Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(8):849-857. doi:10.7150/ijbs.24539 This issue Cite
Research Paper
PRWHMDA: Human Microbe-Disease Association Prediction by Random Walk on the Heterogeneous Network with PSO
Chuanyan Wu1, Rui Gao1 ![]() , Daoliang Zhang1, Shiyun Han2, Yusen Zhang3
, Daoliang Zhang1, Shiyun Han2, Yusen Zhang3
1. School of Control Science and Engineering, Shandong University, Jinan, 250061, China.
2. General Clinic, The No. 2 People's Hospital of Tianqiao, Jinan, 250032, China.
3. School of Mathematics and Statistics, Shandong University, Weihai, 264209, China.
Received 2017-12-24; Accepted 2018-2-28; Published 2018-5-22
Abstract

Microorganisms resided in human body play a vital role in metabolism, immune defense, nutrition absorption, cancer control and protection against pathogen colonization. The changes of microbial communities can cause human diseases. Based on the known microbe-disease association, we presented a novel computational model employing Random Walking with Restart optimized by Particle Swarm Optimization (PSO) on the heterogeneous interlinked network of Human Microbe-Disease Associations (PRWHMDA) (see Figure 1). Based on the known human microbe-disease associations, we constructed the heterogeneous interlinked network with Cosine similarity. The extended random walk with restart (RWR) method was derived to get the potential microbe-disease associations. PSO was utilized to get the optimal parameters of RWR. To evaluate the prediction effectiveness, we performed leave one out cross validation (LOOCV) and 5-fold cross validation (CV), which got the AUC (The area under ROC curve) of 0.915 (LOOCV) and the average AUCs of 0.8875 ± 0.0046 (5-fold CV). Moreover, we carried out three case studies of asthma, inflammatory bowel disease (IBD) and type 1 diabetes (T1D) for the further evaluation. The result showed that 10, 10 and 9 of top-10 predicted microbes were verified by previously published experimental results, respectively. It is anticipated that PRWHMDA can be effective to identify the disease-related microbes and maybe helpful to disclose the relationship between microorganisms and their human host.
Keywords: disease-microbe association prediction, random walk with restart, particle swarm optimization.
Introduction
Microorganisms include bacteria, archaea, protists, fungi and viruses [1]. In human body, there are numerous microorganisms colonizing the gut, skin, uterus, lung, saliva, and so on [2]. They play an important role in human health, metabolism, immune defense, nutrition absorption, cancer control and protection against pathogen colonization [3, 4].
Numerous lab experiments have revealed that many diseases are associated with the changes of microorganisms. For example, evidences from both rodent models and human researches suggested that the composition of the gut microorganisms had a significant influence on the immune system and might influence T1D risks [5]. Fusobacterium was much more abundant in asthmatic patients comparing with healthy group [6]. Some evidences revealed that lecithinase-negative Clostridium and Lactobacillus were detected to be more abundant in colorectal carcinoma patients [7, 8]. More and more clinic studies revealed new associations between microbes and human diseases.
Computational biology can help identify essential gene [9, 10] and secretory protein [11] in disease, identify immunoglobulin [12], reveal the relationship between diseases and environmental factors, predict drug reactions, identify the early diagnostic markers of diseases, and carry out computer-aided drug design and production [13]. Recently, a lot of computational biologic methods provide novel and effective tools for identifying the key associations between microbes and diseases. Ma et al. constructed a human microbe-disease associations database, which facilitated the study of the relationships between microbes and diseases [8]. Based on the Gaussian kernel similarity, some prediction methods were studied as follows. Chen et al. presented a computational model (KATZHMDA) based on KATZ method, the number of walks, and their lengths for calculating the potential association probability between microbes and diseases [14]. For high prediction accuracy, Huang et al. proposed the prediction model (PBHMDA) with a depth-first search algorithm to get all possible paths between microbes and diseases [15]. Wang et al. adopted Laplacian regularized least squares classifier (LRLSHMDA) to construct a prediction model [16]. Shen et al. developed a model utilizing Collaborative Matrix Factorization for Human Microbe-Disease Association prediction (CMFHMDA) [17]. Various Random Walk Methods were utilized to rank the probabilities of predicted microbe-disease association. Based on traditional random walk with three parameters on the heterogeneous network constructed by Spearman correlation, Shen et al. derived a method (RWRHMDA) for prioritization of candidate microbes to predict disease-microbe association [18]. Zou et al. developed a computational model (BiRWHMDA) to predict potential microbe-disease associations by bi-random walk on the heterogeneous network [19]. It is anticipated that various computational prediction models could improve the identification of novel microbe-disease association.
