood concentrations at the advanced CKD stages. These high FGF23 levels have been claimed to be involved not only in the mineral metabolism derangements but also to possibly affect the progression of cardiovascular disease (CVD) and CKD itself, adding negative effects to the poor clinical outcomes of CKD [6-8].
Fetuin-A is a multifunctional protein mainly produced by the liver [9] that plays different roles such as the prevention of vascular calcium deposition and the anti-inflammatory function [10-16].
These two properties are to a certain extent inter-related to each other, since it is acknowledged that Fetuin-A is either up- or down-regulated by different inflammatory cytokines, with a potential negative effect on the CV system in the case of its down-regulation [17-20].
We recently found that FGF23 strictly controls Fetuin-A production in the bone and that the handling of FGF23 activity (addition of recombinant FGF23, gene deletion and overexpression), with the simultaneous modulation of FGF receptors, clearly showed a potential paracrine and endocrine role of FGF23 on Fetuin-A expression and production [21, 22]. A recent paper demonstrated an ectopic FGF23 overexpression in the hepatocytes in two patients with end-stage liver disease [23], another article described the presence of FGF23 in the hepatocytes of CKD mice, where it induced the release of inflammatory factors [24], this being the case we might then hypothesize a potential physiological or pathological role of FGF23 in the liver [25].
The first aim of the present study was to investigate if FGF23 might be involved in Fetuin-A production also in the liver and, if this was the case, through which biochemical mechanism. The second aim was to explore the possible inter-relationship between the FGF23-dependent processes of Fetuin-A and inflammatory cytokine release. Among the inflammatory cytokines, we focused on TNFα, for its recognized pathogenic role in the acute systemic inflammatory process [26-29].
Furthermore, since our previous study showed that FGF23 and Fetuin-A are bound to each other in both the cytoplasm and nucleus of bone cells, and most of all in the bone matrix and possibly in the circulation, we looked for a possible link between these two proteins also in the plasma and in the kidney, as one of the principal targets of FGF23 action [30-32].
Materials and Methods
Tissues, cell culture and plasma
Liver and kidney Tissue Specimens
Frozen kidney tissues were extracted from the non-injured part of the human nephrectomy. Human tissues and plasma (see below) were processed after the patients informed consent was obtained and approved by Director of Nephrology-Urology and Kidney Transplantation Unit (NUTRAR) of Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico. Protocol n°: M.04. NUTRAR. CONS. rev.0. of 30/03/2015.
Frozen liver tissues of 6 weeks old male BALB/c mice (IRCCS San Martino-IST Genoa, IT) were used. All efforts were made to minimize animal suffering and to use only the minimum number of animals necessary. All animals were housed on a 12-h light/dark cycle, allowed free access to food and water and killed by decapitation after anesthesia induced by intraperitoneal injection of 370mg/Kg of Chloral hydrate.
All procedures involving animals were performed in accordance with the European legislation (European Communities Council Directive of 24 November 1986, 86/609/ EEC) and complied with the European Communities Parliament and Council Directive of 22 September 2010 (2010/63/EU) and with the Italian (D.L.vo n. 26/2014), and were reviewed and approved by the licensing and Ethical Committee of the IRCCS-AOU San Martino-IST National Cancer Research Institute, Genoa (Italy) protocol n°371, and by the Italian Ministry of Health.
Hepatocytes and HepG2
Primary human hepatocytes (Lonza, Basel, Switzerland) were thawed and plated on collagen type I Solution (StemCell Technologies, Vancouver, BC, Canada) pre-coated flasks in complete Plating Medium (Lonza). Hepatocytes were then cultured in Maintenance Medium (Lonza) following the manufacturer's instructions. Human hepatocellular carcinoma cell line HepG2 (LGC Standards, Sesto San Giovanni, Italy) was cultured onto collagen-coated flasks in IMDM+Glutamax® supplemented with 20% FBS, 1% non-essential amino acids and 1% penicillin/streptomycin (all from Gibco/Life Technologies, Milan, Italy). Cells were maintained at 37°C in a humidified 5% CO2 incubator.
Podocytes
Conditionally immortalized podocytes SV1 (SV1) from transgenic H-2Kb-tsA58 mice (CLS, Eppelheim Germany) were plated on collagen type IV (Sigma-Aldrich, Milan, Italy) at 33°C with DMEM: F12 medium supplemented with 10% FCS, 1% penicillin/streptomycin, 2mM L-glutamine and 20U/ml recombinant mouse γ interferon (all from Sigma-Aldrich). Subsequently podocytes were thermo-shifted to 37°C for differentiation for 7 days and grown with the complete medium, without γ-interferon, in pre-coated flasks.
Osteocytes
The MC3T3-E1 cell line was obtained from ATCC (LGC Standards S.r.l) and cultured in alpha-MEM (Invitrogen, Milan, Italy), supplemented with 10% FBS (Sigma-Aldrich) and 1% penicillin/streptomycin (Sigma-Aldrich) in 75-cm2 flasks at a density of 400 000 cells/cm2. When cells reached 80% confluence, 50µg/ml ascorbic acid and 3mM glycerol 2-phosphate disodium salt hydrate AA/GP (Sigma-Aldrich) were added and the medium was changed every other day. After 5 days of incubation with AA/GP, the cells were re-plated at a density of 7 000 cells/cm2 and cultured with basal medium plus AA/GP and 10uM All Trans Retinoic Acid (Sigma-Aldrich) to obtain OS [33]. OS were studied at the 4th day of treatment.
Plasma
Cytospin slides were obtained by centrifugation of 200μl of 5 patients (3 with elevated and 2 with low FGF23 concentrations) (1:1 in PBS) on microscope glass slides at 300 rpm for 2min. In parallel, 50μl of undiluted plasma drawn from the same 5 above patients were smeared on slides for further analyzes (see below).
Immunohistochemistry and immunocytochemistry
Liver and kidney tissue
Cold acetone fixed human kidney tissue sections were incubated with anti-FGF23 and anti-Alpha-2-HS-glycoprotein antibodies.
Mouse liver tissue sections were incubated with anti-FGF23 [M251] and, after M.O.M kit blocking (Vector Labotatories, USA), with anti-Alpha-2-HS-glycoprotein for 1h RT followed by relative secondary antibodies for 1h RT. Specificity of Ab labelling was demonstrated by the lack of staining after substituting the primary antibody with the isotype ctrl. Images were acquired by Zeiss AxioObserver microscope equipped with high resolution digital videocamera (AxioCam, Zeiss) and Apotome system for structured illumination and recorded by AxioVision software 4.8. All antibodies information in Table S1.
Hepatocytes and HepG2
Hepatocytes and HepG2 cultured on cover slips for 24h were fixed in cold acetone for 5 min and permeabilized with 0.3 % of Triton X-100 (Sigma-Aldrich) in PBS for 30min RT and then incubated with 1 % of bovine serum blocking solution for 1h RT. Immunocytochemistry was performed with anti-FGF23, anti-Alpha-2-HS-glycoprotein, anti-clathrin and anti-Human serum albumin o/n at 4°C followed by relative secondary antibodies for 1h RT. Specificity of Ab labelling and neg ctrl was performed as described above. All antibodies information in Table S1.
