Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(6):654-666. doi:10.7150/ijbs.24765 This issue Cite
Research Paper
Incorporation of DDR2 clusters into collagen matrix via integrin-dependent posterior remnant tethering
Tingting Li1, Jin'e Liu1, Hao Cai2, Baomei Wang3, Yunfeng Feng4, ![]() , Jun Liu1,
, Jun Liu1, ![]()
1. Jiangsu key lab of Drug Screening, Jiangsu key lab of Drug Discovery for Metabolic Disease, China Pharmaceutical University, Nanjing 210009, China;
2. Research Center for High Altitude Medicine, Qing Hai University, Xining 810001, China
3. Institute of Virology, Wenzhou University, Wenzhou, 325000, China;
4. Innate Gene Inc., Beijing 100085, China.
Received 2018-1-5; Accepted 2018-3-13; Published 2018-4-30
Abstract

Cell-matrix interactions play critical roles in cell adhesion, tissue remodeling and cancer metastasis. Discoidin domain receptor 2 (DDR2) is a collagen receptor belonging to receptor tyrosine kinase (RTK) family. It is a powerful regulator of collagen deposition in the extracellular matrix (ECM). Although the oligomerization of DDR extracellular domain (ECD) proteins can affect matrix remodeling by inhibiting fibrillogenesis, it is still unknown how cellular DDR2 is incorporated into collagen matrix. Using 3-dimentional (3D) imaging for migrating cells, we identified a novel mechanism that explains how DDR2 incorporating into collagen matrix, which we named as posterior remnant tethering. We followed the de novo formation of these remnants and identified that DDR2 clusters formed at the retracting phase of a pseudopodium, then these clusters were tethered to fibrillar collagen and peeled off from the cell body to generate DDR2 containing posterior remnants. Inhibition of β1-integrin or Rac1 activity abrogated the remnant formation. Thus, our findings unveil a special cellular mechanism for DDR2 clusters incorporating into collagen matrix in an integrin-dependent manner.
Keywords: DDR2, integrin, collagen, posterior remnants.
Introduction
Extracellular matrix (ECM)is a complex network of macromolecules including proteins, glycoproteins, proteoglycans and polysaccharides with distinctive biochemical and mechanical properties [1]. Collagens are the most abundant ECM components in both interstitial matrix and basement membrane [2]. Collagen fiber formation via fibrillogenesis is controlled by several factors such as pH, ionic strength and collagen structure [3]. It is also regulated by different collagen-binding proteins and molecules that may directly or indirectly interact with the collagen molecules and fibrils. Several secreted soluble collagen-binding proteins have been shown to affect collagen fibrillogenesis, such as decorin [4], COMP [5], fibromodulin [6] etc.
Collagen-binding transmembrane proteins, discoidin domain receptors 1 and 2 (DDR1 and DDR2) are subfamily members of DDRs. DDRs are powerful regulators of collagen deposition in the extracellular matrix [7-9]. Their extracellular domains (ECD) can reduce the collagen deposition and induce significant changes in fiber morphology and matrix mineralization [8]. Oligomeric DDR2 inhibits fibrillogenesis of collagen better than dimeric or monomeric forms [7][8]. Both purified DDR1 and DDR2 ECD oligomeric proteins are shown to affect matrix remodeling [9][10]. Although DDR1 ECD is shown to be released from the cells to the matrix by collagen dependent ectodomain shedding [11-13], how DDR2 molecules are released and incorporated into collagen matrix is unknown.
Cells cultured in 3D ECM environment behave quite differently from those on 2D substrate. Tumor cells exhibit two distinct migration patterns when invading 3D matrix, with either mesenchymal or amoeboid patterns. Cells showing mesenchymal motility have an elongated spindle-like shape with one or more pseudopodia. Integrin activation and ECM remodeling, which rely on the proteolytic activity of degrading enzymes, are required for mesenchymal movement [14][15]. Integrin-based focal adhesion components exhibit distinct roles in regulating mesenchymal migration, some regulate pseudopodium activities [16][17] while others recruit MT1-MMP for proximal matrix degradation [18]. DDRs and integrins bind to distinct motifs of collagens and activate their downstream signaling pathways respectively [19]. Although DDR1 activation in epithelial cells has been shown to suppress α2β1-integrin triggered cell migration and spreading [20-22], the functional interaction of DDRs with integrins in migrating cells in 3D cultures remains largely unknown.
Membrane tethers are nanotubes constituted by lipid bilayers. They can be induced by localized external forces [23] or formed in vivo when neutrophils rolling under shear stress. The adhesion molecules are concentrated at the tips of microvilli and initiate adhesion contact with endothelial cells on the vessel wall, when the force applied to a microvillus exceed the threshold, the plasma membrane will be peeled from the cytoskeleton and form a membrane tethers or slings [23][24]. These membrane tethers are important in inflammation, thrombosis and atherosclerosis [25][24].
DDR oligomerization states are associated with their affinity to collagen fibrils [10][19]. Although a single discoidin(DS) domain of DDRs contains the collagen binding site, DS domain dimerization is required for high affinity binding to collagen [26]. DDR homodimer formation is mediated by the transmembrane domain in a ligand independent manner [27]. Collagen interacts with DDR dimers on cell surface, which induces oligomerization of DDR dimers and conformational changes, leading to increased affinity to its ligand collagen fibers and its kinase activation [10][19]. Since the DDRs' affinity to collagen is dynamically associated with their oligomerization states, it is pertinent that high affinity DDRs may be released to collagen matrix via the mechanism of membrane tether formation. In the present study, we used 3D confocal imaging approaches to investigate how clustered DDR2 is released and incorporated into collagen matrix in migrating cells, we also explored the coordination between integrin and DDR2 in regulating DDR2 positive remnant formation.
Materials and Methods
Cell culture: HT-1080 fibrosarcoma cells, MDA-MB-231 human breast cancer cells (from ATCC) were cultured in DMEM-10 (Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum (FBS, Hyclone) and glutamine, pen/strp (Sigma)). Medium for HT-1080 cells transduced with lentiviral vectors containing DDR2-Ypet and mutants, mCherry-CAAX, Ypet-CAAX or shRNA expression cassettes also included 10 μg/ml blasticidine or 3μg/ml puromycin, based on the availability of drug selection marker in the lentiviral vectors. Cells were maintained at 37°C and 5% CO2 in a humidified incubator during culture.
Reagents: rat anti human β1-integrin antibody (mAb13) was purchased from BD Pharmingen. Rabbit anti-DDR2 antibody (ab5520) was purchased from Abcam. Rac inhibitor was purchased from Calbiochem. MMP inhibitor (Marimastat), actin polymerization inhibitor (latrunculin B), myosin II inhibitor (blebbistatin), were purchased from Sigma-Aldrich.