Researching the interaction of the microorganism with disease will provide critical insight into the pathogenesis of disease and the development of strategies to prevent and treat disease. However, the knowledge in this field is far from satisfied. The aim of this article is to present a computational model to predict potential microbe-disease associations based on the known ones. In this study, we put forward a novel computational model of extended Random Walking with Restart optimized by PSO on the heterogeneous interlinked network of Human Microbe-Disease Associations (PRWHMDA) (see Figure 1). Firstly, under the hypothesis that microbes involved in similar disease tend to be functionally similar, we computed the similarities of diseases and microbes with Cosine measurement, respectively. With the disease-disease similarity matrix, the network of diseases was constructed. The microbe network was dealt with the same method. Combing the above-mentioned networks with the known microbe-disease network, we got the heterogeneous interlinked network. Then, the extended RWR method was presented to get the potential associations. In RWR method, we set a disease and the known associated microbes as seed nodes, after several steps, the probabilities of reaching each node (microbe) could be calculated. The higher the probability of reaching the node was, the more believable the association was. PSO was utilized to get the four optimal parameters of RWR. With PSO, PRWHMDA model performed well with 325 of 450 associations ranked in top 10. To evaluate the prediction performance, we performed the LOOCV and 5-fold CV, made the comparison with some state-of-the-art methods and analyzed the three case studies on the proposed model. The CV verified the effectiveness of the proposed model with the AUC of 0.915 (LOOCV) and average AUCs of 0.8875 ± 0.0046 (5-fold CV), which are higher than some other existing methods. In our case studies, 10, 10 and 9 of top-10 inferred microbes have been confirmed to have associations with asthma, IBD and T1D according to the literature evidences. The results indicate that PRWHMDA is effective to identify the disease-related microbes and could be helpful in future to disclose the relationship between microorganisms and their human host.
Flowchart of PRWHMDA.
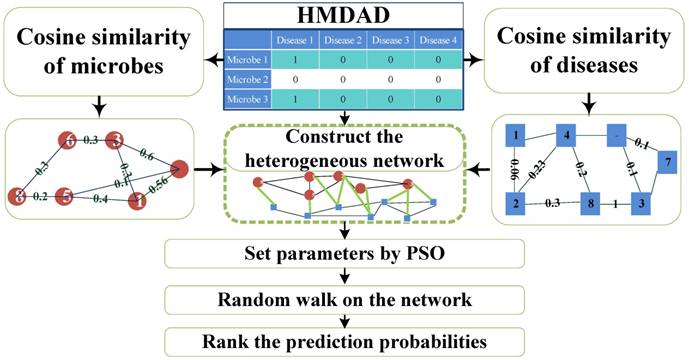
The paper is organized as follows. In Materials and Methods section, we describe the similarity calculation using Cosine measurement. The construction of networks of microbes, diseases and microbe-disease associations are also presented. Furthermore, the random walk with restart method is outlined in this section. In Results and Discussion section, we show the optimized parameters by PSO of RWR. In addition, the results of CV, comparison results with state-of-the-art methods and the top 10 most potential microbes for asthma, IBD and T1D are presented, respectively. In Conclusion part, we discuss the results succinctly and conclude the paper.
Materials and Methods
Materials
We adopted the microbe-disease association data set called HMDAD which was constructed by Ma et al. [8]. HMDAD contains manually curated 483 microbe-disease associations, which involves 39 diseases and 292 microbes. As some associations have more than one evidences, we extracted 450 distinct disease-microbe associations. Based on these known microbe-disease associations, we constructed the network of diseases, the network of microbes, and the network of microbe-disease associations, respectively. In this paper, Nd = 39 denotes the number of diseases, and Nm = 292 denotes the number of microbes.
Construction of the disease network
In the disease-disease similarity network, there are 39 nodes representing the diseases. The value of the edge between two nodes represents the similarity of the two diseases. We utilized the Cosine similarity measurement to calculate disease similarity with the known microbe-disease associations.
The disease-disease similarity matrix was
(1)
where sd(di,dj) calculated by (2) denoted the similarity of the two diseases di and dj.
The similarity of two diseases di and dj was calculated by Cosine similarity as
(2)
where
(3)
MS(di) denoted the associations of disease i with all the microbes,
(4)
Mm(di) denoted whether microbe m was associated with disease i.
Therefore, the disease-disease similarity network could be constructed by nodes representing the diseases and edges representing the similarities defined by (1).
Construction of microbe network
Similarly, in the microbe-microbe similarity network, there are 292 nodes representing the microbes. The value of the edge between two nodes is the similarity of the two microbes.
The microbe-microbe similarity matrix was
(5)
where sm(mi,mj) calculated by (6) denoted the similarity of the two microbes mi and mj.
The similarity of two microbes mi and mj was defined by Cosine similarity as
(6)
where
(7)
DS(mi) denoted the associations of microbe i with all the diseases,
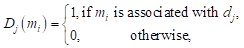
which denoted whether disease j was associated with microbe i.
Therefore, the microbe-microbe similarity network could be constructed according to adjacency matrix defined by (5).