Plasma of CKD patients
Cytospin of plasma and plasma smear were fixed in cold acetone for 3 min and incubated in 1 % of bovine serum blocking solution in PBS for 30min RT. Immunochemistry was performed as described above following human tissues procedure.
Immunoprecipitation (IP) and western blot (WB)
1mg of HepG2 lysate and 700µg of plasma of five patients (3 with high and 2 with low FGF23 concentrations) were IP-ed with anti-FGF23 [M-251] (5ug antibody/1mg protein) using the standard protocol (Thermofisher, USA). Positive control was performed using the 10% protein lysate of HepG2 and the plasma at high and low FGF23 level to indicate the specific band, negative control was performed on samples IP-ed with normal rabbit IgG to evaluate the IP efficiency.
IP of HepG2 and plasma were then separated on a SDS-PAGE and transferred by electroblotting on a PVDF membrane (ImmunBlot PVDF membrane, Bio-Rad). After blocking solution (BM Chemiluminescence WBting Kit Roche, USA), each membrane was incubated with the primary antibodies anti-FGF23 and anti-Alpha-2-HS-glycoprotein diluted in TBS followed by the HRP-conjugated secondary antibodies in blocking solution. Positive reaction products were identified by chemiluminescence (BM Chemiluminescence WBting Kit). Loading controls for HepG2 were conducted with anti-cofilin antibody.
To confirm that IP was successful, we repeated the same WB also on the component non FGF23/IP with FGF23 antibody. The same WB was performed with mouse anti-Alpha-2-HS-glycoprotein on the same component evaluated to see if Fetuin-A could circulate without FGF23 binding.
To avoid the interference of background signals caused by heavy and light chains from IgGs, we repeated the experiment also with Clean-Blot IP detection reagent Kit (following the manufacter's instructions of Thermofisher, USA) and no difference was observed. In this last method we blocked the membrane with StartingBlock T20 (TBS), we used the same primary antibodies concentration diluted in TBS blocking buffer and for the secondary antibody we used a dilution of 1:200 of clean-blot IP detection reagent HRP in TBS. Images were digitally acquired by Chemidoc XRS instrument (Bio-Rad) and analyzed by Quantity One software (Bio-Rad). All antibodies information in Table S1.
FGF23 addition
For evaluation of FGF23 effect on FGF23, Fetuin-A, TNFα, IL6 and Fetuin-A promoter expression, HepG2 were stimulated with different concentrations of human recombinant FGF23 (100-12500pg/mL) (Immunological Sciences, Roma, Italy) for 24h.
FGF23 overexpression
20000 cells/cm2 of HepG2 were transfected with 10nM human ORFclone cDNA clone FGF23 (Myc-DDK tagged) (OriGene Technologies Inc.MD, USA) using Lipofectamine2000 (Invitrogen) as transfection agent for 24h. As control, only Lipofectamine2000 was applied.
mRNA extraction and qRT-PCR
Total RNA of primary Hepatocytes, HepG2, OS and podocytes were extracted by Trizol (Invitrogen), precipitated by chloroform and isopropanol, washed in ethanol 75%, treated with DNase (Invitrogen) and then suspended in nuclease free-water. cDNA was prepared from 1μg RNA using the iScript Select cDNA Synthesis Kit and oligo(dt)20 primers (Bio-Rad, Segrate, Milan, Italy). After assessment of primer specificity, mRNA extracted was used to evaluate human FGF23, AHSG, TNFα, IL6, AHSG promoter and mouse FGF23. To verify the absence of genomic DNA in the samples, qRT-PCR was performed also on the original RNA (minus-reverse transcriptase). Data were normalized against the expression of human RPL4 or mouse RPL13. Real Time RT-PCR were run with iQ Sybr Green Supermix (Bio-Rad) on a MyIQ instrument (Bio-Rad), and data were analyzed by the IQ5 Bio-Rad Software. All primers information in Table S2.
Enzyme-linked immunosorbent (ELISA) tests
FGF23 levels in the cultured cell supernatants were evaluated by ELISA Kit (KAINOS laboratories, Japan). The minimal detectable concentration is 3pg/mL. The FGF23 levels of both hepatocytes and HepG2 supernatant were compared to OS (positive ctrl) and to both cells culture media (negative ctrl). Replicate background measurements were subtracted to all 450nm measures. To normalize Janus Green Whole-Cell Stain (Thermofisher) was added for 5min. Careful washing was followed by addition of Elution Buffer and absorbance was read at 630nm. The resulting A450 values were then normalized to the A630 values to account for differences in numbers of cells. Plasma level of FGF23 and Fetuin-A were measured by Human FGF23 (Intact) (Immunotopics, USA) and Human Fetuin-A (BioVendor, Heidelberg Germany) ELISA kits. The lowest concentration of Human FGF23 measurable is 1.5pg/mL. Limit of detection of Human Fetuin-A is 3.5µg/mL.
Protein interaction
To detected Fetuin-A/FGF23 interactions in the HepG2 and in the plasma, Duolink (Sigma-Aldrich) in situ experiment was performed following the manufacturer's instructions. Cells were fixed in PFA for 10min, blocked for 30min (Sigma-Aldrich) and stained with anti-FGF23 and anti-Alpha-2-HS-glycoprotein o/n at 4°C. A second incubation with PLUS/MINUS PLA probe was performed and after ligation and amplification (both from Sigma) we visualized the interaction by immunofluorescence. As negative ctrl, we chose to omit one of the two antibodies alternatively.
Chromatine immunoprecipitation (CHIP) and semi quantitative PCR
CHIP of 106 cells of HepG2 were performed using the standard protocol (Thermofisher, USA) with antibodies rabbit anti-FGF23, anti-normal rabbit IgG as negative control and anti-RNA polymerase II as positive control (Thermofisher). PCR were performed on 10% on DNA as INPUT, on CHIP with FGF23 and on negative and positive ctrl.
The primers tested for Fetuin-A promoter are designed from a sequence declared by IDT as promoter sequence of Fetuin-A and verified by Blast, on the contrary for the sample immunoprecipitated with RNA polymerase II, the primers came from GAPDH provided by Thermoscientific KIT. Amplification conditions were PCR buffer with 2.5mM MgCl2, 200nM each primer and fast Start Taq in the presence of 200nM dNTPs (Roche). Samples were amplified step 1: 6' at 95°C, step 2: 35'' at 95°C, step 3: 35''at 57,5°C-60°C, step 4: 50''at 72°C, step 5: from step 2 (x12), step 6: 35''at 95°C, step 7: 35'' at 57°C, step 8: 50'' at72°C, step 9: from step 6 (x23), step 10: 9'at 72°C, step 11: 4°C. The presence of PCR products (146bp) Fetuin-A promoter were resolved with loading dye on 2% agarose gels containing 3µl ethidium bromide in 0.5X Tris Borate EDTA (TBE) buffer alongside a low molecular weight DNA ladder (Invitrogen) and photographed under UV light using a GelDoc system (Bio-Rad, Segrate, Milan, Italy).