Lentiviruses mediated RNAi or gene expression
The shRNA sequences targeting human DDR2 were as described [28]. shRNA sequence targeting β1-integrin was: 5'-TGCCTACTTCTGCACGATGT-3'. shRNA sequence targeting p130Cas was as described [17]. We used a GFP shRNA sequence 5'-TCTCGGCATGGACGAGCTGTA-3' as a nonspecific control shRNA. shRNA expression cassettes were constructed as described and were cloned into lentiviral vector pFLRu-puro [29]. mCherry-CAAX or Ypet-CAAX lentiviral expression plasmids were constructed by C-terminal in-frame fusion of mCherry and Ypet with CAAX sequence (5'-AAA AAG AAG AAA AAG AAG TCA AAG ACA AAG TGT GTA ATT ATG TAA-3') and cloned to pFLRcmv-FH-bsr (vector with blasticidine resistance). Human DDR2-Ypet, DDR2-FHexpression lentiviral vectors were constructed by in-frame cloning DDR2 into pFLRcmv-Ypet-bsr (vector with blasticidine resistance) and pFLRcmv-FH-puro (FH: Flag-His6 tag) between EcoRI/AgeI sites. Ypet tagged DDR2 mutants including K608E, W52A, E113K were generated by overlapping PCR and sequence validation. All constructs had a GGGGS linker between DDR2 variants and the tags. Vinculin-Ypet lentiviral expression vectors were constructed by in-frame cloning into MluI/XmaI of pFLRg with a GGGGS linker in between also. Lentivirus production and lentivirus mediated cell transduction were performed as described [29]. HT1080 cells expressing mCherry-CAAX and DDR2-Ypet were obtained by sequential lentiviral transduction, e. g. HT1080 parental cells were transduced with mCherry-CAAX expression lentiviruses, selected with 10 μg/ml of blasticidine for 4 days, the remaining cells were transduced with DDR2-Ypet expression lentiviruses, selected with 1. 5 μg/ml of puromycin for two days.
3D collagen I matrix. Cell-impregnated 3D collagen matrices were prepared by mixing cells suspended in culture medium (5X104 cells encapsulated for 0. 5 ml gel scale for imaging) and 10X reconstitution buffer (1. 1g sodium bicarbonate, 2. 4g HEPES, 50ml H2O)1:1 (v/v), with soluble rat tail type I collagen in acetic acid (BD Biosciences) to achieve a final concentration of 2 mg/ml collagen or as indicated. 1 M NaOH was then added to neutralize pH (pH 7. 0, 10-20 μl 1 M NaOH), and the mixture was placed in multi-well, coverslip-bottomed culture plates (LabTek). In integrin blocking experiments, mouse anti human β1 integrin monoclonal antibody (mAb13, inactive β1-integrinBD Pharmingen, 1:100) or a non-specific mouse IgG antibody (Sigma-Aldrich) was added to cell suspension and incubated for 10 min before mixing with collagen. All ingredients were kept chilled and care was taken to avoid bubbles forming. Collagen gels solidified for 30 minutes in an incubator at 37°C and 5% CO2, then 500 μl of pre-warmed DMEM-10 (phenol red free, Sigma) was added. Drug inhibitors used were MMP inhibitor Marimastat (Sigma-Aldrich, final: 2 μg/ml), myosin II inhibitor blebstatin (Sigma-Aldrich, final: 30 μM), actin polymerization inhibitor Latrunculin B (Sigma-Aldrich, final: 30 μM), Rac inhibitor (Calbiochem, final: 50 μm). Drugs were added to the DMEM-10 on the gels for 16 hours before fixation and staining.
3D time lapse cell migration imaging with confocal microscopy
To visualize live cell migration in 3D, HT-1080 cells transduced with DDR2-Ypet, mCherry-CAAX or DDR2-mCherry lentiviral vectors were embedded in 3-D collagen matrices were imaged using a Nikon Elements A1 Confocal Microscope, excitation and emission laser wavelength were selected based on the fluorescent proteins imaged, for 3D time lapse imaging, pinhole at 250 μm, Z stack scan setting, step at 0. 5 μm, 40-65 steps for each cell, 2-3min for one time point. The 3D migration movies were reconstituted and analyzed with Nikon Elements A1 software. Average remnant length was obtained by averaging the maximum length of remnants around the cell, number of stable remnants formed per cell were counted as semi-quantitative parameters. For fluorescence resonance energy transfer (FRET) imaging, cells were transduced sequentially with DDR2-Ypet and DDR2-Turquoise lentiviral vectors with blasticidine and puromycin drug resistances and selections. Cells were encapsulated in collagen gel and imaged. CFP and raw FRET signals was obtained by excitation with 458 nm and emission at 480 and 530 nm respectively, FRET signals were calculated as described [30].
Results
1. Generation of filamentous structures around cells cultured in 3D matrix is DDR2 dependent
To detect DDR2 subcellular localization and interaction with matrix in cells cultured in 3D collagen matrix, we used HT1080 human fibrosarcoma cells, which is commonly used to model 3D tumor cell motility [31][16]. To illuminate cell surface, cells were stably labeled withYpet-CAAX or mCherry-CAAX by lentivirus transduction. The mCherry-CAAX positive cells were subsequently transduced with another lentivirus expressing C-terminal Ypet tagged DDR2. Cells stably expressing DDR2-Ypet/mCherry-CAAX proteins were embedded in type I collagen gel for confocal imaging. To avoid potential edge effect from stiff glass bottom, only cells >150 μm above the glass bottom were selected for imaging. In control cells illuminated with Ypet-CAAX, there were two distinct types of protrusions exhibited, the major protrusions and hair-like protrusions around the major one (Figure 1A, indicated by yellow and white arrows respectively), we designated these two types of protrusions as pseudopodia and hairy protrusions respectively (Figure 1A). In cells expressing DDR2-Ypet, DDR2 containing punctate filamentous structures around the cell body were apparent (Figure 1B indicated by white arrows, supplemental movie 1).
Generation of filamentous structures in DDR2 dependent manner in 3D. overexpression of DDR2 in HT1080 cells leads to formation of filamentous structures. HT1080 cells expressing Ypet-CAAX (A, yellow arrow indicated pseudopodium, white arrow pointed to hairy protrusions) or co-expressing mCherry-CAAX and DDR2-Ypet(B) were encapsulated in collagen gel, confocal imaging scanning was performed, 3D image of each cell was reconstituted with Element A1 software, white arrow pointed to filamentous structures. C. Depletion of DDR2 in MDA-MB-231 cells by shRNAs, immunoblot using DDR2 and β1-tubulin antibodies were performed as indicated. D. Depletion of DDR2 leads to loss of filamentous structures in MDA-MB-231 cells. Cells were sequentially lentiviral transduced for expression of Ypet-CAAX and GFP-shRNA, DDR2-shRNAs as illustrated. Transduced cells were encapsulated in collagen gel for imaging. 3D images were obtained as described in A, scale bar represent 20 μm.
