Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(9):1033-1040. doi:10.7150/ijbs.25589 This issue Cite
Research Paper
Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution
Min Feng1*, Jie Ding1*, Min Wang2, Jie Zhang1,2, Xinhua Zhu1 ![]() , Wenxian Guan1
, Wenxian Guan1 ![]()
1. Department of General Surgery, Nanjing Drum Tower Hospital, the Affiliated Hospital of Nanjing University Medical School, Nanjing 210008, China;
2. Department of General Surgery, the Affiliated Hospital of Yangzhou University Medical School, Yangzhou 225001, China;
*These authors contributed equally to this work
Received 2018-2-17; Accepted 2018-5-3; Published 2018-6-3
Abstract

Kupffer cells (KCs) contribute to liver fibrosis resolution by production of a large spectrum of matrix metalloproteinases (MMPs). MMP9 is a major MMP expressed by KCs. However, its role in liver fibrosis resolution remains unclear. In this study, rodent liver fibrosis was induced by intraperitoneal thioacetamide (TAA) and the resolution process was initiated by TAA withdrawal. The role of KC-derived MMP9 in fibrolysis was investigated by adoptive transfer of KCs with or without MMP9 following their depletion. The levels of serum alanine aminotransferase (ALT) and hepatic cytokines were measured during fibrosis regression. The mRNA levels of MMPs and tissue inhibitor of metalloproteinases (TIMPs) were analyzed as well. It was found that removing KCs delayed fibrosis resolution. Adoptive transfer of KCs from WT animals promoted liver fibrosis resolution, compared with transfer of KCs from MMP9-/- mice. Depletion of KCs also resulted in prolonged liver wound healing, which was reversed partially by transferred KCs from either WT or MMP9-/- mice. Likewise, the absence of KCs led to reduction in MMPs mRNA levels and elevation in TIMPs mRNA levels. The expression patterns of MMPs or TIMPs were restored by adoptive transfer of the wild-type but not MMP9-/- KCs. In addition, liver fibrosis resolution was accelerated in MMP9-/- mice by adoptive transferred KCs from WT animals, compared to the KCs from MMP9-/- mice. Overall, KC-derived MMP9 plays a critical role in fibrosis resolution, which might serve as the foundation for developing anti-fibrosis therapy.
Keywords: Kupffer cells, matrix metalloproteinase, fibrosis resolution, liver fibrosis
Introduction
Liver fibrosis is caused by chronic or iterative hepatic insult and advanced chronic fibrosis is often associated with functional failure and attendant life-threatening complications [1]. Increasing evidence from rodent models and clinic studies indicates that if the injury is removed liver fibrosis can be reversible [1-3]. The immune cells, particularly macrophages, participates in resolution of liver fibrosis critically [4, 5].
KCs are resident hepatic macrophages that localize within the lumen of the liver sinusoids, accounting for about 30% of hepatic non-parenchymal cells (HNPCs) [6, 7]. Through expression of matrix metalloproteinases (MMPs), KCs carry out an important function in liver fibrosis resolution [6, 8]. However, KCs can be classified into several subsets based on different markers or functions, and these subsets, controlled by environmental and inflammatory factors, undergo transdifferentiation [4]. The KC subset characterized by the phenotype of CD11bhiF4/80intLy-6Clo, is abundant in hepatic scar during fibrosis resolution. Depletion of this population leads to a failure of scar remodeling [6]. Further studies indicate that KC-derived MMP13 plays a critical role in extracellular matrix (ECM) degradation [8]. Apart from MMP13, KCs have been shown to produce a serie of proteolytic enzymes, including gelatinases (MMP2, MMP9), metalloelastase (MMP12), matrilysin (MMP7), and collagenases (MMP1 and MMP13) in different conditions [5, 8], leading to degradation of ECM.
MMP9 is the critical gelatinase produced by KCs and is implicated in playing a role in the pathological process of acute or chronic hepatic injury [9, 10]. Our previous studies indicated that the absence of MMP9 inhibits activation of TGFβ and reduces accumulation of collagen and α-Smooth muscle actin in liver during fibrogenesis [11]. Recent studies showed that MMP9 expression was increased in KCs during fibrosis resolution [6]. However, the role of KC-derived MMP9 in liver fibrosis resolution is still unclear.