Construction of heterogeneous interlinked network
Based on the two networks, i.e., microbe-microbe network and disease-disease network, we used the known microbe-disease associations to construct the heterogeneous interlinked network. In the heterogeneous interlinked network, there are two types of node sets defined as D = [d1, d2, …, dNd] and M = [m1, m2, …, mNm] corresponding to diseases and microbes, respectively.
The associations between microbes and diseases was
(8)
where
(9)
md(di,dj) denoted the association or non-association pattern between microbe mi and disease dj.
The adjacency matrix of the network was
(10)
where SM denoted the similarity of microbes inferred by (5), SD denoted the similarity of diseases inferred by (1), MD defined by (8) denoted the associations of microbes and diseases, DM denoted the transpose of the matrix MD. In this way, the heterogeneous interlinked network was constructed.
Random walk with restart on heterogeneous interlinked network
RW was first put forward by Karl Pearson in 1905 [20]. In Bioinformatics, it has been applied to disease-gene prediction [21] and inferring gene-phenotype relationship [22]. To emphasize the different weights of jumping between different networks, we extended the traditional RW method to predict microbe-disease association.
The walker can walk in disease network and microbe network, respectively and jump to the other network by disease-microbe network. Thus, at each step, the probabilities of reaching the nodes could be calculated. For the (s+1)-th step, the probability was
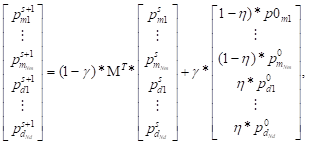
where 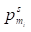 denoted the s-th iteration probability of reaching microbe mi,
denoted the s-th iteration probability of reaching microbe mi, 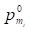 denoted the initial probability of microbe mi, γ∈(0, 1) denoted the restart probability, η denoted the weights of the two networks. MT defined by (11) denoted the transition matrix as
denoted the initial probability of microbe mi, γ∈(0, 1) denoted the restart probability, η denoted the weights of the two networks. MT defined by (11) denoted the transition matrix as
(11)
SM′ that represented the transition probability from a microbe to a microbe was defined by
(12)
where
,
sm′(i,j) denoted the transition probability from microbe mi to mj, λ∈(0, 1) denoted that the walker stayed in the microbe network with the weight (1-λ).
MD′ that represented the transition probability from microbe network to disease network was defined by
(13)
where
,
md′(i,j) denoted the transition probability from microbe mi to disease dj, λ∈(0, 1) denoted that the walker jumped from the microbe network to disease network with the weight λ.
DM′ that represented the transition probability from disease network to microbe network was defined by
(14)
where
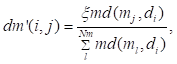
dm′(i,j) denoted the transition probability from disease di to microbe mj, ξ∈(0, 1) denoted the weight of jumping from disease to microbe network.
SD′ represented the transition probability from a disease to a disease, defined by
(15)
where
,
sd′(i,j) denoted the transition probability from disease di to disease dj, (1-ξ) denoted the weight of staying in disease network.
In (12)-(15), the value was set to be 0 if the denominator was 0. After several iterations, the difference between the vectors ps+1 and ps become negligible such as 1e-10. Thus, we got a stationary probability distribution which represented a proximity measure from the seed node(s) to each node.
Determination of parameters based on PSO with mutation
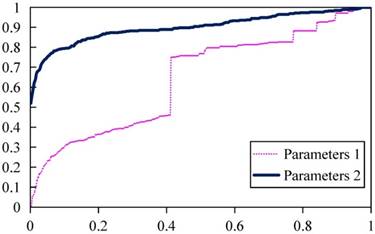
In the RW algorithm, the parameters λ, γ, η, and ξ are important to the prediction performance. For example, in Figure 2, if with parameters 1 (λ = 0.99, γ = 0.078, η = 0.44, ξ = 0.10), the ROC of LOOCV is shown as the pink curve with AUC = 0.49. While with parameters 2 (λ = 0.35, γ = 0.2, η = 0.25, ξ = 0.6), the ROC of LOOCV is shown as the blue curve with 0.8996. In order to get the optimal parameters i.e. λ, γ, η, and ξ, we utilized the PSO method.
Prediction performance of PRWHMDA with different parameters.
PSO is a population-based stochastic optimization technique, proposed by Kennedy and Eberhart (1995) [23]. In PSO, the solution was thought of as a bird, called a particle. In this study, all particles were searched in a four dimensional space with values between 0 and 1. All particles (parameters values) were evaluated by a fitness-function (16) to judge whether the current solution was good. Considering the accuracy and high AUC, the fitness function F(xi) was defined as
, (16)
where NActual = 450 denoted the number of actual disease-microbe associations, NPredicted(xi) denoted the number of tested associations ranked top 10 with parameter xi, xi = [λi, γi, ηi, ξi]T. The target was to get a suitable parameters vector to get the minimum value of F(x).