Statistical analyses
Experiments were conducted on at least 3 replicates per each condition. Data were expressed as mean ± standard deviation, Student's t-test was applied to determine significance (p<0.05). For qRT-PCR, relative RNA abundance was determined using the comparative Ct method. The fold-change was calculated by the software [34].
Results
Fetuin-A and FGF23 expression in the liver
FGF23 and Fetuin-A expression in mouse liver tissues
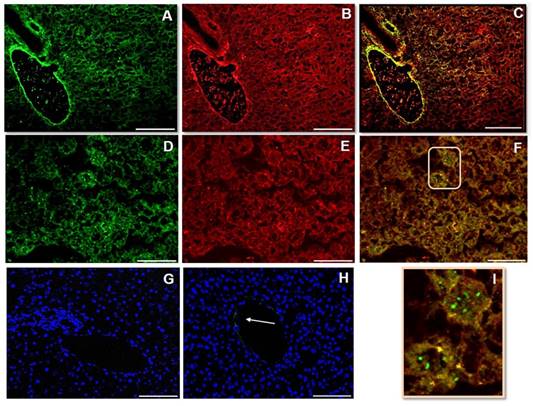
Fetuin-A (Fig 1A, D) and FGF23 (Fig 1B, E) were highly represented in mouse liver tissue. However, while the FGF23 appeared always colocalized with Fetuin-A, the immunopositivity for Fetuin-A was also present separately (Fig 1C, F, I). The Neg Ctrl was performed to verify a possible non-specific signal. While no positivity was present around the bile duct and the hepatic artery (Fig 1G), a faint positivity for Fetuin-A was present on the branch of portal vein (Fig 1H). Dapi staining has been deliberately shown to demonstrate the tissue presence.
IS (immunostaining) of Fetuin-A (A, D), FGF23 (B, E) and MERGE (C, F) visualized respectively by Fitc and rhodamine in mouse liver tissue; In panel I, a magnification of the part indicated by the rectangle in panel F is shown. The Neg Ctrl (G, H) exhibits only nuclear DAPI (4',6-diamidino-2-phenylindole) staining, attesting the cell presence. The arrow shows a faint parietal positivity for Fetuin-A, in a portal vein branch (H), but not in the bile duct and hepatic artery branch (G). Scale bars A, B, C: 100µm; D, E, F, G, H: 50µm.
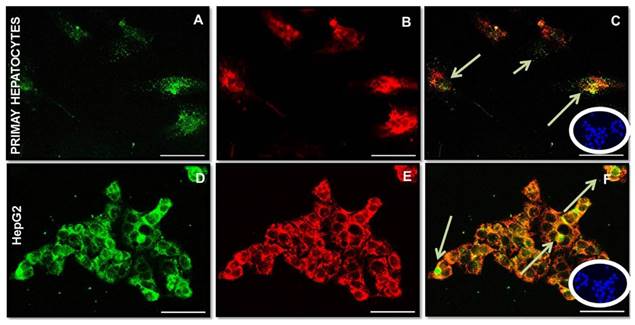
Fetuin-A, FGF23 and Clathrin expression in the primary Human Hepatocytes and HepG2
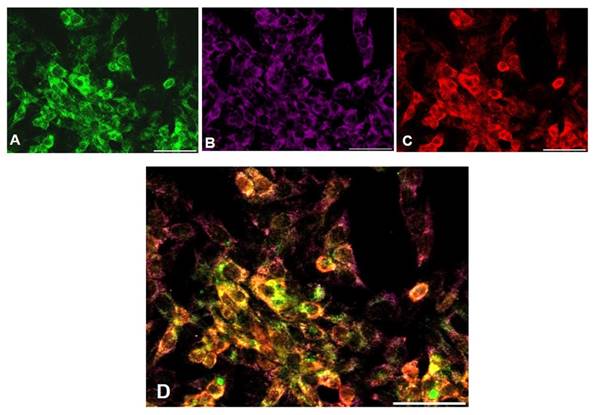
Preliminarily, albumin expression was tested in cultured hepatocytes to evaluate their functional activity (data not shown). A high expression of Fetuin-A (Fig 2A, D) and FGF23 (Fig 2B, E) was detected in both primary hepatocytes and HepG2 cells, in basal conditions, with a close co-localization at the IS. It was also evident that Fetuin- A was in part bound and in part unbound to FGF23 (Fig 2C, F). The negative Ctrl was performed to verify a possible non-specific signal (Blue Fig 2C, F). Since a punctuate staining of Fetuin-A in HepG2 may suggest an endosome localization of Fetuin-A/FGF23 complexes, we decided to perform a Fetuin-A (Fig 3A), FGF23 (Fig 3B), and Clathrin (Fig 3C), experiment of co-localization. A good co-localization of Fetuin-A, FGF23 and Clathrin was evident visualized Fig 3D.
Fetuin-A (A, D), FGF23 (B, E) and merge (C, F) expression visualized respectively by Fitc and rhodamine IS in primary hepatocytes and in HepG2. Arrows show Fetuin-A immunopositivity not co-localized. The negative Ctrl exhibits only nuclear DAPI staining (C, F in blue). Scale Bars: 50μm.
Fetuin-A (A), FGF23 (B), Clathrin (C) and MERGE expression (D) visualized respectively by Fitc, CY5 and rhodamine IS in HepG2. Scale Bars: 50μm.
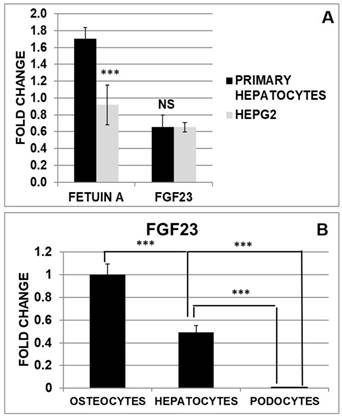
Fetuin-A and FGF23 gene expression in human hepatocytes and HepG2; FGF23 expression in primary hepatocytes compared to osteocytes and podocytes
To compare the expression of Fetuin-A and FGF23 in hepatocytes and HepG2 we performed a Quantitative Real Time RT-PCR (qRT PCR). While Fetuin-A was expressed at a higher extent in primary hepatocytes compared to HepG2, FGF23 expression was not significantly different in the two cell lines (Fig 4A). Then we explored FGF23 production in hepatocytes in comparison with osteocytes (well-known FGF23 secreting cells) and podocytes (negative control). Primary hepatocytes expressed a significant amount of FGF23 compared to podocytes (negative Ctrl), but far less than OS (positive Ctrl) (Fig 4B).
mRNA expression of Fetuin-A and FGF23 in primary hepatocytes and HepG2 (A). qRT PCR of FGF23 in primary hepatocytes compared to podocytes (negative control) and OS (Positive Ctrl) (B). Asterisks indicate significant differences: ***=p<0.001 by Student's t-test. n=3/group.