In this study, KCs from wild type (WT) or MMP9-/- mice were adoptive transferred following KCs depletion in the host animals. It was found that depletion of KCs delayed fibrosis resolution, while adoptive transfer of KCs with MMP9 promoted this progress, compared to the treatment by KCs without MMP9. Meanwhile, KC-derived MMP9 treatment led to elevation in MMP9 mRNA level and reduction in TIMP1 mRNA level. In MMP9-/- animal model, we found that adoptive transferred KCs with MMP9 promoted fibrosis resolution as well.
Materials and Methods
Animal work
MMP9-/- (FVB background) mice were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China), and then crossed into the C57BL/6 background for six generations. The homozygous wild types were used as controls [12]. All animals were treated according to the guidelines for the use of experimental animals, with protocols approved by the Institutional Animal Care and Research Advisory Committee of Nanjing University (Nanjing, China). Liver fibrosis was induced by repeated intraperitoneal administration of thioacetamide (TAA, 0.1 mg/g body weight, Sigma, shanghai, PRC) every two days for 8 weeks. Control mice were injected with the same volume of saline. For KC or PMN depletion, liposome-encapsulated clodronate (Cl2MDP, 100 ml/mouse, iv, Roche, shanghai, PRC) or anti-Gr-1 (200 mg/mouse, i.p., BD, shanghai, PRC) was administrated 24 hours after the final TAA treatment. Adoptive transfer experiments were performed intravenously at indicated time point using 2×106 FACS-sorted (Fluorescence-Activated Cell Sorting) F4/80+ cells from HNPCs.
Serum transaminase and ELISA
Serum was collected by ocular blood extraction at the indicated time points. Liver injury was estimated by increased activity of serum alanine aminotransferase (ALT), which was measured in a clinical biochemical laboratory of Nanjing Drum Tower Hospital. The levels of tumor necrosis factor-α (TNFα), interleukin-10 (IL10) and transforming growth factor-β (TGFβ) in the liver homogenate were determined using commercial ELISA kits (R&D System, shanghai, PRC) according to the manufacturer's instructions. All samples and standards were measured in duplicate.
Cells isolation and analysis
Hepatic non-parenchymal cells (HNPCs) were prepared as previously described with minor modifications [11]. Briefly, the liver was perfused with Ca 2+ and Mg 2+ free Hanks' Balanced Salt Solution (HBSS) and ethylene glycol tetraacetic acid (EGTA) for 5.0 min. Then it was digested by 0.4 mg/ml pronase and 0.5 mg/ml collagenase for 5.0 min each. The liver was further digested by shaking in a 0.5 mg/ml pronase/collagenase solution at 37°C for 20 min. The digested cells were passed through a 70-μm cell strainer and washed with HBSS. At this stage, the cells were referred to as HNPCs. KCs, PMNs and HSCs were sorted or analyzed by flow cytometry. The PE-anti-Cr1 and PE-anti-F4/80 antibodies were purchased from BioLegend (San Diego, CA). The PE-anti-desmin and FITC-anti-GFAP antibodies were from R&D Systems (Minneapolis, MN).
Western blot and zymography
Proteins were extracted from liver tissues for western blot 1, 5 or 9 days after TAA withdrawal. The primary antibodies targeting collagen I, III, IV and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from Abcam Biotechnology Company (shanghai, PRC). The horseradish peroxidase conjugated secondary antibodies (Santa Cruz Biotechnology, CA) were used. Proteins were visualized with Super Signal West Femto Chemiluminescent substrate (Pierce Protein Biology, PRC). The intensity of the protein bands in Western blot were quantified using the software Image Quant version 5.2. Data were normalized to GAPDH level. For gelatin zymography, proteins prepared from KCs or fibrotic tissues were incubated with gelatin-conjugated Sepharose 4B (GE Health Science, PRC) at 4°C for 30 min, and then used for zymography test according to a previous report [13].