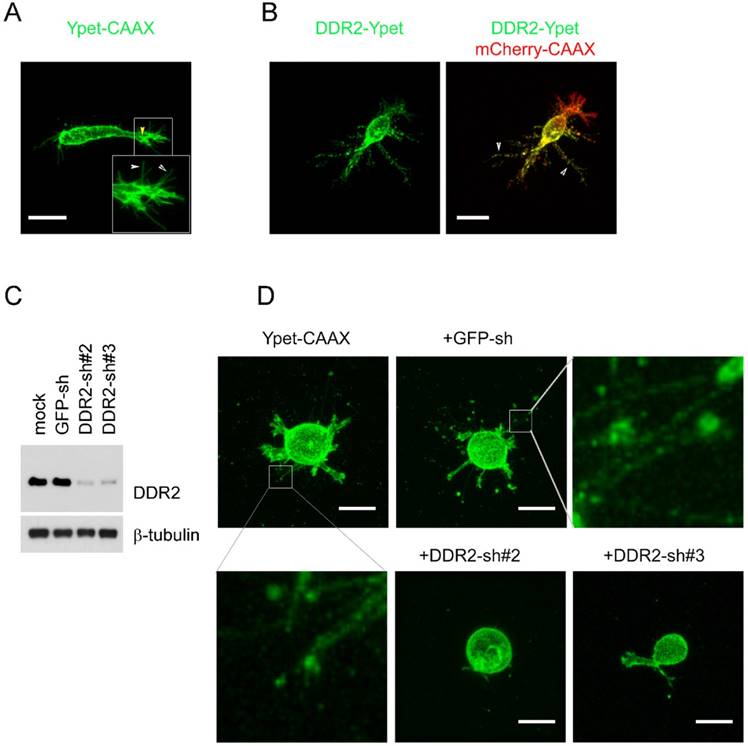
To determine whether DDR2 is required for the filamentous structure formation, we selected human breast cancer cell line MDA-MB-231 for loss of function study, this is an invasive cell line commonly used for 3D culture [32]. DDR2 was depleted in MDA-MB-231 cells with lentivirus expressing both DDR2 shRNA and Ypet-CAAX [29] (Figure 1C). The mock, GFP-sh control and DDR2 depleted cells were embedded into collagen gel for imaging. In mock and control MDA-MB-231 cells, filamentous tether structures around the cell body were readily observed. In cells with DDR2 depletion, the filamentous structures were abrogated, and two distinct shRNAs generated similar phenotype indicating it was not caused by off-target effects of RNAi (Figure 1D). These data suggest that formation of these filamentous structures is DDR2 dependent.
2. DDR2-dependent filamentous structures in 3D matrix are posterior remnants formed in the retracting phase of pseudopodia
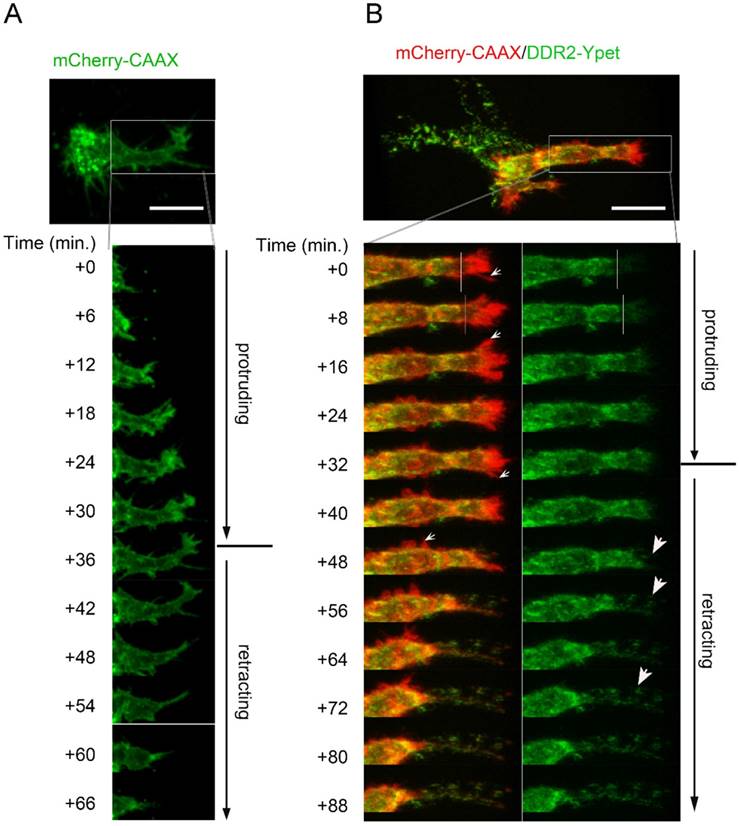
To determine what subcellular structures were these DDR2 containing filamentous structures, and to monitor the de novo formation of the structures, we set up timelapse live cell imaging in 3-D cultures. In control cells expressing mCherry-CAAX, we observed that pseudopodium was the invasive cell domain that kept penetrating the collagen matrix (Suppl. movie2). The dynamics of a pseudopodium displayed two typically migrating phases, a protruding phase and a retracting phase which took about 1-2 hours (Fig. 2A). In the protruding phase, we found that relative low amount of DDR2 was recruited to the top region of the growing pseudopodium, which did not form clusters (right side of white lines in Fig. 2B, green panel). In the retracting phase, however, DDR2 was recruited to the end of retracting pseudopodium, which formed clusters (Figure 2B indicated by white arrows). These clusters might adhere to the collagen fibers and served as anchors that peeled off the cell body when the pseudopodium was retracting. Cell membrane pieces were also stripped, leading to the formation of the posterior filament-like remnants between the cell body and the anchors on collagen fibers (Figure 2B). These DDR2 positive posterior remnants did not show contractility (suppl. Movie3). In control cells, we did not see posterior remnant formation at the rear of pseudopodia (Figure 2A). In conclusion, radial filamentous structures around the cells are posterior remnants made up of DDR2 clusters and membrane tethers, which were formed at the retracting phase of pseudopodia.
Filamentous structures are DDR2 based posterior remnants formed at retracting phase of pseudopodia. A. a pseudopodium dynamic has typical protruding phase and retracting phase. Lentiviral vector transduced HT1080 cells with mCherry-CAAX expression were embedded in collagen,3D time lapse cell migration imaging with confocal microscopy was performed. B. Dynamic formation of DDR2 based posterior remnants. Legend described in A was followed except HT1080 cells carrying DDR2-Ypet and mCherry-CAAX expression cassettes were used, lower left panel showed DDR2-Ypet/mCherry-CAAX merge, small white arrows pointed to hairy protrusions, right panel showed DDR2-Ypet channel alone, big arrow pointed to DDR2 clusters, scale bar represent 20 μm.