In the canonical PSO, each particle had position and velocity. For example, the position xi and velocity vi of particle i was updated at each iteration according to
(17)
(18)
where vi denoted the velocity of the i-th particle, w denoted the weight of current velocity, c1 and c2 denoted two positive constants representing learning parameters, r1, r2 denoted a random number between 0 and 1, pbi denoted the optimal parameters value of the i-th particle had searched for, gb denoted global best particle, xi denoted the current value of the i-th particle. In order to avoid the locally optimal solution, we allowed particles to mutate with a certain probability, i.e. they could reset their positions sometimes. Thus, each particle's velocity was updated according to (17) and (18) until the convergence criterion was satisfied. After the convergence criterion was satisfied, we got global best particle to be the optimal parameters.
Results and Discussion
Determination of parameters based on PSO with mutation
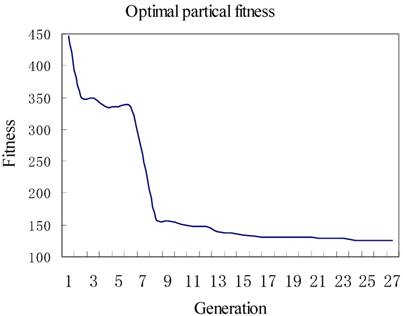
We set c1 = 1.49445, c2 = 1.49445, and set iteration times to be 27, the size of population to be 2, respectively. The iteration result is shown in Figure 3. The x-axis represented the iteration times, while the y-axis represented the fitness or called predicted error number of associations. Figure 3 showed that at the first iteration, there were 447 wrongly predicted associations, after 26 generations the number reduced to 125. Finally, the parameters calculated by PSO were λ = 0.6, γ = 0.5, η = 0.76, ξ = 0.79.
The optimal fitness of each iteration.
Cross validation
To effectively evaluate the prediction performance of PRWHMDA, LOOCV was implemented on the known disease-microbe associations from HMDAD database. Each known microbe-disease association was taken out in turns as a test association and the others were used for training to reconstruct the prediction model. Each test association was ranked in all unverified microbe-disease associations with their final probabilities. Receiver-operating characteristics (ROC) curve is widely used in binary classification problems. By changing the thresholds, the true positive rate (TPR, sensitivity) versus false positive rate (FPR, 1-specificity) can be calculated to plot the ROC curves. In the proposed method, TPR refers to the percentage of the positive test samples with higher ranks than the specific threshold, and FPR refers to the percentage of negative test samples with higher ranks than the specific threshold. AUC was calculated to further evaluate the prediction performance. As a result, our model obtained the AUC of 0.915 (see Figure 4 in red line).
The ROC curves and AUC values of different methods.
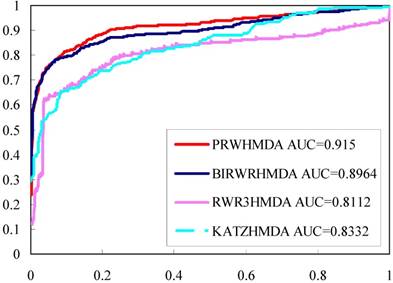
Furthermore, 5-fold CV was carried out. All the known microbe-disease associations were randomly divided into five disjoint groups, in which one group was for testing in turn and the other four groups were for training model. To reduce the bias brought by sample divisions, we executed the 5-fold CV 100 times to evaluate the robustness of PRWHMDA. As a result, the average AUC value was 0.8875 with the standard deviation of 0.0046.
Comparison with other methods
KATZHMD [14], RW3RHMDA [18] and BiRWHMDA [19] are state-of-the-art methods for predicting microbe-disease associations. All these methods are based on a heterogeneous network which was constructed by connecting the microbe similarity network and the disease similarity network via the known microbe-disease associations. To further validate the performance of the proposed method, we implemented these three methods using the same data sets and calculated the LOOCVs (see Figure 4). Comparing to other methods, the proposed method got a better ROC and higher AUC, while RWR3HMDA, KATZHMDA and BiRWHMDA had AUC values of 0.8112, 0.8332, and 0.8964, respectively.
Case studies
To further evaluate the prediction performance of PRWHMDA, we also implemented case studies of asthma, IBD, and T1D to predict novel associated microbes. The association strength score calculated by the proposed method and the benchmark was also presented, respectively. We used the RWR with three parameters as the benchmark.
Asthma
Asthma is a common chronic lung disease. To evaluate the prediction performance on asthma, we implemented a case study of asthma with our approach. In the prediction list, 10 of top-10 predicted microbes have been verified to have an impact on the asthmatic patients (see Table 1).