Quantitative RT-PCR analysis
Total RNA was extracted from liver tissue by TRIZOL (Life Technologies, shanghai, PRC), and the first-strand cDNA was synthesized with Superscript II Reverse Transcriptase (Promega, Shanghai, PRC) according to manufacturer's instructions. qRT-PCR was carried out in ABI Prism 7900HT (Applied Biosystems, USA) using SYBR Green master mix (Eurogentec, Shanghai, PRC) according to manufacturer's instructions. Primer sequences used for PCR amplification of mouse genes are as follow [14]: MMP9 F: '5-CGT GTC TGG AGA TTC GAC TTG A-3', R: '5-TGG AAG ATG TCG TGT GAG TTC C-3'; MMP13 F: '5-CCT TCT GGT CTT CTG GCA CAC-3', R: '5-GGC TGG GTC GTC ACA CTT CTC TGG-3'; MMP2 F: '5-CAA CGG TCG GGA ATA CAG CAG-3', R: '5-CCA GGA AAG TGA AGG GGA AGA-3'; MMP14 F: '5-ATC TCA CAG CTC GGT GTG TGT TCA-3', R: '5-AAG GTC AGA GGG TCT TGC CTT CAA-3'; TIMP1 F: '5-GCA TGG ACA TTT ATT CTC CAC TGT-3', R: '5-TCT CTA GGA GCC CGA TCT G-3'; TIMP2 F: '5-GCC AAA GCA GTG AGC GAG AAG-3', R: '5-GGG GAG GAG ATG TAG CAA GGG-3'; GAPDH F: '5-AAC TTT GGC ATT GTG GAA GG-3',R: '5-ACA CAT TGG GGG TAG GAA CA-3'.
Statistical analyses
The data were expressed as mean ± SD. The differences between two groups were analyzed using two-tailed Student's t-test. Differences between multiple groups were tested with Kruskal-Wallis nonparametric analysis of variance with multiple group comparisons. P<0.05 was considered significant.
Results
Delayed liver fibrosis resolution following KCs depletion
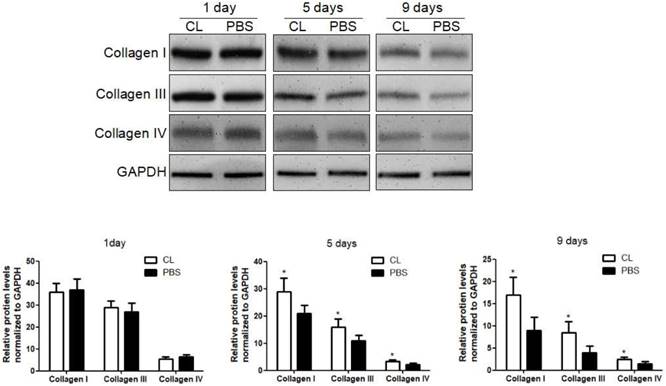
We first examined the role of KCs in liver fibrosis resolution by depleting KCs with Cl2MDP. The efficiency of depletion was determined by immunofluorescence at 24 hours after administration (Figure S1) [10]. Liver fibrosis was induced following the protocol described above, and Cl2MDP was given at 24 hours after the last TAA challenge. The hepatic protein levels of collagen type I, III and IV were determined by western blotting at 1, 5 or 9 days following TAA withdrawal. It was found that the expression of collagen I was increased by 33% after 5 days in Cl2MDP treated mice, compared with control. Additionally, the levels of collagen III and IV were increased by 38% and 27%, respectively, after KC depletion. At 9 days after TAA withdrawal, the protein levels of collagen I, III and IV were increased by 47%, 51% and 32% respectively in KC-depleted mice, compared with control. (Figure 1).
Delayed liver fibrosis resolution following KC depletion. WT mice were treated with TAA (0.1 mg/g body weight, ip) every 2 days for 8 weeks and then KCs was depleted at 24 hours after TAA withdrawal. Then the hepatic total protein was extracted at 1 day, 5 days and 9 days following TAA withdrawal. The levels of collagen type I, III and IV in livers tissues were determined by western blotting. Data were normalized to GAPDH and expressed as mean ± standard deviation. n=5, *p<0.05 (compared with control). CL, The mice treated with liposome-encapsulated clodronate (Cl2MDP).