3. DDR2 in the posterior remnants remains clustered in the matrix
To determine if DDR2 in the posterior remnants still remains in clusters and adheres to the collagen fiber, we traced the remnants and proximal collagen fibers that were formed for at least 24 hours, and the cell bodies migrated out of our scope. We observed that DDR2 clusters in the posterior remnants co-localized with collagen fibers (Fig 3A).
DDR2 in the remnants remain clustered in the matrix. A. DDR2 based posterior remnants on collagen fibers when maternal cell left. HT1080 cells expressing DDR2-mCherry were sparsely embedded in collagen gel and remnants were imaged 48 hours later, collagen fibers were imaged by 488nm laser reflection and was pseudocolored as green. B. DDR2 cluster FRET signals at the retracting phase of a pseudopodium. HT1080 cells were sequentially transduced with DDR2-Turqoise and DDR2-Ypet expressing lentiviral vectors, embedded in collagen gel and imaged with450 nm laser excitation, Turquoise and Ypet emission signals were collected, FRET signals were calculated and analyzed as described in methods, scale bar represent 10 μm.
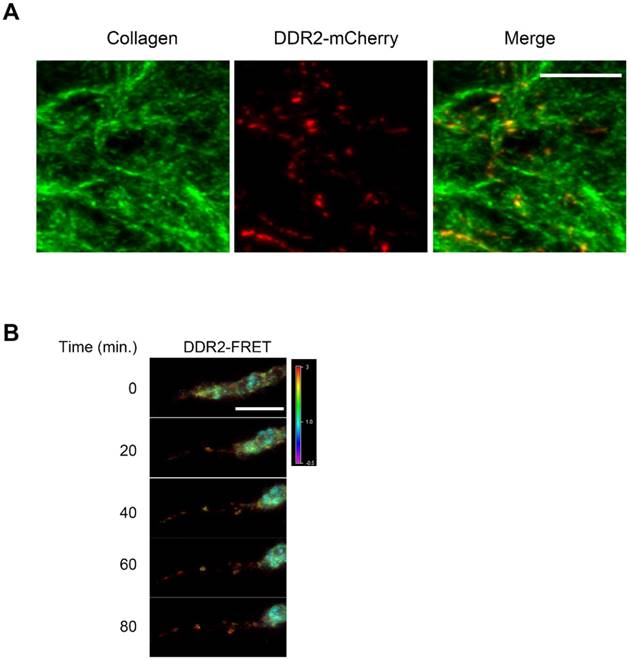
To investigate the DDR2 clustering status in a retracting pseudopodium and in the posterior remnants in 3-D culture, we established a FRET imaging system. HT1080 cells were sequentially transduced with the lentiviral vectors expressing DDR2-Turquoise and DDR2-Ypet proteins. Double positive cells were then embedded in 3D for FRET imaging. As shown in Fig. 3B, strong DDR2 FRET signals were detected at the top part of a retracting pseudopodium and in the posterior remnants, indicating that the DDR2 remnants sustained clusters after they peeled off the cell body. Based on these imaging data and Fig. 2B, it reveals that at the retracting phase of a pseudopodium, DDR2 in cluster state generates adhesions to the collagen fibers that forms posterior remnants and remains clustered in the matrix, it is not just released to the matrix as free molecules.
4. Matrix concentration negatively affects formation of DDR2-based posterior remnants
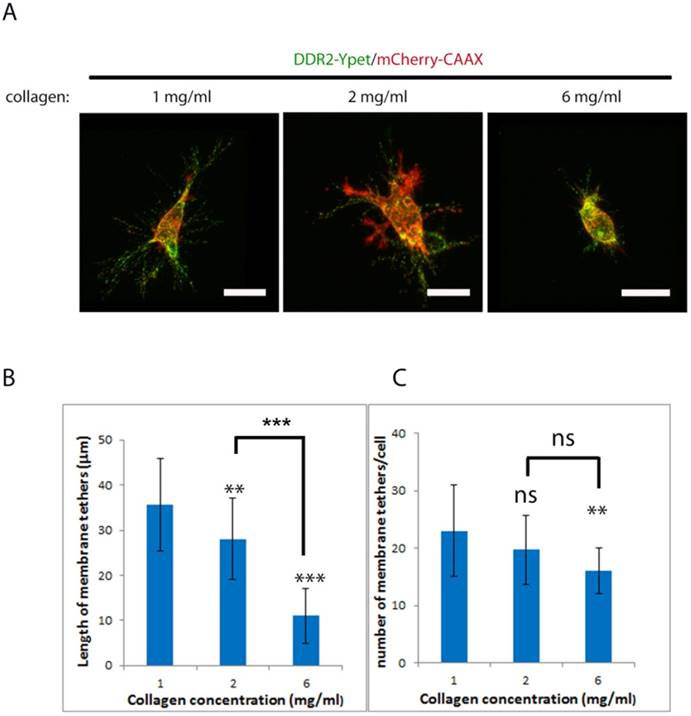
Physical properties of collagen matrix including stiffness play a critical role in cell migration, the requirement of MMPs for invasion [33][34] and oncogenic signaling [35]. Cancer cell invadopodium formation has been shown to be affected by the rigidity of matrix [36]. To investigate if altering collagen matrix stiffness affects DDR2 based posterior remnant formation and properties, we encapsulated DDR2-Ypet and mCherry-CAAX co-expressing HT1080 cells in three different concentrations of collagen gels, 1, 2 and 6 mg/ml (Figure 4A), the average remnant length around the cell and number of tethers from 20 discreet cells were imaged and measured with NIS-Element AR software (Figure 4B, C). It was noticed that the average remnant length in both 2mg/ml and 6 mg/ml of collagen gel was significantly shorter than that formed in 1mg/ml. The number of tethers formed in 6 mg/ml of collagen gel was also significantly reduced. This result suggests that stiff matrix shortens the DDR2 based remnant length; the higher the collagen gel concentration, the shorter the remnants formed and less DDR2 incorporation into the matrix.
Collagen concentration negatively affect formation of DDR2 based posterior remnants. Lentiviral vector transduced HT1080 cells with DDR2-Ypet and mCherry-CAAX expressions were embed in 1, 2, 6 mg/ml of collagen gels, imaged (A, scale bar represents 20 μm), average remnant tether length(B) and number of DDR2 based tethers(C) of each cell were calculated with NIS-Elements AR software, plotted (statistical analysis performed, ***P<0. 001 compare with 1 mg/ml, ns not significant, n=20).