FGF23 and Fetuin-A protein expression and link in HepG2
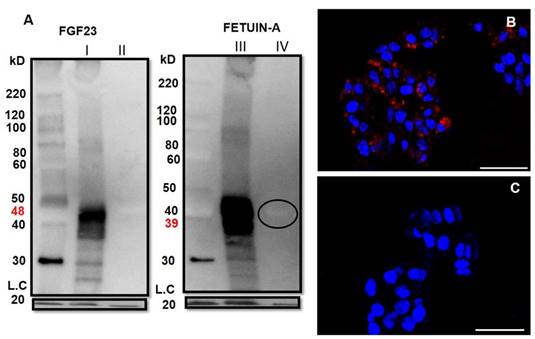
To investigate more in depth the interaction between Fetuin-A and FGF23 in the liver cells, we performed a Fetuin-A and FGF23 WB analysis in HepG2 (Fig 5) and primary hepatocytes (data not shown) immunoprecipitated with FGF23. We found, as expected, a strong positivity for FGF23 mature fragment (48kD) (Fig 5AI) in the FGF23 WB but not in the non-FGF23/IP fraction (Fig 5AII). A clear band of Fetuin-A (39kD) was evident in the FGF23/IP fraction (Fig 5AIII) and to a by far lesser extent in the non-FGF23/IP fraction (Fig 5AIV). The paucity of this last free component could be due to the dilution requested by the manufacturer's instructions. Positive control confirmed the correctness of the bands, negative control avoided the presence of false positivity (Fig S1), a very faint signal for IgG was present. The interaction between Fetuin-A and FGF23 was confirmed by the Duolink assay which was highly positive in the HepG2 cells (Fig 5B) and completely negative in the Ctrl (Fig 5C).
WB of FGF23 on HepG2 FGF23/IP (AI) and on the portion non-FGF23/IP (negative Ctrl) with FGF23 (AII). WB of Fetuin-A on FGF23/IP (AIII) and on the aliquot non- FGF23/IP (AIV). Cofilin (bottom of the gel) acted as loading control (LC). Cell localization of Fetuin-A and FGF23 interactions detected by Duolink in situ experiment in HepG2 (B) and negative Ctrl (C) Scale bars: A, B: 50µm.
FGF23 liver release
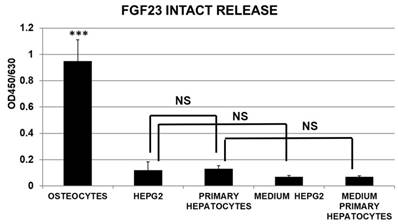
The release of intact FGF23 was substantially null in hepatocytes cell cultures and comparable to media negative Ctrl with no significant difference between primary hepatocytes and HepG2, at variance with the positive control of OS cultures (Fig 6).
Cells Cultured medium harvested from primary hepatocytes, HepG2, and OS for measurement of intact FGF23 release, assessed by ELISA. (n=3/group; mean ±SD; data were normalized by Janus Green Nuclear marker; ***=p<0.001 OS vs all the other groups).
Fetuin-A and FGF23 interaction in the liver cells
FGF23, Fetuin-A, TNFα and IL6 expression after FGF23 recombinant addition and overexpression on HepG2
The addition of increasing doses (from 100 up to 12500 pg/mL) of recombinant FGF23 induced a progressive increase in FGF23 mRNA in HepG2 cells (Fig 7A). The same doses of FGF23 induced a biphasic effect on Fetuin-A mRNA, with a significant increase for doses up to 600 pg/mL, followed by a progressive reduction at higher doses (Fig 7B).
Changes in FGF23, Fetuin-A and TNFα mRNA expression after 24h from the addition of scalar FGF23 dose (A, B, C). qRT PCR of FGF23, Fetuin-A and TNFα in HepG2 after 24h from FGF23 overexpression (D, E, F). Asterisks indicate significant differences versus HepG2 (ctrl): *=p<0.01, **=p<0.01, ***=p<0.001 by Student's t-test. n=3/group.
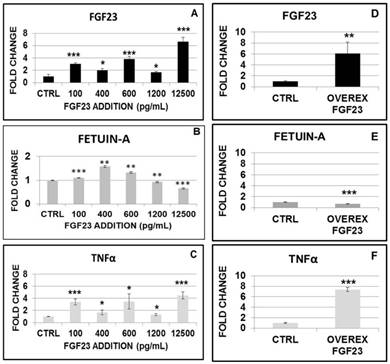
Then we analysed the effects of FGF23 overexpression on Fetuin-A expression in HepG2 transfected with mouse cDNA clone FGF23. After we verified the expected increase of FGF23 mRNA (Ctrl) (Fig 7D), we found that FGF23 overexpression was followed by a reduction of Fetuin-A mRNA (Fig 7E).
We explored also the effects of FGF23 addition and overexpression on TNFα, a classic inflammatory cytokine. 24h after either FGF23 addition or overexpression, TNFα showed a progressive increase with a figure that resembled very closely that observed for the FGF23 behavior (Fig 7C, F). To support the hypothesis of inflammatory system implication, we checked for IL6 expression at low and high concentrations of added and overexpressed FGF23: in all these cases, IL6 upregulations was evident (Fig S2).
Fetuin-A Promoter activation and biochemical mechanism
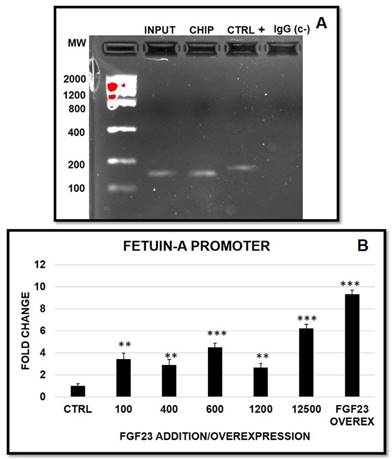
In order to investigate a possible nuclear FGF23-Fetuin-A promoter interaction, we performed a CHIP with FGF23 on HepG2 followed by a polymerase chain reaction (PCR) amplification for the identification of the Fetuin-A promoter we chose to investigate. The positivity for the chosen band (146 bp) indicated that the fragment immunoprecipitated by FGF23 corresponds to the Fetuin-A promoter (Fig 8A). A false positivity was excluded by the IgG control, while the correctness of the PCR was confirmed by the positive control and the input. Then, we decided to investigate the behaviour of Fetuin-A promoter after scalar additions of FGF23 and after FGF23 overexpression. In all these conditions the Fetuin-A promoter activation occurred (Fig 8B).
Semi-quantitative PCR of Fetuin-A promoter of HepG2 after FGF23/CHIP. Product size of human Fetuin-A promoter 146bp (A). Changes in Fetuin-A promoter expression after 24h from the addition of scalar FGF23 dose and after 24h from FGF23 overexpression (B). Asterisks indicate significant differences versus HepG2 (ctrl): **=p<0.01, ***=p<0.001 by Student's t-test. n=3/group.