Candidate microbes of Asthma
| Rank | Microbe | Evidence | Score of PRW- HMDA (%) | Score of RWR3- HMDA (%) |
|---|---|---|---|---|
| 1 | Actinobacteria | PMID: 23265859 | 0.23 | 0.08 |
| 2 | Firmicutes | PMID: 23265859 | 0.22 | 0.09 |
| 3 | Clostridium coccoides | PMID: 21477358 | 0.15 | 0.05 |
| 4 | Streptococcus | PMID: 17950502 | 0.13 | 0.05 |
| 5 | Lactobacillus | PMID: 20592920 | 0.12 | 0.05 |
| 6 | Lachnospiraceae | Validated by [24] | 0.12 | 0.04 |
| 7 | Veillonella | PMID: 25329665 | 0.10 | 0.03 |
| 8 | Propionibacterium | PMID: 27433177 | 0.10 | 0.04 |
| 9 | Propionibacterium acnes | PMID: 27433177 | 0.10 | 0.04 |
| 10 | Fusobacterium | Validated by [25] | 0.09 | 0.03 |
For example, Actinobacteria, Firmicutes, Lachnospiraceae, and Veillonella (1st, 2nd, 6th and 7th in the prediction list) were found to have a lower proportion in asthmatic patients when compared to non-asthmatic people [24, 26, 27]. Clostridium coccoides (3rd in the prediction list) was found significantly associated with a positive Asthma Predictive Index (API) [28]. Based on the effect of Streptococcus pneumonia (4th in the prediction list) on the development of asthma, effective immune-modulatory therapies were hoped to be presented [29]. Lactobacillus (5th in the prediction list) could inhibit airway inflammation in an ovalbumin (OVA)-induced murine model of asthma [30]. Propionibacterium acnes (9th in the prediction list) was more prevalent in asthma patients; therefore, Propionibacterium (8th in the prediction list) was also considered to be associated with asthma [31]. Fusobacterium (10th in the prediction list) was much more abundant in asthmatic patients relative to healthy people [25]. These predictions may give an insight into the potential prevention action of asthma.
Inflammatory bowel disease (IBD)
In the prediction list of IBD, 10 of top-10 predicted microbes have been validated by experimental literatures (see Table 2).
Candidate microbes of IBD
| Rank | Microbe | Evidence | Score of PRW- HMDA (%) | Score of RWR3- HMDA (%) |
|---|---|---|---|---|
| 1 | Clostridium coccoides | PMID: 19235886 | 0.33 | 0.15 |
| 2 | Firmicutes | PMID: 25307765, 28842640 | 0.27 | 0.12 |
| 3 | Bacteroidetes | PMID: 25307765, 28842640, 24013298 | 0.27 | 0.11 |
| 4 | Prevotella | PMID: 25307765, 24013298 | 0.20 | 0.08 |
| 5 | Staphylococcus | PMID: 28174737 | 0.19 | 0.08 |
| 6 | Streptococcus | PMID: 23679203, 28842640 | 0.15 | 0.06 |
| 7 | Haemophilus | PMID: 24013298 | 0.14 | 0.05 |
| 8 | Lactobacillus | PMID: 26340825, 17897884 | 0.14 | 0.05 |
| 9 | Veillonella | PMID: 28842640, 24013298 | 0.13 | 0.05 |
| 10 | Bacteroidaceae | PMID: 17897884 | 0.13 | 0.05 |
Clostridium coccoides (1st in the prediction list) were less represented in A-IBD patients, compared to healthy subjects [32]. In the IBD group, Firmicutes (2nd in the prediction list) was significantly increased, whereas Bacteroidetes (3rd in the prediction list) was decreased. At genus level Streptococcus (6th in the prediction list) and Veillonella (9th in prediction list) were increased [33]. The results showed that Bacteroidetes (3rd in the prediction list) was significantly increased in Proteobacteria in the salivary microbiota of IBD patients. The dominant genera Prevotella (4th in the prediction list), Haemophilus (7th in the prediction list) and Veillonella (9th in prediction list) were found to largely contribute to dysbiosis (dysbacteriosis) observed in the salivary microbiota of IBD patients [34]. Conventional culture showed that the counts of total obligate anaerobes and of obligate anaerobes such as Bacteroidaceae (10th in the prediction list) was decreased in the faeces of IBD patients. On the other hand, the counts of total facultative anaerobes such as Lactobacillus (8th in the prediction list) were increased [35]. Research showed that dysbiosis within deeper layers of the ileum of CD patients, there were specifically enrichment of enterotoxigenic Staphylococcus aureus. Staphylococcus(5th in the prediction list) was thus validated [36].
Type 1 diabetes
In the prediction list of T1D, 9 of predicted microbes in the top 10 have been validated by experimental literatures (see Table 3).