5. DDR2-based clusters and integrin-based focal adhesions are distinct cell adhesive domains in 3D cultures
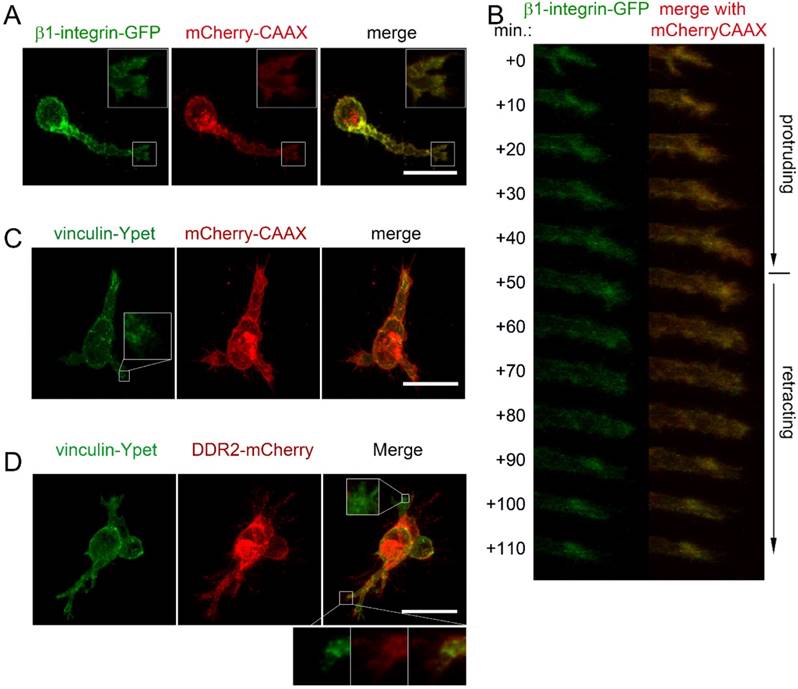
To determine if focal adhesions and DDR2 clusters are distinct cell adhesive domains in 3D cultures, we first explored the subcellular localization of integrin by transfecting HT1080-mCherry-CAAX labeled cells with β1-integrin-GFP expressing plasmid. As shown in figure 5A, β1-integrin-GFP labeled focal adhesions were detectable at the cell surface of the migrating cells, particularly at the tip of pseudopodia. Unlike DDR2, β1-integrin-GFP labeled focal adhesions were at the tips of the pseudopodium at both protruding and retracting phase. There were no obvious β1-integrin positive posterior remnants formed at the rear of a retracting pseudopodium (Figure 5B). Next, we monitored focal adhesions with vinculin-Ypet and cell surface with mChery-CAAX. DDR2 based clusters were illustrated with DDR2-mCherry by sequential lentiviral vector transductions for stable expressions. As expected, vinculin-Ypet illuminated focal adhesions (Figure 5C), DDR2-basedposteriorclusters and vinculin labeled focal adhesions were apparently different. The two domains did not overlap with each other. Interestingly, unlike focal adhesions that are localized at all pseudopodia, DDR2 based clusters were present only at certain pseudopodia (Figure 5D). It is likely that DDR2-negative pseudopodia were at protruding phase, whereas others were at retracting phase. This result suggests that DDR2 based posterior clusters and integrin based focal adhesions are distinct adhesive domains in 3D culture.
DDR2 based posterior clusters and integrin based focal adhesions are distinct cell adhesive domains. Lentiviral vector transduced HT1080 cells carrying β1-integrin-GFP and mCherry-CAAX expressions were collagen embedded and imaged. β1-integrin based focal adhesions at the tip of a pseudopodium was reconstituted (A). β1-integrin recruited to the end of pseudopodia at both protruding and retracting phase (B). HT1080 with vinculin-Ypet/mCherry (C) or with vinculin-Ypet/DDR2-mCherry (D) expressions were collagen embedded and imaged, scale bar represents 20 μm.
6. Formation of DDR2 based posterior remnants requires integrin-dependent pseudopodium activities
The matrix stiffness depends on collagen concentration, it has a direct impact on invadopodium formation and cell invasion [33][36][37]. Meanwhile, DDR2-based posterior remnants are formed at the rear of a retracting pseudopodium. Therefore, it is pertinent that the pseudopodium activities, including both the depth of pseudopodia penetrated and the frequency of active pseudopodia formed in 3D conditions, determine the length and number of DDR2-containing membrane tether. If so, blocking pseudopodium activities could inhibit DDR2-based posterior remnant formation. To test this hypothesis, we carried out two independent approaches to block pseudopodia activities. First, we inhibited integrin function by knocking down β1-integrin expression with shRNA or by using antibody that specifically blocksβ1-integrin. Second, we inhibited Rac activity by depleting p130Cas with shRNA or by using Rac inhibitor. Focal adhesion proteins have been shown to play distinct roles in 3D migration by regulating pseudopodium protruding activity and matrix deformation [16]. Blocking β1-integrin in invading HT1080 cells leads to loss of motility [38] and pseudopodia activities [16]. To determine if inhibiting protruding activity affect DDR2-based posterior remnant formation, we first depleted β1-integrin in DDR2-Ypet transduced HT1080 cells with lentiviral vector mediated RNAi. The depletion efficiency was about 80% as detected by immunoblot (Figure 6A). Depletion of β1 integrin significantly inhibited pseudopodium formation (Figure 6B). The DDR2 positive posterior remnant formation was also inhibited (Figure 6C). Similar effects were observed with an alternative method to block integrin activity by using β1-integrin neutralizing antibody (mAb13). Interestingly, blocking β1-integrin did not abrogate hairy protrusions or pseudopodium initiation, but halted the maturation of pseudopodia from their precursor. DDR2 did not co-localize with actin in hairy protrusions or pseudopodium precursors (Figure 6C, mAb13 treated). These results suggest that the maturation of integrin-dependent pseudopodiumis required for the formation of DDR2 positive posterior remnants. DDR2 does not regulate pseudopodium maturation.
Formation of DDR2 based posterior remnants requires integrin dependent pseudopodium activities. (A). Immunoblot to test β1-integrin shRNA depletion efficiency. (B). HT1080 cells with DDR2-Ypet expression were transduced with lentiviruses carrying mCherry-CAAX and concurrentβ1-integrin-shRNA or p130Cas-shRNA expression cassette as indicated. Cells were collagen embedded and imaged. (C). HT1080 cells with DDR2-Ypet expression were collagen embedded, β1-integrin blocking antibody (10 μg/ml), MMP inhibitor Marimastat (2 μg/ml), myosin II inhibitor blebstatin (30 μM), actin polymerization inhibitor Latrunculin B (30 μM), Rac inhibitor (50 μm) were added for overnight culture, cells were fixed, stained with Phalloidin-Alexa-568, and imaged, white arrow pointed to DDR2-Ypet, red arrow pointed to actin, average remnant tether length and number of DDR2 based tethers (D) of each cells were calculated with NIS-Elements AR software, plotted (statistical analysis performed,*P<0. 05, **P<0. 01, ***P<0. 001 compare with control cells, n=20), scale bar represents 20 μm.