FGF23 and Fetuin-A protein expression and interaction in the circulation
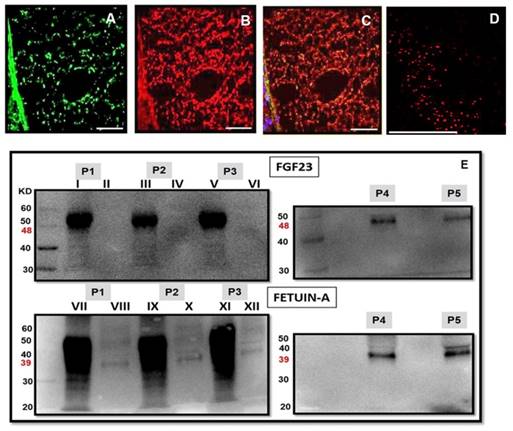
Since the strong expression of Fetuin-A (Fig 9A) and FGF23 (Fig 9B) in the liver tissue was characterized by a strict co-localization (Fig 9C) not only in the liver cells but also in the interstice of the portal vein, we looked for a possible co-localization of these two proteins also in the circulation. In fact, a strong positive interaction of FGF23 and Fetuin-A was observed by Duolink in situ experiments in the plasma of CKD patients obtained by cytospin (Fig 9D). To avoid the possibility of false positive results in the duolink assay due to the cytospin centrifugation method, the co-localization of FGF23 and Fetuin-A was also assessed with IS in the plasma of CKD patients prepared by both cytospin and smear methods. Both methods confirmed either a strong co-localization of the two proteins and the presence of bound and unbound Fetuin-A (Fig 10). To further confirm this interaction, we performed FGF23 and Fetuin-A WB in the plasma of 3 FGF23 plasma samples, a strong visible band of FGF23 at 48kD (Fig 9E P1I, P2III, P3V, FGF23 P4, P5) and of Fetuin-A at 39kD (Fig 9E P1VII, P2IX, P3XI, Fetuin-A P4, P5) were detected in the FGF23/IP samples. In the non-FGF23/IP aliquot we did not find any FGF23 band (Fig 9E P1II, P2IV, P3VI), whereas a clear band of Fetuin-A was evident also in this aliquot (Fig 9E P1VIII, P2X, P3XII). The correctness of the explored bands is confirmed by the negative and positive controls, as shown in Fig S1.
Fetuin-A (A), FGF23 (B) and merge (C) IS in the periportal vein interstice (human liver). Cell co-localization of Fetuin-A and FGF23 detected by Duolink in situ experiment in the plasma of a CKD patient (D). Scale Bars: 50μm. FGF23 WB in the FGF23/IP plasma samples of 3 CKD patients with high FGF23 levels (E P1I, P2III, P3V) and 2 CKD patients with low FGF23 levels (E P4, P5) and in the non-FGF23/IP plasma samples of the first three high FGF23 patients (E P1II, P2IV, P3VI). Fetuin-A WB in the FGF23/IP plasma samples of 3 CKD patients with high FGF23 levels (E P1VII, P2IX, P3XI, P4, P5) and 2 CKD patients with low FGF23 levels (E P4, P5) and in the non-FGF23/IP plasma samples of the first three high FGF23 patients (E P1VIII, P2X, P3XII).
IS of Fetuin (A), FGF23 (B) and merge (C) after cytospin centrifugation, IS of Fetuin (D), FGF23 (E) and merge (F) after plasma smear in human plasma. Scale bars: 50μm.
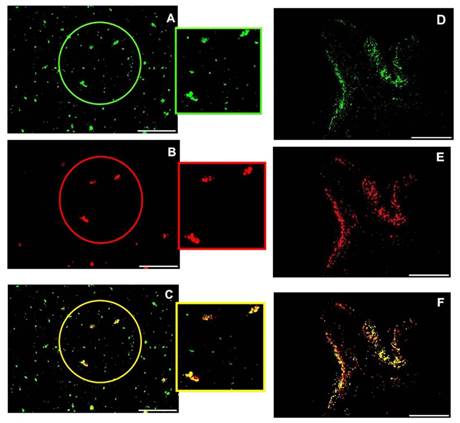
The quantitative measurement of FGF23 in plasma of 5 CKD patients in pg/ML was: (P1: 841.2, P2:603.4, P3:547 P4:26.9, P5: 18.1) and the dosage of Fetuin-A in G/L was (P1: 0.372, P2:0.342, P3:0.335, P4:0.266, P5: 0.222).
FGF23 and Fetuin-A interaction in the renal tubules
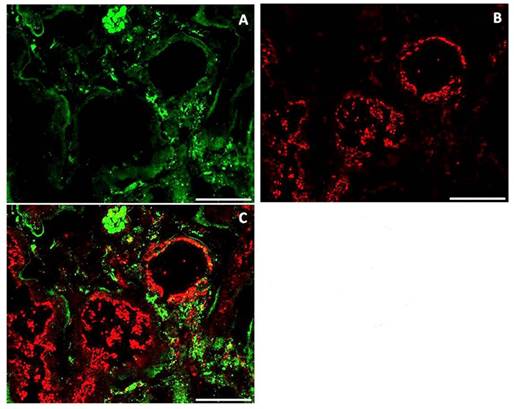
Given this strict inter-relationship between FGF23 and Fetuin-A also in the circulation, we looked for a similar interaction also in the renal tissue, which is one of the main target sites of FGF23. Although a strong IS of both Fetuin-A (Fig 11A) and FGF23 (Fig 11B) was observed, no co-localization of both molecules was detected (Fig 11C).
IS of Fetuin (A), FGF23 (B) and merge (C) in human kidney tissue. Scale bars: 50μm.
Discussion
In our recent articles [21, 22] we focused on FGF23 and Fetuin-A proteins that are involved in both bone and vascular system physiopathology, the first being a well-recognized bone-derived protein and the second being produced mainly by the liver. Our results demonstrated not only that Fetuin-A is produced also by bone cells and that its production is controlled by FGF23, but that both proteins are strictly linked to each other in the bone tissue.
Then, we wondered: a) if these characteristics are present also in the liver, the main site of Fetuin-A production; b) if this was the case, which were the biochemical mechanism(s); c) if the strict interaction of FGF23 and Fetuin-A found in the bone is also present in the circulatory system.
Starting from the experimental data obtained in the liver, in addition to the expected expression of Fetuin-A, we found also a strong FGF23 expression with a tight co-localization of the two proteins to each other in the liver tissue and in cultures of either primary hepatocytes and HepG2 cells. We chose to study also HepG2 since it is well known that human hepatocytes, though considered the most appropriate in vitro model to study human liver cells, often face a rapid functional loss combined with a poor proliferative speed [35], while HepG2 have a high proliferative rate and are easier to handle. Anyway, we proved the viability of our cultured primary hepatocytes showing a clear positivity for albumin IS.
It is worth underlining that while all the detectable FGF23 was completely co-localized with Fetuin-A, the latter was also present as unbound protein. To further characterize the cytoplasmic site of FGF23/Fetuin-A co-localization, we looked for a possible interaction of this complex with an endosomal protein (Clathrin). In fact, the IF appearance of the 3 immunostainings in HepG2 cells strongly suggests a co-localization of FGF23/ Fetuin-A complex in the endosomes. The physiopathological meaning of this co-localization, if any, (intracellular trafficking, lysosome, secretory) deserves future studies.