Acceleration of fibrolysis by KC-derived MMP9
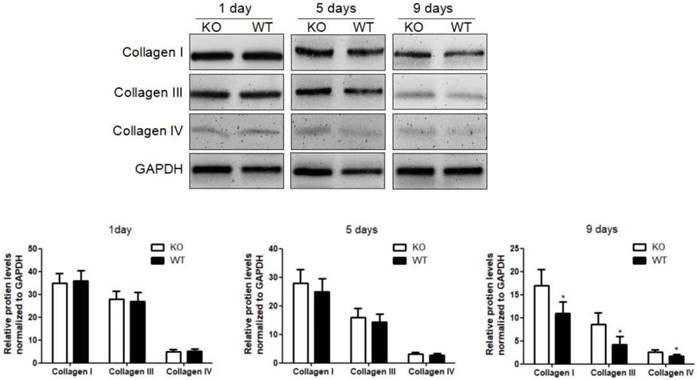
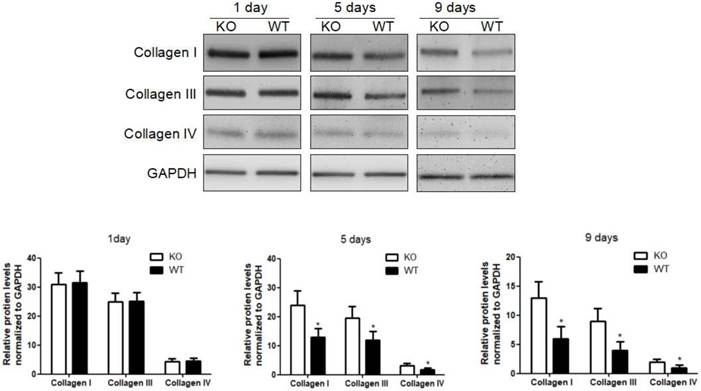
To investigate the role of KC-derived MMP9 in fibrosis resolution, KCs from WT or MMP9-/- were adoptively transferred to liver fibrotic mice following KC depletion. The challenge course was shown in Figure S2. Compared with that of KCs from MMP9-/- mice, adoptive transfer of KCs from WT mice significantly reduced the levels of collagen I, III and IV by 34%, 48% 23% respectively at 9 days (6 days after adoptive transfer), although no significant difference was found at 5 days (2 days after adoptive transfer) or 1 day (Figure 2).
Acceleration of fibrosis resolution by KC-derived MMP9. After TAA withdrawal, mice were treated with Cl2MDP (100 ml/mouse, iv) at 1 day, and then were adoptive transferred KCs (2×106/mice) from WT or MMP9-/- mice at 3 days. The hepatic total protein was extracted from at 1 day, 5 days and 9 days, after TAA withdrawal. The levels of collagen type I, III and IV in livers tissues were determined by western blotting. Data were normalized to GAPDH and expressed as mean ± standard deviation. n=5, *p<0.05 (compared with the treatment by KCs from MMP9-/- animals). WT, treatment with KCs from wild type mice; KO, treatment with KCs from matrix metalloproteinase-9 deficient mice.
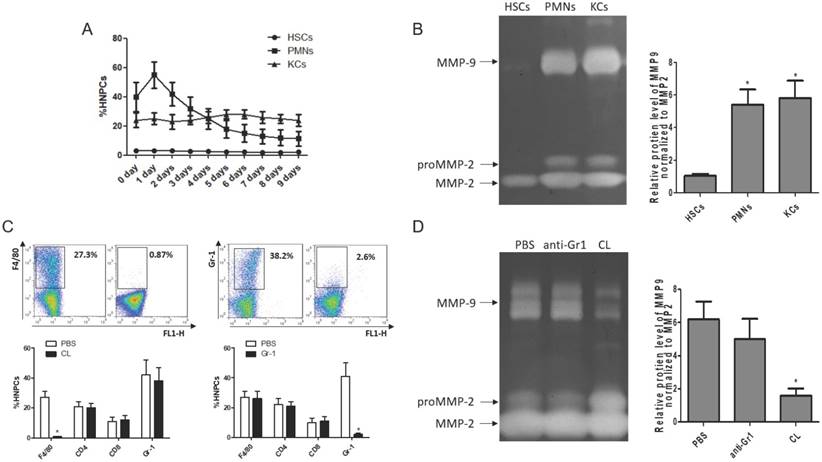
To clarify the cellular sources of MMP9 in the fibrosis resolution, several cell types (KCs, HSCs and PMNs) were analyzed by flow cytometry. The frequency of PMNs was gradually reduced with liver wound healing during fibrosis resolution, while the amounts of KCs and HSCs remained stable (Figure 3A). Then, these cells were sorted by FACS (the purity level of yielded cell over 95% routinely) for zymography at 24 hours after TAA withdrawal. The results indicated that HSCs lost the ability to produce MMP9, rather than MMP2. KCs and PMNs still expressed MMP9 (Figure 3B). To further confirm the role of KCs in MMP9 production, KCs or PMNs were depleted respectively at 24 hours after TAA withdrawal. The efficiency of depletion was determined by FACS (Figure 3C). Meanwhile, MMP9 and MMP2 levels were analyzed by zymography at 24 hours after depletion. It was found that depletion of KCs resulted in a significant reduction of MMP9 level, while depletion of PMNs had little effects (Figure 3D).