Candidate microbes of T1D
| Rank | Microbe | Evidence | Score of PRW- HMDA (%) | Score of RWR3- HMDA (%) |
|---|---|---|---|---|
| 1 | Clostridium coccoides | PMID: 23433344 | 0.09 | 0.02 |
| 2 | Enterobacteriaceae | PMID: 24475780 | 0.05 | 0.01 |
| 3 | Staphylococcus | PMID: 26068542 | 0.04 | 0.01 |
| 4 | Faecalibacterium prausnitzii | PMID: 20613793 | 0.03 | 0.01 |
| 5 | Clostridium | PMID: 23433344 | 0.03 | 0.01 |
| 6 | Clostridium leptum | PMID: 22046124 | 0.03 | 0.01 |
| 7 | Enterococcus | PMID: 26176548 | 0.03 | 0.01 |
| 8 | Desulfovibrio | Confirmed by [37] | 0.03 | 0.01 |
| 9 | Tannerella | PMID: 24236037 | 0.03 | 0.01 |
| 10 | Bacilli | Not confirmed | 0.03 | 0.01 |
At the genus level, a significant increase in the number of Clostridium (Genus of 1st in the prediction list) was found in the children with diabetes [38]. It was found that Enterobacteriaceae (2nd in the prediction list) was increased in patients with type 1 diabetes compared to the control group. A concomitant increase in Enterobacteriaceae may lead to a disturbance in the ecological balance of intestinal flora, which could be a triggering factor in type 1 diabetes etiology [39]. Furthermore, subjects with autoantibodies, seronegative FDRs, and new-onset patients had different levels of the genera Firmicutes, Lactobacillus, and Staphylococcus (3rd in the prediction list) compared with healthy control subjects with no family history of autoimmunity [40]. An increase of Faecalibacterium prausnitzii (4th in the prediction list) might result in a disturbance in the ecological balance, which could cause type 1 diabetes [41]. At genus level, Mora Murri et al. [38] found a significant increase in the number of Clostridium (5th in the prediction list) in the children with diabetes. A study was performed on in non-obese prediabetic (NOD) mice. Metabolic changes of NOD mice were accompanied by diminished gut microbial diversity of the Clostridium leptum group (6h in the prediction list) [42]. Two species in the genus Staphylococcus were abundantly presented in the conjunctiva of healthy rats, and were replaced by Enterococcus species (7th in the prediction list) and other bacterial species in diabetic rats [43]. Members of the genus Desulfovibrio (8th in the prediction list) were identified in the colon samples of the control rats, with <0.5% abundance. It was noted that the insulin treatment was accompanied by fundamental changes within the phylum Proteobacteria: their overall abundance decreased spectacularly, and the genus Klebsiella was basically eradicated, being replaced by the genera Desulfovibrio (8th in the prediction list) and Bifidobacterium, with a combined abundance of 4% [37]. GCC-fed NOD mice had the expected high incidence of hyperglycemia whereas NOD mice fed with a GFC had significantly reduced incidence of hyperglycemia. When the fecal microbes were compared, Tannerella species (9th in the prediction list) were increased in the microbes of GCC mice[44].
Moreover, as shown in Tables 1-3, the association strength score of the proposed method is higher than that of the benchmark. In summary, these case studies further demonstrate that the proposed approach is effective to predict novel microbe-disease associations. The top-10 potential associated microbes for all the 39 diseases are listed in Table s1 of Supplementary File.
Conclusion
In this paper, we proposed a novel computational model based on the known microbe-disease associations. Cosine similarity was adopted to construct the microbe similarity network and disease similarity network. Using the experimental validated associations, we connected the two networks. The extended RWR method was utilized to walk on the network to get the association probability, which represented candidate microbe-disease associations. PSO was incorporated to get the optimal parameters for optimizing the prediction performance. As a result, the proposed model achieved a reliable prediction performance of LOOCV with AUC of 0.915 and 5-fold CV with average AUCs of 0.8875 ± 0.0046. In our case studies, 10, 10, and 9 of top-10 inferred microbes have been confirmed to have associations with asthma, IBD, and T1D according to the literature evidences. Given the promising prediction performance, it is believed that PRWHMDA could be an effective tool accelerating the progress of biomedical identification of potential disease-related microbes.
The good performance of the new method benefits from several major factors as the following. (1) We used Cosine similarity to extract the potential similarity for microbes and diseases by making use of known microbe-disease associations. (2) The proposed model is based on semi-supervised learning method, i.e., the training data is regarded as labeled samples while other test data as unlabeled samples. (3) Based on the training data set, PSO with mutation was adopted to get the optimal parameters. (4) We used the extended RW method that has adjustable parameters to emphasize the probabilities of transitions of different networks.
There are some limitations in the performance of PRWHMDA. Because the experimentally verified microbe-disease associations used in our approach are relatively insufficient, the sparse association network could affect the predictive capability. It is anticipated that this problem will be solved when collecting more microbe-disease associations in the future. Furthermore, PRWHMDA cannot be applied to diseases and microbes which are not in HMDAD. It is believed that the network would be powerful with the introducing of other types of disease/microbe similarity based on different kinds of data, such as symptom-based disease similarity, etc. We think that this method can guide medical experiments to get the potential associations.