To qualify the nature of FGF23 positivity in the liver, we looked for its expression by qRT-PCR. In fact, we found that both primary hepatocytes and HepG2 cells highly expressed not only Fetuin-A, but, though at a lesser extent, also FGF23 mRNA. It is also worth underlining that while Fetuin-A showed a higher expression in primary hepatocytes compared to HepG2; no difference was evident for FGF23 expression between the two diverse types of cells. The lack of any difference between the primary hepatocytes and the HepG2 cells is an important finding, since it is known that tumoral liver cells can non-physiologically express many FGFs (FGF8, FGF17, FGF18) [36, 37]. In any case, our results are confirmatory of previous studies, which have demonstrated that FGF23 is expressed also in the liver cells [23].
We also compared the entity of the FGF23 expression in the liver cells with that of osteocytes (Positive Ctrl) and podocytes (Negative Ctrl) expression. In fact, we found that the amount of FGF23 expression in liver cells is almost in the midway between osteocytes and podocytes. This finding is confirmatory of a sizeable FGF23 expression in the liver, which was however consistently lower than that found in OS.
To further explore the degree of closeness of FGF23 and Fetuin-A molecules observed in the IF studies in the liver, we investigated their co-localization by both WB and Duolink analyses.
We found a strong FGF23 band presence in the FGF23/IP fraction while a complete absence in the non-FGF23/IP fraction, confirming the correctness of the procedure.
We also found a clear Fetuin-A band in the FGF23/IP fraction, in both primary Hepatocytes and even more evidently in the HepG2. The presence of a small Fetuin-A band in non-FGF23/IP suggests the presence of an FGF23 unbound component in agreement with the findings obtained by IS. The paucity of this free component could be due to the dilution requested by the manufacturer's instructions. The presence of a strict interaction between Fetuin-A and FGF23 was also suggested by the Duolink in situ results in HepG2 cells. Then, from the WB first results we may suppose that covalent disulfide bridge is one of the possible bonds between Fetuin-A and FGF23 and that, while FGF23 is strictly linked to Fetuin-A, the last one could also circulate in an unbound manner. It is well known that all the members of the FGF family contain some cysteines protein which are involved in structural stability and that these cysteines are conserved as vestigial epitope though with an unclear role [38]. It has been suggested that cysteine residues could enhance the affinity of some FGFs (FGF1 and 2) for the FGF-binding protein (FGF-BP1), forming a disulfide bond between the carrier protein and these FGFs [39]. In the same way, it could be hypothesized that Fetuin-A might interact with the cysteine residues of FGF23 vestigial site. The high propensity to closely link between Fetuin-A due to its molecular properties with other factors is already known, so that it is difficult to obtain crude Fetuin-A with a purification process [9]. Furthermore, the capacity of Fetuin-A to link various growth factors creating “enigmatic properties” was already hypothesized [40]. However, further research is needed to better clarify the meaning of the strict link between Fetuin-A and FGF23.
Subsequently, we evaluated if FGF23, in addition to being expressed, is also released by the liver cells. No relevant amount of FGF23 was present in the medium of both cell cultures, without significant difference between the two cell types. This finding could be explained by a fast degradation of the protein due to specific proteases (furin) as suggested by a recent study where transgenic mice, characterized by a conditional block of furin proteases in the liver tissue, could secrete FGF23 in the circulation [24]. We cannot exclude that the complete proteolysis of the intact FGF23, which very likely happens in the normal liver cells, is present also in the CKD liver cells: this point will deserve specific research in in vivo CKD experimental studies. However, it is worth underlining that we dosed only the mature form of FGF23 (intact FGF23), and it cannot be excluded that intact FGF23 might have been cleaved into C-terminal fragment(s).
Since we previously found that the addition of FGF23 to the OS cultures resulted in consistent changes of FGF23 and Fetuin A expression [21], we aimed to explore if these findings were present in liver cells as well. After stimulating HepG2 with escalating FGF23 doses, we observed a clear, though not completely dose dependent, increase of FGF23 expression, but only a slightly increased and biphasic expression of Fetuin-A, which was stimulated by the lowest and inhibited by the highest FGF23 doses. There is no counterintuitive explanation for this expression pattern in the liver cells. However, it has been recently demonstrated that the FGF23 dependent stimulation of FGFR4, which is the only FGF receptor expressed in the liver, induces the release of inflammatory cytokines through a Klotho-independent pathway [26, 28, 32]. It is also well recognized that Fetuin-A, which plays also an anti-inflammatory role [41-45], can be either up- or down-regulated by other inflammatory cytokines [17, 20]. Since it has been demonstrated that TNFα is an anti-inflammatory cytokine with inhibitory effects on Fetuin-A expression [42], we looked for the TNFα behavior after FGF23 stimulation. In fact, TNFα changes mirrored the FGF23 variations and at the highest TNFα mRNA levels corresponded to the lowest Fetuin-A expression. Therefore, we could hypothesize that a primary stimulatory effect of FGF23 on Fetuin-A expression might be dampened by the simultaneous increased levels of TNFα which has a predominant inhibitory effect.
This hypothesis is indirectly confirmed by the results that we obtained after autocrine strong overexpression of FGF23 in hepatocytes. In fact, we found that FGF23 overexpression induced a striking up-regulation of TNFα expression associated with a deep downregulation of Fetuin-A expression.
This finding might be of relevance, particularly in CKD patients, since the very high levels of FGF23 often observed in this clinical setting could explain the contemporary increase in the inflammatory cytokines and the reduction of Fetuin-A levels [46, 47].
First, we tried to clarify the biochemical mechanism(s) by which FGF23 stimulates Fetuin-A. To this end, after verifying that no known transcription factor (TF), common to FGF23 and Fetuin-A, has been as yet reported (Gene Cards® data base), we performed a CHIP of HepG2 with FGF23 followed by a PCR amplification using the primers of human Fetuin-A promoter. In the nucleus, we observed that FGF23 affects nuclear transcription of the Fetuin-A promoter gene, possibly acting as a transcriptional factor. Our data cannot prove if FGF23 acts alone or in cooperation with other TFs. Thereafter, we investigated if the Fetuin-A promoter activity was changed by the addition or overexpression of FGF23. In all these experimental conditions, the Fetuin-A promoter was significantly stimulated by FGF23. This finding is at variance with the biphasic effects of FGF23 on Fetuin-A protein levels (see above). At the present time, we could only hypothesize that this apparently discrepant effect on Fetuin-A promoter gene and Fetuin-A protein levels induced by progressively increasing levels of FGF23 could be due to some unexplored epigenetic post-transcriptional regulation and/or to a counter-regulatory effect secondary played by inflammatory cytokines (TNFα/ NF-κB mechanism) which also are stimulated by FGF23[48, 49]. This intriguing question could be only answered by future studies. We know that CKD is characterized by high levels of FGF23 and a generalized inflammatory status. Both conditions have been put in relationship with the overwhelming incidence of cardio-vascular complications. We could hypothesize that, while relatively low-high levels of FGF23 could be protective for the CV system, given the stimulation of a “cardio-vascular” protective agent (Fetuin-A), the highest doses of FGF23 could act in a opposite direction, due to the contemporary stimulation of inflammatory cytokines which, in addition to their specific CV negative role, could also reverse the effects of FGF23 on Fetuin-A.