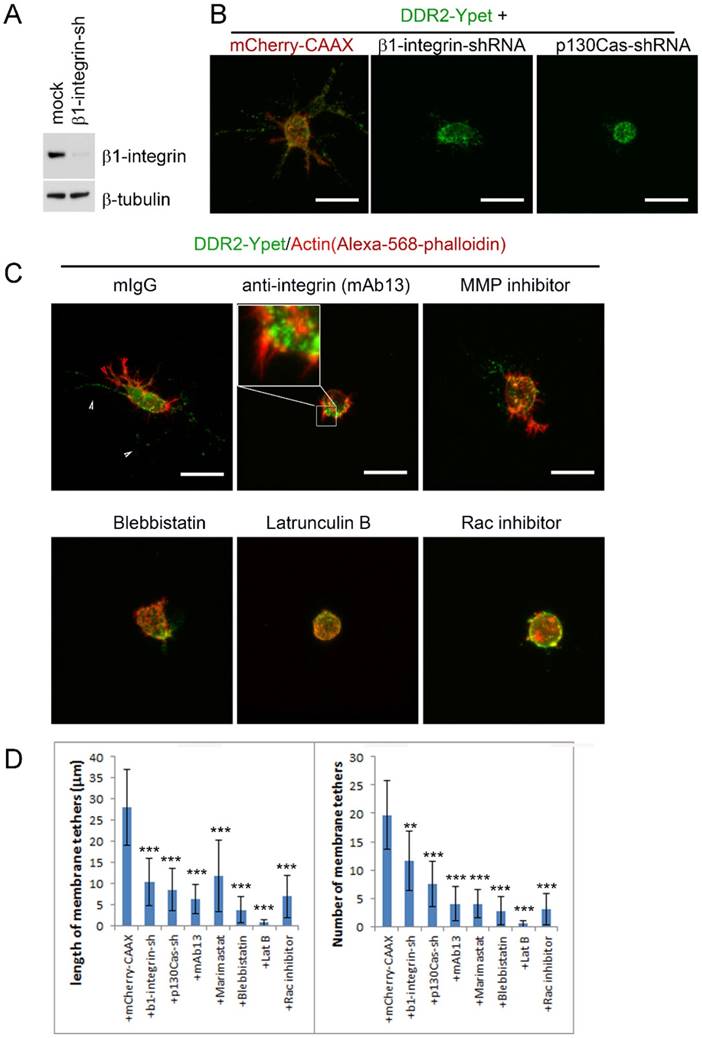
Rho family small GTPase Rac has been shown critical for mesenchymal migration by promoting lamellipodium formation through actin polymerization via WAVE2-Arp2/3 at the leading edge [39][40]. The protein p130Cas forms complex with Crk to activate Rac1 through DOCK180 [41]. Depletion of p130Cas inhibits pseudopodium formation in 3D cultured HT1080 cells [16]. We next blocked pseudopodium formation by inhibiting Rac through either knocking down p130Cas with RNAi or using pharmaceutical inhibitor of Rac. In both maneuvers, DDR2 positive posterior remnant formation was inhibited, no remnants were observed around the DDR2-Ypet transduced cells. In contrast to β1-integrin, Rac activity seemed to be critical for pseudopodium initiation, neither pseudopodium precursors nor hairy protrusions were observed in Rac inhibited cells (Figure 6B, C, D).
Actin polymerization and actomyosin-mediated contraction are associated with invadopodium and the metastasis of cancer cells [37][42][43]. Although actin is not a component of DDR2 positive posterior remnants (Figure 6B, red arrows vs white arrows in mIgG treated), actin polymerization inhibitor latrunculin B (latB) abrogated DDR2 based posterior remnant formation, Pseudopodium and hairy protrusion initiations were also blocked, similar to those observed in Rac inhibitor treated cells. Neither pseudopodium precursors nor hairy protrusions were observed around these cells (Figure 5C, D, latB and Rac inhibitor treated), whereas inhibiting myosin II activity by blebbistatin suppressed remnant formation and pseudopodium maturation (Figure 6C, D, blebbistatin treated).
In addition to the regulatory roles of β1-integrin and Rac in pseudopodium activity and tumor metastasis, MT1-MMP is also required for mesenchymal migration and morphology in 3D cultures [31], [38]. We investigated the effects of MT1-MMP inhibition on posterior remnants. Cells treated with Marimastat (a specific inhibitor for MT1-MMP) showed inhibitory effect on DDR2 positive posterior remnant formation (Figure 6C, D).
Taken together, DDR2 positive posterior remnants are trajectory of pseudopodium activity in collagen gel in 3D. Their formation requires pseudopodium maturation. Rac mediated actin polymerization is critical for pseudopodium initiation, whereas integrin activation, myosin II mediated contraction and MT1-MMP mediated proteolysis of ECM are required for pseudopodium maturation. Aberration of this pseudopodium activity impairs the posterior remnant formation. DDR2 is not able to rescue the loss of function of integrin.
7. DDR2 collagen binding instead of kinase activity is required for DDR2 based posterior remnant formation
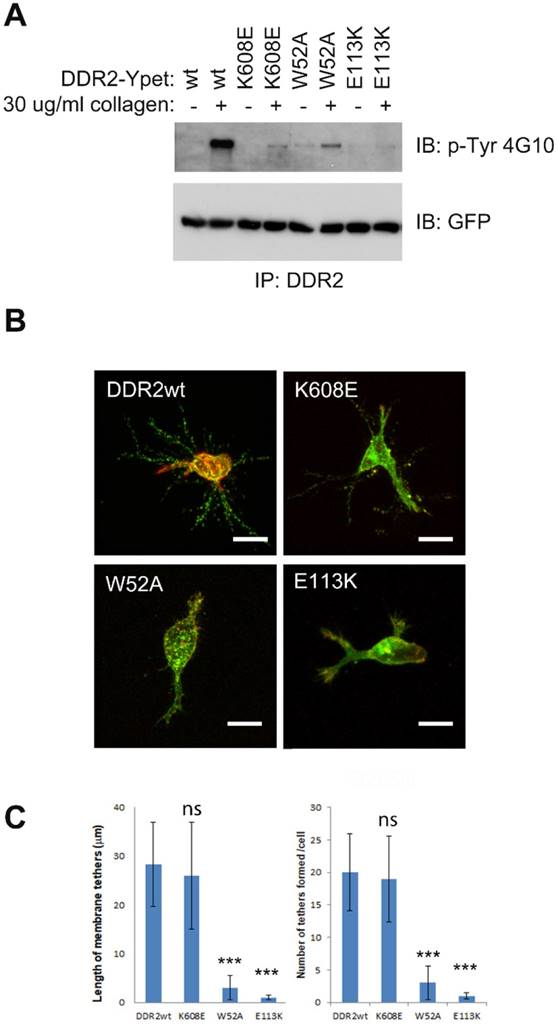
Although DDR dimerization is mediated by the leucine zipper motif in the transmembrane domain [27], oligomerization is instead mediated by their ECD upon collagen binding. DDR2 oligomerization mediates high-affinity interactions with their ligand [9][10]. DDR2 mutant lacking collagen binding activity is defective in collagen induced receptor activation [44]. To determine whether kinase or collagen binding activities are required for DDR2 based posterior remnant formation, we generated several DDR2 mutants including kinase dead (K608E) [45], collagen binding inactive (W52A) [46], and DDR2 mutant (E113K) associated with Spondylo-meta-epiphyseal dysplasia (SMED) with short limbs and abnormal calcifications (SMED-SL) [44]. In contrast to wild type DDR2, all the DDR2 mutants showed diminished or abrogated kinase activation upon binding to collagen (Figure 7A). However, cells with kinase dead DDR2 mutation (K608E) formed DDR2-positive posterior remnants normally, whereas cells with W52A, E113K DDR2 mutations exhibited abrogated posterior remnants (Figure 7B, 7C). These results suggest that DDR2 kinase activity is dispensable forDDR2 based posterior remnant formation, while the DDR2 collagen binding activity for oligomerization is crucial.