Cellular source of MMP9 in the fibrotic liver. (A) The rates of HSCs, PMNs and KCs were determined by flow cytometry based on specific fluorescent antibodies (PE-anti-desmin and FITC-anti-GFAP, PE-anti-Cr1, PE-anti-F4/80). (B) MMPs levels were determined by zymography at the time point of TAA withdrawal. (C) Efficiency rates of KC or PMN depletion were determined by flow cytometry at 24 hours after administration. (D) MMPs levels were determined by zymography in liver tissues after KC or PMN depletion. Data were expressed as mean ± SD; n= 5, *P <0.05 (compared with control). HNPCs, hepatic non-parenchymal cells; HSCs, hepatic stellate cells; PMNs, polymorphonuclear neutrophils; KCs, Kupffer cells; CL, liposome-encapsulated clodronate.
The effect of KC-derived MMP9 on hepatic inflammation and MMPs/TIMPs expression
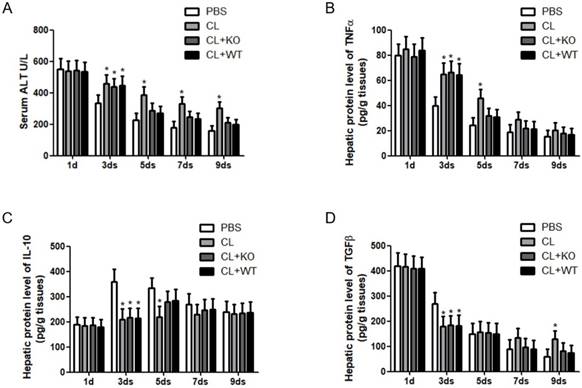
Immune cells and inflammatory factors cooperate in the establishment and resolution of liver fibrosis [4, 5]. To address the role of KC-derived MMP9 in hepatic inflammation during fibrosis resolution, KCs with or without MMP9 were adoptively transferred to liver fibrotic mice. It was found that the levels of serum ALT (Figure 4A) and hepatic TNFα (Figure 4B) were increased, while the expressions of hepatic IL-10 (Figure 4C) and TGFβ (Figure 4D) were decreased after KC depletion. However, the levels of these cytokines had no significant change after the transfer of KCs with MMP9, compared to that of KCs without MMP9.
Attenuation of chronic injury by KC-derived MMP9. (A) Serum was collected at indicated time points. ALT level was measured by a clinical biochemical laboratory. The levels of (B) TNF-α, (C) IL-10, (D) TGFβ in the liver homogenate were determined using commercial ELISA kits. All samples and standards were measured in duplicate. Data were expressed as mean ± standard deviation. *p<0.05 (compared with control). WT, treatment with KCs from wild type mice; KO, treatment with KCs from matrix metalloproteinase-9 deficient mice.
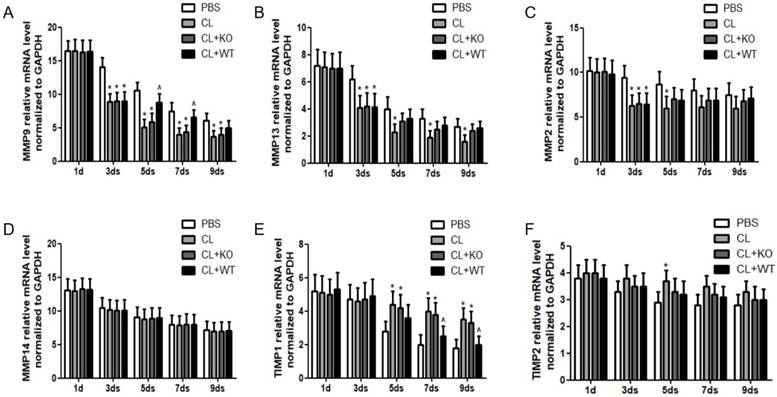
The ultimate step for achieving reversibility of fibrosis is the degradation of ECM, which is carried out by the balance between activities of MMPs and their inhibitors, TIMPs [15, 16]. In this study, it was found that, compared to the treatment by KCs without MMP9, the mRNA level of MMP9 was increased significantly at 5days or 7days after the treatment of KCs with MMP9 (Figure 5A), while the mRNA levels of MMP13 (Figure 5B), MMP2 (Figure 5C) and MMP14 (Figure 5D) were not increased. In addition, the mRNA levels of TIMP1 (Figure 5E) but not those of TIMP2 (Figure 5F) were decreased by 27% or 33% at 7 or 9 days, respectively, following treatment of KCs with MMP9.