To reinforce this information, we decided to add also information on IL6, another major inflammatory cytokine. In fact, IL6 behaves in a way almost overlapping to that of TNF alpha.
Since in the present study and in our previous studies [21, 22] we found a strict co-localization of Fetuin-A and FGF23 molecules in the liver interstice of portal vein and in bone matrix, we wonder if this strict binding was present also in the circulatory system.
In fact, we found a strict binding of these two proteins to each other in the blood of CKD patients, as highlighted by both FGF23/IP and Duolink in situ experiments. It is also worth noting that this strict interrelationship was evident at both high and low FGF23 levels. To avoid a possible artifact link due to the cytospin method, we compared the IF images of Fetuin-A and FGF23 co-localization obtained in the cytospin samples with plasma smears. Both experiments gave almost overlapping results, excluding potential technical factitious results.
Overall, these results, reinforcing those observed in the tissue and cell cultures, strongly suggest a strict interrelationship between FGF23 and Fetuin-A.
Then, to try to give an explanation to the meaning of the circulating complex FGF23-Fetuin-A, we examined whether this dimeric complex is present also at the sites where FGF23 displays its canonical effects, such as in the renal tubular cells. However, we were not able to find any co-localization of these proteins in renal tubules, excluding that the dimer could play a cooperative role in FGF23 renal activity. We did not explore other canonical and non-canonical sites of FGF23 activity, so we cannot exclude that at the other levels the FGF23-Fetuin-A dimer could play any effective role.
Therefore, the meaning of this strict interaction remains largely to be clarified. We could just limit ourselves to hypothesize that Fetuin-A could play the role of carrier for FGF23, which in turn controls the production of the carrier itself.
Conclusions
In conclusion, our data show that FGF23 is expressed also in the liver, but, at variance with bone, it is not secreted, likely playing only autocrine and/or paracrine roles. The addition or overexpression of FGF23 in the liver cells have a biphasic effect on Fetuin-A production, with this behaviour potentially being explained by the contemporary stimulatory effect on inflammatory cytokines. We also found that FGF23 directly acts on the Fetuin-A promoter gene, possibly as a transcription factor. FGF23 and Fetuin-A are also present in the circulation, strictly bound to each other, while no co-localization is found in the kidney, which suggests a possible Fetuin-A role as a carrier protein for FGF23. Overall, we can hypothesize that the biphasic regulation of Fetuin-A production by FGF23 could add a piece in the puzzle of the intricate inter-relationships linking CKD to inflammation and possibly to CVD.
Abbreviations
CKD: Chronic kidney disease; OS: osteocytes; CVD: Cardio-vascular disease; WB: Western blot; MBD: Mineral bone disease; ELISA: Enzyme-linked immunosorbent assay; TGFβ: Transforming growth factor beta; TNFα: Tumor necrosis factor alpha; FGF23: Fibroblast growth factor 23; qRT-PCR: Quantitative polymerase chain reaction; IP: Immunoprecipitation, IS: Immunostaining.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding was provided by IRCCS Ospedale Maggiore Policlinico.
Contributors
Study Conception and design: MP; Acquisition of data: MD, IM; Analysis: MD, IM, TK; Data interpretation: MP, MD, ACM, TE, LL, PA; Article drafting: MD, MP; final approval of the version to be submitted MP, MD, IM, DP, VL. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kaneko I, Tatsumi S, Segawa H, Miyamoto KI. Control of phosphate balance by the kidney and intestine. Clin Exp Nephrol. 2016 Review
2. Messa P. FGF23 and vascular calcifications: another piece of the puzzle? Nephrol Dial Transplant. 2014;29(8):1447-9
3. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T. et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561-8
4. Kawakami K, Takeshita A, Furushima K, Miyajima M, Hatamura I, Kuro-O M. et al. Persistent fibroblast growth factor 23 signalling in the parathyroid glands for secondary hyperparathyroidism in mice with chronic kidney disease. Sci Rep. 2017;7:40534
5. Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T. et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010;78(10):975-80
6. Wolf M. FGF23: fashion or physiology? M Clin J Am Soc Nephrol. 2010;5(10):1727-9
7. Moe SM, Chertow GM, Parfrey PS, Kubo Y, Block GA, Correa-Rotter R. et al. Cinacalcet, Fibroblast Growth Factor-23, and Cardiovascular Disease in Hemodialysis: The Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) Trial. Circulation. 2015;132(1):27-39
8. Humalda JK, Seiler-Muler S, Kwakernaak AJ, Vervloet MG, Navis G, Fliser D. et al. Response of fibroblast growth factor 23 to volume interventions in arterial hypertension and diabetic nephropathy. Medicine (Baltimore). 2016;95(46):e5003
9. Mori K, Emoto M, Inaba M. Fetuin-A: a multifunctional protein. Recent. Pat. End. Metab. Immune. Drug. Discover. 2011;5(2):124-46
10. Brylka L, Jahnen-Dechent W. The role of fetuin-A in physiological and pathological mineralization. Calcif Tissue Int. 2013;93(4):355-64
11. Heiss A, DuChesne A, Denecke B, Grötzinger J, Yamamoto K, Renné T. et al. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J Biol Chem. 2003;278(15):13333-41
12. Kubota T, Yamamoto T, Ichioka H, Yamamoto K, Kanamura N, Kamegai A. et al. Biological implications of fetuin for bone remodeling system and possible evidence for its use in heterotopic ossification. Journal of Oral and Maxillofacial Surgery, Medicine, and Pathology. 2012;24(1):36-41
13. Wang H, Li W, Zhu S, Wang P, Sama AE. Acute Phase Proteins: Regulation and Functions of Acute Phase Proteins. Intech Open Access Publisher. 2011:329-344 Role of Fetuin-A in Injury and Infection
14. Li W, Zhu S, Li J, Huang Y, Zhou R, Fan X. et al. A hepatic protein, fetuin-A, occupies a protective role in lethal systemic inflammation. PLoS ONE. 2011;6(2e):16945
15. Ombrellino M, Wang H, Yang H, Zhang M, Vishnubhakat J, Frazier A. et al. Fetuin, a negative acute phase protein, attenuates TNF synthesis and the innate inflammatory response to carrageenan. Shock. 2001;15(3):181-185
16. Lord JM. A physiological role for alpha2-HS glycoprotein: stimulation of macrophage uptake of apoptotic cells. Clin Sci (Lond). 2003;105(3):267-268