DDR2 collagen binding instead of kinase activity is required for DDR2 based posterior remnant formation. (A). DDR2 tyrosine phosphorylation of K608E, W52A, E113K mutants was impaired upon collagen stimulation. Serum starved HT1080 cells expressing Ypet tagged DDR2 or mutants were stimulated with 30 μg/ml collagen I for 2 hours, washed with cold PBS and lysed, immunoprecipitated with DDR2 antibody pre-charged to protein G agarose, and washed, immunobloted with p-Tyr 4G10 or GFP antibody. (B). DDR2 W52A, E113K mutants show diminished posterior remnant formation in 3D. Cells coexpressing Ypet tagged DDR2 or mutants and mCherry-CAAX were embedded in 2 mg/ml of collagen I for 16 hours before fixation and imaging. Average remnant tether length and number of DDR2 based tethers (C) of each cell were calculated with NIS-Elements AR software, plotted (statistical analysis performed, ***P<0. 001 compare with wt control cells, n=20), scale bar represents 20 μm.
Discussion
In this study, we determined that DDR2 clusters are released and incorporated into collagen matrix via posterior remnant tethering in an integrin-dependent manner. The remnants form at the distal end of retracting pseudopodia. Their formation depends on the availability of integrin initiated and Rac-regulated pseudopodium activity. The collagen binding activity of DDR2 is required for the tethering, while the kinase activity of DDR2 is not required for this process.
We showed that DDR2 at the retracting phase of a pseudopodium clusters before tethering. Since membrane tether formation requests pulling force applied to lipid bilayers [47], this pulling force has to exceed certain threshold before the cell membrane eventually separates from the cytoskeleton [23]. The DDR2 cluster adhesive strength to the collagen fiber needs to be firm enough, and the pulling force generated from the pseudopodium retraction needs to exceed the threshold to stripe the DDR2 clusters off the maternal cells. Therefore, DDR2 is not only a RTK signaling molecule to convey extracellular matrix cues, but also an adhesion molecule once clustered at the rear of a retracting pseudopodium, these adhesive molecules mediate posterior tethering for DDR2 incorporation into collagen matrix, they are not just released to the matrix as free molecules.
Our results suggest that human disease associated DDR2 mutants (W52A and E113K) have impaired posterior remnant formation. These mutants lack collagen binding activity and oligomerization [44][46]. It reveals that lack of DDR2 oligomerization is associated with deficient remnant formation and RTK signaling/human development, in which high affinity to collagen fibrosis required. As we observed DDR2 clusters when tethering, these clustered DDR2 could be oligomeric form of DDR2.
Though activated integrins are also adhesive to collagen fibers, we did not observe formation of integrin-based posterior remnants in the collagen matrix. In addition, our results suggest that activeβ1 integrin is required for pseudopodium maturation. Although DDR2 is also a collagen adhesive molecule, it is not able to rescue the loss of function of β1-integrin, suggesting these two different collagen receptors play distinct roles in cell adhesion and migration.
DDR ECDs are powerful regulators of collagen deposition and fibrillogenesis. It is reported that DDR ECD expression reduces the rate and quantity of collagen deposition and induces significant changes in fiber morphology and matrix mineralization [8], oligomeric DDR2 inhibits fibrillogenesis of collagen better than dimeric or monomeric forms [7][8]. Our results suggest that DDR2 clusters formed by posterior tethering can firmly adhere to the collagen fibers with sustained persistence, the high-density incorporation and persistence of DDR2 positive remnants in the matrix may generate sustained perturbation to ECM structure. Overall, our findings unveil a special mechanism for incorporation of DDR2 clusters into collagen matrix by posterior remnant tethering in an integrin-dependent manner, this provides cellular basis for the role of DDR2 in regulating matrix remodeling.
Supplementary Material
Movie S1.
Movie S2.
Movie S3.
Acknowledgements
The authors gratefully acknowledge Drs. Gregory D. Longmore at Washington University in St. Louis, Denis Wirtz at Johns Hopkins University and Stephanie I. Fraley at University of California in San Diegofor their helps. This work was supported by National Natural Science Foundation of China (grants 81673443 and 81373303).