Effect of KC-derived MMP9 on transcription of MMPs and TIMPs. Total mRNA was extracted from liver at indicated time points. The levels of (A) MMP9, (B) MMP13, (C) MMP2, (D) MMP14, (E) TIMP1 and (F) TIMP2 were analyzed by qRT-PCR. Data were normalized to GAPDH and expressed as mean ± standard deviation. n=5, *p<0.05 (compared with control); ^p<0.05 (compared with the treatment by KCs from MMP9-/- animals). WT, treatment with KCs from wild type mice; KO, treatment with KCs from matrix metalloproteinase-9 deficiency mice.
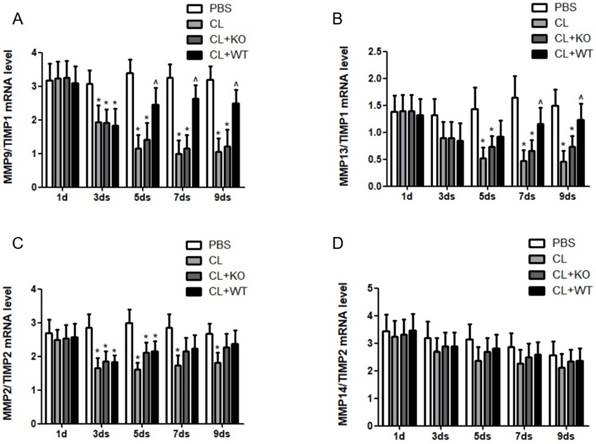
The actual proteolytic activity of MMPs depends on the ratio of MMPs vs their corresponding TIMPs [14]. In a recent study, we found that the ratio of MMP9/TIMP1 (Figure 6A) and MMP13/TIMP1 (Figure 6B) was increased after adoptive transfer of KCs with MMP9, while there was no significant change in the ratio of MMP2/TIMP2 (Figure 6C) or MMP14/TIMP2 (Figure 6D), compared to the mice treated by KCs without MMP9.
Effect of KC-derived MMP9 on ratio of MMPs/TIMPs. The ratios of (A) MMP9/TIMP1, (B) MMP13/TIMP1, (C) MMP2/TIMP2 and (D) MMP14/TIMP2 were calculated based on MMPs and TIMP mRNA levels. Data were expressed as mean ± standard deviation. n=5, *p<0.05 (compared with control); ^p<0.05 (compared with the treatment by KCs from MMP9-/- animals). WT, treatment with KCs from wild type mice; KO, treatment with KCs from matrix metalloproteinase-9 deficiency mice.
The promotive fibrolysis role of KCs-derived MMP9 in MMP9-/- mice
To confirm the role of KC-derived MMP9 in fibrosis resolution, KCs with or without MMP9 were adoptively transferred to the liver fibrotic model established in MMP9-/- mice. The challenge course was shown in Figure S3. The efficiency of adoptive transfer of KCs was determined by FACS. The frequency of KCs was increased by 23.9% after KCs treatment (Figure S4). Compared to the mice treated by KCs without MMP9, collagen I level was reduced by 42% at 5days and 53% at 9days in the animals treated by KCs with MMP9. Collagen III or IV showed the same variation following adoptive transfer of KCs with MMP9, and decreased 34% or 25% respectively at 5 days, 54% or 51% at 9days (Figure 7).
The fibrolysis promoting role of KC-derived MMP9 in MMP9-/- mice. MMP9-/- mice were treated with TAA (0.1 mg/g body weight, ip) every 2 days for 8 weeks and then KCs (2×106/mice) from WT or MMP9-/- mice was adoptive transferred at 1 day. The hepatic total protein was extracted at 1 day, 5 days and 9 days, after TAA withdrawal. The levels of collagen type I, III and IV in livers tissues were determined by western blotting. Data were normalized to GAPDH and expressed as mean ± standard deviation. n=5, *p<0.05 (compared with the treatment by KCs from MMP9-/- animals). WT, treatment with KCs from wild type mice; KO, treatment with KCs from matrix metalloproteinase-9 deficient mice.