17. Weikert C, Stefan N, Schulze MB, Pischon T, Berger K, Joost HG. et al. Plasma fetuin-a levels and the risk of myocardial infarction and ischemic stroke. Circulation. 2008;118(24):2555-2562
18. Sullivan G.W, Sarembock IJ Linden. J. The role of inflammation in vascular diseases. Leukoc. Biol. 2000;67:591-602
19. Keçebaş M, Güllülü S, Sağ S, Beşli F, Açikgöz E, Sarandöl E. et al. Serum fetuin-A levels in patients with systolic heart failure. Acta Cardiol. 2014;69(4):399-405
20. Maréchal C, Schlieper G, Nguyen P, Krüger T, Coche E, Robert A. et al. Serum fetuin-A levels are associated with vascular calcifications and predict cardiovascular events in renal transplant recipients. Clin J Am Soc Nephrol. 2011;6(5):974-85
21. Mattinzoli D, Rastaldi MP, Ikehata M, Armelloni S, Pignatari C, Giardino LA. et al. FGF23-regulated production of Fetuin-A (AHSG) in osteocytes. Bone. 2016;83:35-47
22. Mattinzoli D, Ikehata M, Alfieri CM, Messa P. Authors' reply to the comments on the paper: "FGF23-regulated production of Fetuin A (AHSG) in osteocytes". Bone. 2016;93:225-229
23. Wasserman H, Ikomi C, Hafberg ET, Miethke AG, Bove KE, Backeljauw PF. Two Case Reports of FGF23-Induced Hypophosphatemia in Childhood Biliary Atresia. Pediatrics. 2016:138 (2)
24. Bai X, Miao D, Li J, Goltzman D, Karaplis AC. Transgenic Mice Overexpressing Human Fibroblast Growth Factor 23 (R176Q) Delineate a Putative Role for Parathyroid Hormone in Renal Phosphate. Endocrinology. 2004;145(11):5269-79
25. Singh S, Grabner A, Yanucil C, Schramm K, Czaya B, Krick S. et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016;90(5):985-996
26. Mendoza MJ, Isakova T, Ricardo AC, Xie H, Navaneethan SD, Anderson AH. et al. Fibroblast growth factor 23 and Inflammation in CKD. Clin J Am Soc Nephrol. 2012;7(7):1155-62
27. Meijer C, Huysen V, Smeenk RT, Swaak AJ. Profiles of cytokines (TNF alpha and IL-6) and acute phase proteins (CRP and alpha 1AG) related to the disease course in patients with systemic lupus erythematosus. Lupus. 1993;2(6):359-65
28. Jain S, Gautam V, Naseem S. Acute-phase proteins: As diagnostic tool. J Pharm Bioallied Sci. 2011;3(1):118-127
29. Tonelli M, Sacks F, Pfeffer M, Jhangri GS, Curhan G. Cholesterol and Recurrent Events (CARE) Trial Investigators: Biomarkers of inflammation and progression of chronic kidney disease. Kidney Int. 2005;68:237-245
30. Wanner C, Zimmermann J, Schwedler S, Metzger T. Inflammation and cardiovascular risk in dialysis patients. Kidney Int Suppl. 2002(80):99-102
31. Dervisoglu E, Kir HM, Kalender B, Caglayan C, Eraldemir C. Serum fetuin-a concentrations are inversely related to cytokine concentrations in patients with chronic renal failure. Cytokine. 2008;44(3):323-7
32. Usama AA, Sharaf El Din, Mona M Salem, Dina O Abdulazim. FGF23 and inflammation. World J Nephrol. 2017;6(1):57-58
33. Mattinzoli D, Messa P, Corbelli A, Ikehata M, Zennaro C. et al. A novel model of in vitro osteogenesis induced by retinoic acid treatment. Eur Cell Mater. 2012;24:403-25
34. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(- delta delta C(T)) method. Methods. 25. 2001;25:402-408
35. Wilkening S, Stahl F, Bader A. Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metab Dispos. 2003;31(8):1035-42
36. Itoh N, Nakayama Y, Konishi M. Roles of FGFs As Paracrine or Endocrine Signals in Liver Development, Health, and Disease. Front Cell Dev Biol. 2016;13:4-30
37. Sauder A, Wiernek S, Dai X, Pereira R, Yudd M, Patel C. et al. FGF23-Associated Tumor-Induced Osteomalacia in a Patient with Small Cell Carcinoma: A Case Report and Regulatory Mechanism Study. Int J Surg Pathol. 2016;24(2):116-20
38. Lee J, Blaber M. Structural basis of conserved cysteine in the fibroblast growth factor family: Evidence for a vestigial half-cystine. J Mol Biol. 2009;393:128-139
39. Xie B, Tassi E, Swift MR, McDonnell K, Bowden ET, Wang S. et al. Identification of the fibroblast growth factor (FGF)-interacting domain in a secreted FGF-binding protein by phage display. AJ Biol Chem. 2006;281(2):1137-44
40. Jahnen-Dechent W, Heiss A, Schäfer C, Ketteler M. Fetuin-A regulation of calcified matrix metabolism. Circ Res. 2011;108(12):1494-509
41. Mukhopadhyay S, Mondal SA, Kumar M, Dutta D. Proinflammatory and anti-inflammatory attributes of Fetuin-A: a novel hepatokine modulating cardiovascular and glycemic outcome in metabolic syndrome. Endocr Pract. 2014;20(12):1345-51
42. Wang H, Sama AE. Anti-inflammatory role of fetuin-A in injury and infection. Curr Mol Med. 2012;12(5):625-33
43. Zhang P, Shen H, Huang J, Wang H, Zhang B. et al. Intraperitoneal Administration of Fetuin-A Attenuates d-Galactosamine/Lipopolysaccharide-Induced Liver Failure in Mouse. Dig Dis Sci. 2014;59(8):1789-1797
44. Sato M, Kamada Y, Takeda Y, Kida S, Ohara Y, Fujii H. et al. Fetuin-A negatively correlates with liver and vascular fibrosis in nonalcoholic fatty liver disease subjects. Liver Int. 2015;35(3):925-35
45. Daveau M, Christian D, Julen N, Hiron M, Arnaud P, Lebreton JP. The synthesis of human alpha-2-HS glycoprotein is down-regulated by cytokines in hepatoma HepG2 cells. FEBS Lett. 1988;241(1-2):191-4
46. Cottone S, Nardi E, Mulè G, Vadalà A, Lorito MC, Riccobene R. et al. Association between biomarkers of inflammation and left ventricular hypertrophy in moderate chronic kidney disease. Clin Nephrol. 2007;67(4):209-16
47. Wilund KR. Is the anti-inflammatory effect of regular exercise responsible for reduced cardiovascular disease? Clin Sci (Lond). 2007;112(11):543-55
48. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31-42
49. Rossaint J, Unruh M, Zarbock A. Fibroblast growth factor 23 actions in inflammation: a key factor in CKD outcomes. Nephrol Dial Transplant. 2017;32(9):1448-1453
Author contact
![]() Corresponding author: Prof. Piergiorgio Messa, Phone: +39 02 55034552; FAX: +39 02 55034550; E-mail: piergiorgio.messait; Unit of Nephrology, Urology, Dialysis and Renal Transplantation, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Università degli studi di Milano, Via Commenda 15, 20122 Milan, Italy.
Corresponding author: Prof. Piergiorgio Messa, Phone: +39 02 55034552; FAX: +39 02 55034550; E-mail: piergiorgio.messait; Unit of Nephrology, Urology, Dialysis and Renal Transplantation, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Università degli studi di Milano, Via Commenda 15, 20122 Milan, Italy.