Author Contributions
YF, JL and TL designed the study, performed the imaging, data analysis and wrote the paper. JEL, HC, BMW constructed part of reagents and assisted some of the experiments. All authors analyzed the results and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. OzbekS BalasubramanianPG, Chiquet-Ehrismann R et al. The evolution of extracellular matrix. Molecular biology of the cell. 2010;21:4300-5
2. Leitinger B. Transmembrane collagen receptors. Annual review of cell and developmental biology. 2011;27:265-90
3. Pawelec KM, Best SM, Cameron RE. Collagen: a network for regenerative medicine. J Mater Chem B Mater Biol Med. 2016;4:6484-6496
4. Gubbiotti MA, Vallet SD, Ricard-Blum S. et al. Decorin interacting network: A comprehensive analysis of decorin-binding partners and their versatile functions. Matrix Biol. 2016;55:7-21
5. Halasz K, Kassner A, Morgelin M. et al. COMP acts as a catalyst in collagen fibrillogenesis. J Biol Chem. 2007;282:31166-73
6. Kalamajski S, Oldberg A. Fibromodulin binds collagen type I via Glu-353 and Lys-355 in leucine-rich repeat 11. J Biol Chem. 2007;282:26740-5
7. Blissett AR, Garbellini D, Calomeni EP. et al. Regulation of collagen fibrillogenesis by cell-surface expression of kinase dead DDR2. J Mol Biol. 2009;385:902-11
8. Flynn LA, Blissett AR, Calomeni EP. et al. Inhibition of collagen fibrillogenesis by cells expressing soluble extracellular domains of DDR1 and DDR2. J Mol Biol. 2010;395:533-43
9. Wang C, Yeung D, Wellerming J. et al. Role of DDR2 ECD Oligomerization in Binding to Collagen. Microscopy and Microanalysis. 2016;22:1126-1127
10. Yeung D, Chmielewski D, Mihai C. et al. Oligomerization of DDR1 ECD affects receptor-ligand binding. J Struct Biol. 2013;183:495-500
11. Slack BE, Siniaia MS, Blusztajn JK. Collagen type I selectively activates ectodomain shedding of the discoidin domain receptor 1: involvement of Src tyrosine kinase. J Cell Biochem. 2006;98:672-84
12. Shitomi Y, Thogersen IB, Ito N. et al. ADAM10 controls collagen signaling and cell migration on collagen by shedding the ectodomain of discoidin domain receptor 1 (DDR1). Mol Biol Cell. 2015;26:659-73
13. FuHL Sohail A, Valiathan RR et al. Shedding of discoidin domain receptor 1 by membrane-type matrix metalloproteinases. J Biol Chem. 2013;288:12114-29
14. Pankova K, Rosel D, Novotny M. et al. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol Life Sci. 2010;67:63-71
15. Mierke CT. Physical view on migration modes. Cell Adh Migr. 2015;9:367-79
16. Fraley SI, Feng Y, Krishnamurthy R. et al. A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat Cell Biol. 2010;12:598-604
17. FraleySI Feng Y, Giri A et al. Dimensional and temporal controls of three-dimensional cell migration by zyxin and binding partners. Nature communications. 2012;3:719
18. Wang Y, McNiven MA. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. The Journal of cell biology. 2012;196:375-85
19. Borza CM, Pozzi A. Discoidin domain receptors in disease. Matrix Biol. 2014;34:185-92
20. WangCZ Su HW, Hsu YC et al. A discoidin domain receptor 1/SHP-2 signaling complex inhibits alpha2beta1-integrin-mediated signal transducers and activators of transcription 1/3 activation and cell migration. Molecular biology of the cell. 2006;17:2839-52
21. Yeh YC, Wang CZ, Tang MJ. Discoidin domain receptor 1 activation suppresses alpha2beta1 integrin-dependent cell spreading through inhibition of Cdc42 activity. Journal of cellular physiology. 2009;218:146-56
22. Eswaramoorthy R, Wang CK, Chen WC. et al. DDR1 regulates the stabilization of cell surface E-cadherin and E-cadherin-mediated cell aggregation. J Cell Physiol. 2010;224:387-97
23. Pospieszalska MK, Lasiecka I, Ley K. Cell protrusions and tethers: a unified approach. Biophysical journal. 2011;100:1697-707
24. Sundd P, Gutierrez E, Koltsova EK. et al. 'Slings' enable neutrophil rolling at high shear. Nature. 2012
25. SperandioM Smith ML, Forlow SB et al. P-selectin glycoprotein ligand-1 mediates L-selectin-dependent leukocyte rolling in venules. The Journal of experimental medicine. 2003;197:1355-63
26. Leitinger B. Molecular analysis of collagen binding by the human discoidin domain receptors, DDR1 and DDR2. Identification of collagen binding sites in DDR2. The Journal of biological chemistry. 2003;278:16761-9
27. Noordeen NA, Carafoli F, Hohenester E. et al. A transmembrane leucine zipper is required for activation of the dimeric receptor tyrosine kinase DDR1. J Biol Chem. 2006;281:22744-51
28. Zhang K, Corsa CA, Ponik SM. et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nature cell biology. 2013;15:677-87
29. Feng Y, Nie L, Thakur MD. et al. A multifunctional lentiviral-based gene knockdown with concurrent rescue that controls for off-target effects of RNAi. Genomics Proteomics Bioinformatics. 2010;8:238-45
30. Kardash E, Bandemer J, Raz E. Imaging protein activity in live embryos using fluorescence resonance energy transfer biosensors. Nat Protoc. 2011;6:1835-46
31. Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. The Journal of cell biology. 2009;185:11-9
32. Lee GY, Kenny PA, Lee EH. et al. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4:359-65
33. ZamanMH Trapani LM, Sieminski AL et al. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10889-94
34. Fraley SI, Wu PH, He L. et al. Three-dimensional matrix fiber alignment modulates cell migration and MT1-MMP utility by spatially and temporally directing protrusions. Sci Rep. 2015;5:14580
35. Levental KR, Yu H, Kass L. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891-906
36. Parekh A, Ruppender NS, Branch KM. et al. Sensing and modulation of invadopodia across a wide range of rigidities. Biophysical journal. 2011;100:573-82
37. Alexander NR, Branch KM, Parekh A. et al. Extracellular matrix rigidity promotes invadopodia activity. Current biology: CB. 2008;18:1295-9
38. Wolf K, Mazo I, Leung H. et al. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. The Journal of cell biology. 2003;160:267-77
39. Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. The EMBO journal. 1998;17:6932-41
40. Suetsugu S, Miki H, Yamaguchi H. et al. Enhancement of branching efficiency by the actin filament-binding activity of N-WASP/WAVE2. Journal of cell science. 2001;114:4533-42
41. Cheresh DA, Leng J, Klemke RL. Regulation of cell contraction and membrane ruffling by distinct signals in migratory cells. The Journal of cell biology. 1999;146:1107-16
42. Koch TM, Munster S, Bonakdar N. et al. 3D Traction forces in cancer cell invasion. PloS one. 2012;7:e33476
43. Poincloux R, Collin O, Lizarraga F. et al. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1943-8
44. Ali BR, Xu H, Akawi NA. et al. Trafficking defects and loss of ligand binding are the underlying causes of all reported DDR2 missense mutations found in SMED-SL patients. Human molecular genetics. 2010;19:2239-50
45. Labrador JP, Azcoitia V, Tuckermann J. et al. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO reports. 2001;2:446-52
46. Carafoli F, Bihan D, Stathopoulos S. et al. Crystallographic insight into collagen recognition by discoidin domain receptor 2. Structure. 2009;17:1573-81
47. Baoukina S, Marrink SJ, Tieleman DP. Molecular structure of membrane tethers. Biophysical journal. 2012;102:1866-71
Author contact
![]() Corresponding authors: Yunfeng Feng, PhD. Email: fengyunfeng3com or Jun Liu, PhD. Email: junliuedu.cn
Corresponding authors: Yunfeng Feng, PhD. Email: fengyunfeng3com or Jun Liu, PhD. Email: junliuedu.cn