Supplementary Material
Table S1.
Abbreviations
PSO: Particle Swarm Optimization; RWR: random walk with restart; LOOCV: leave one out cross validation; AUC: The area under ROC curve; IBD: inflammatory bowel disease; T1D: type 1 diabetes; NOD: non-obese prediabetic.
Acknowledgements
This work was supported by the Natural Science Foundation of China [Grant Numbers 61473335, 61533011].
Competing Interests
The authors have declared that no competing interest exists.
References
1. The Human Microbiome Project C. A framework for human microbiome research. Nature. 2012;486:215
2. Sommer F, Backhed F. The gut microbiota-masters of host development and physiology. Nat Rev Microbiol. 2013;11:227-38
3. Guarner F, Malagelada JR. Gut flora in health and disease. The Lancet. 2003;361:512-9
4. Pickard JM, Zeng MY, Caruso R, Núñez G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. 2017;279:70
5. Davis-Richardson AG, Ardissone AN, Dias R, Simell V, Leonard MT, Kemppainen KM. et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front Microbiol. 2014;5:678
6. Qiu R, Li N, Yang Z, He M, Fen X, Li J. et al. Analysis of the Sputum Microbiome in the Severe Asthma. Chest. 2016;149:A14
7. Rowland IR. The role of the gastrointestinal microbiota in colorectal cancer. Curr Pharm Des. 2009;15:1524-7
8. Ma W, Zhang L, Zeng P, Huang C, Li J, Geng B. et al. An analysis of human microbe-disease associations. Brief Bioinform. 2016;18:85-97
9. Hua ZG, Lin Y, Yuan YZ, Yang DC, Wei W, Guo FB. ZCURVE 3.0: identify prokaryotic genes with higher accuracy as well as automatically and accurately select essential genes. Nucleic Acids Res. 2015;43:W85-W90
10. Hua HL, Zhang FZ, Labena AA, Dong C, Jin YT, Guo FB. An Approach for Predicting Essential Genes Using Multiple Homology Mapping and Machine Learning Algorithms. BioMed Research International. 2016;2016:9
11. Yang H, Tang H, Chen XX, Zhang CJ, Zhu PP, Ding H. et al. Identification of Secretory Proteins in Mycobacterium tuberculosis Using Pseudo Amino Acid Composition. BioMed Research International. 2016;2016:7
12. Tang H, Chen W, Lin H. Identification of immunoglobulins using Chou's pseudo amino acid composition with feature selection technique. Molecular Biosystems. 2016;12:1269
13. Liu K, Huang J, Luo D, Xu K, Wu Z, Xu X. Analysis and quality control of carbohydrates in therapeutic proteins with fluorescence HPLC. Biochem Biophys Res Commun. 2016;478:864-7
14. Chen X, Huang YA, You ZH, Yan GY, Wang XS. A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics. 2017;33:733-9
15. Huang ZA, Chen X, Zhu Z, Liu H, Yan GY, You ZH. et al. PBHMDA: Path-Based Human Microbe-Disease Association Prediction. Front Microbiol. 2017;8:233
16. Wang F, Huang ZA, Chen X, Zhu Z, Wen Z, Zhao J. et al. LRLSHMDA: Laplacian Regularized Least Squares for Human Microbe-Disease Association prediction. Sci Rep. 2017;7:7601
17. Shen Z, Jiang Z, Bao W. CMFHMDA: Collaborative Matrix Factorization for Human Microbe-Disease Association Prediction. Intelligent Computing Theories and Application: Springer International Publishing. 2017 p. 261-9
18. Shen X, Chen Y, Jiang X, Hu X, He T, Yang J. Predicting disease-microbe association by random walking on the heterogeneous network. IEEE International Conference on Bioinformatics and Biomedicine. 2016 p. 771-4
19. Zou S, Zhang J, Zhang Z. A novel approach for predicting microbe-disease associations by bi-random walk on the heterogeneous network. PLoS One. 2017;12:e0184394
20. Pearson K. The Problem of The Random Walk. Nature. 1905;72:342
21. Köhler S, Bauer S, Horn D, Robinson PN. Walking the Interactome for Prioritization of Candidate Disease Genes. Am J Hum Genet. 2008;82:949-58
22. Li Y, Patra JC. Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network. Bioinformatics. 2010;26:1219-24
23. Eberhart R, Kennedy J. A new optimizer using particle swarm theory. MHS'95 Proceedings of the Sixth International Symposium on Micro Machine and Human Science: IEEE. 1995 p. 39-43
24. Ciaccio CE, Barnes C, Kennedy K, Chan M, Portnoy JM, Rosenwasser LJ. Home Dust Microbiota is Disordered in Homes of Low-Income Asthmatic Children. J Asthma. 2015;52:873-80
25. Dang HT, Song AK, Park HK, Shin JW, Park SG, Kim W. Analysis of Oropharyngeal Microbiota between the Patients with Bronchial Asthma and the Non-Asthmatic Persons. J Bacteriol Virol. 2013;43:270
26. Marri PR, Stern DA, Wright AL, Billheimer D, Martinez FD. Asthma-associated differences in microbial composition of induced sputum. J Allergy Clin Immunol. 2013;131:346-52.e1 -3
27. Park H, Shin JW, Park SG, Kim W. Microbial Communities in the Upper Respiratory Tract of Patients with Asthma and Chronic Obstructive Pulmonary Disease. PLoS One. 2014;9:e109710
28. Vael C, Vanheirstraeten L, Desager KN, Goossens H. Denaturing gradient gel electrophoresis of neonatal intestinal microbiota in relation to the development of asthma. BMC Microbiol. 2011;11:68
29. Preston JA, Essilfie AT, Horvat JC, Wade MA, Beagley KW, Gibson PG. et al. Inhibition of allergic airways disease by immunomodulatory therapy with whole killed Streptococcus pneumoniae. Vaccine. 2007;25:8154-62
30. Yu J, Jang SO, Kim BJ, Song YH, Kwon JW, Kang MJ. et al. The Effects of Lactobacillus rhamnosus on the Prevention of Asthma in a Murine Model. Allergy Asthma Immunol Res. 2010;2:199-205
31. Jung JW, Choi JC, Shin JW, Kim JY, Park IW, Choi BW. et al. Lung Microbiome Analysis in Steroid-Naїve Asthma Patients by Using Whole Sputum. Tuberc Respir Dis. 2016;79:165-78
32. Sokol H, Seksik P, Furet JP, Firmesse O, Nionlarmurier I, Beaugerie L. et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15:1183-9
33. Santoru ML, Piras C, Murgia A, Palmas V, Camboni T, Liggi S. et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci Rep. 2017;7:9523
34. Said HS, Suda W, Nakagome S, Chinen H, Oshima K, Kim S. et al. Dysbiosis of Salivary Microbiota in Inflammatory Bowel Disease and Its Association With Oral Immunological Biomarkers. DNA Res. 2014;21:15-25
35. Takaishi H, Matsuki T, Nakazawa A, Takada T, Kado S, Asahara T. et al. Imbalance in intestinal microflora constitution could be involved in the pathogenesis of inflammatory bowel disease. Int J Med Microbiol. 2008;298:463-72
36. Pedamallu CS, Bhatt AS, Bullman S, Fowler S, Freeman SS, Durand J. et al. Metagenomic Characterization of Microbial Communities In Situ Within the Deeper Layers of the Ileum in Crohn's Disease. Cell Mol Gastroenterol Hepatol. 2016;2:563-6.e5
37. Wirth R, Bódi N, Maróti G, Bagyánszki M, Talapka P, Fekete É. et al. Regionally Distinct Alterations in the Composition of the Gut Microbiota in Rats with Streptozotocin-Induced Diabetes. PLoS One. 2014;9:e110440
38. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F. et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 2013;11:46
39. Soyucen E, Gulcan A, Aktuglu-Zeybek AC, Onal H, Kiykim E, Aydin A. Differences in the gut microbiota of healthy children and those with type 1 diabetes. Pediatr Int. 2014;56:336-43
40. Alkanani AK, Hara N, Gottlieb PA, Ir D, Robertson CE, Wagner BD. et al. Alterations in Intestinal Microbiota Correlate With Susceptibility to Type 1 Diabetes. Diabetes. 2015;64:3510-20
41. Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G. et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5:82-91
42. Sysi-Aho M, Ermolov A, Gopalacharyulu PV, Tripathi A, Seppänen-Laakso T, Maukonen J. et al. Metabolic Regulation in Progression to Autoimmune Diabetes. PLoS Comput Biol. 2011;7:e1002257
43. Yang C, Fei Y, Qin Y, Luo D, Yang S, Kou X. et al. Bacterial Flora Changes in Conjunctiva of Rats with Streptozotocin-Induced Type I Diabetes. PLoS One. 2015;10:e0133021
44. Marietta EV, Gomez AM, Yeoman C, Tilahun AY, Clark CR, Luckey DH. et al. Low Incidence of Spontaneous Type 1 Diabetes in Non-Obese Diabetic Mice Raised on Gluten-Free Diets Is Associated with Changes in the Intestinal Microbiome. PLoS One. 2013;8:e78687
Author contact
![]() Corresponding author: Rui Gao, School of Control Science and Engineering, Shandong University, No.17923 of Jingshi Road, Jinan 250061, China. E-mail: gaoruiedu.cn.
Corresponding author: Rui Gao, School of Control Science and Engineering, Shandong University, No.17923 of Jingshi Road, Jinan 250061, China. E-mail: gaoruiedu.cn.