Discussion
Although KC-derived MMP9 has been reported to play an important role in various forms of liver injury [17-19], its effect on fibrosis resolution is largely unknown. In this study, we employed the liver fibrotic model of TAA-induced chronic liver injury established by us and others to investigate the role of KC-derived MMP9 in liver fibrosis resolution. Our results show that gross depletion of KCs delays liver fibrolysis, which is characterized by prolonged high levels of collagen deposition and persistent hepatic inflammation. Notably, we show that adoptive transfer of KCs from WT mice, but not those of the MMP9-/- mice, can restore the process of fibrolysis in the KC-depleted mice. These findings indicate that MMP9s play a critical role KC-mediated degradation of ECM during liver fibrosis regression. Interestingly, our work also demonstrated that the ability of the adoptively transferred KCs to restore the immune homeostasis is not dependent of their MMP9 status. These results cued that KC-derived MMP9 promoting fibrosis resolution should be independent on inflammatory factors.
We further investigated the effect of KC depletion and reconstitution on expression levels of MMPs or TIMPs. Our data show that adoptive transfer of KCs from WT mice resulted in elevation in MMP9 mRNA level and reduction in TIMP1 mRNA level, compared to that of KCs from MMP9-/- animals. In addition, we found significant elevation in the ratio of MMP9/TIMP1 and that of MMP13/TIMP1, following adoptive transfer of WT versus MMP9-/- KCs. These results established that the role of KCs-derived MMP9 in fibrosis resolution is associated with elevation in ratio of MMPs/TIMPs.
Recent studies have demonstrated that after cessation of liver injury, the regression of liver fibrosis has three steps, including shifting the balance from inflammation to restoration, elimination of active myofibroblasts and ECM degradation. KC-derived MMPs are implicated in playing a critical role in all these processes [1, 5]. It has been reported that MMP8 can promote the transformation of KC phenotype, which plays an important role in shifting the balance from inflammation to restoration [20]. In addition, VEGF was found to cause KC phenotypic changes. Meanwhile, the expression of VEGF is increased by treatment of TGFβ, which is activated by MMP9 or MMP13 [21, 22]. KC-derived MMP9 might also play important role in elimination of active myofibroblasts. A recent study found that transgene of MMP9 resulted in apoptosis of myofibroblasts [23]. Other studies also found that TIMPs and collagen I prevented myofibroblasts from apoptosis, while MMP9 or MMP13 derived from KCs has the able to reduce the levels of TIMPs and collagen I [22]. These studies, together, suggested that KCs-derived MMPs contribute to elimination of active myofibroblasts. The degradation of ECM was the ultimate step of liver fibrosis resolution. KC-derived MMP13 was found to be involved in this progress [6]. In this study, we found that KC-derived MMP9 plays a critical role in liver fibrosis resolution.
We have investigated the cellular source of MMP9 during liver fibrosis resolution. We found that, similar to that of KCs and HSCs, the frequency of PMNs was also decreased gradually after TAA withdrawal. However, our zymography study indicated that HSCs lost the ability to produce MMP9 in the fibrotic liver. Our in vivo study further demonstrated that the expression of MMP9 reduced dramatically after diminishing KCs but not PMNs. These findings support the notion that KCs represent the main source of MMP9 during fibrosis resolution.
The role of KC-derived MMP9 on liver fibrosis resolution was investigated in MMP9-/- animal model as well. We found that the degradation of collagen was enhanced, following adoptive transfer of KCs from WT mice. These results confirmed the role of KC-derived MMP9 in promoting fibrosis resolution.
In summary, we have demonstrated that MMP9 play a critical role in KC-mediated liver fibrolysis. Our results indicate that KC-derived MMP9 is a key regulatory factor of matrix remodeling and architectural repair during fibrosis regression. These findings provide the basis for the development of therapeutic approaches to accelerate regression as well as novel diagnostic tools.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors thank Prof. Qiang Wang, from Cedars-Sinai Medical Center in Los Angeles, for critical review and language editing of this paper.
This work was supported by the National Nature Science Foundation of China (81670561, 81300336), the Nature Science Foundation of Jiangsu Province (QNRC2016022, 2014-WSW-046, BK20130086, 2016-WSW-067, 2017015) and the Nature Science Foundation of Nanjing (JQX14003).
Author's contributions
Min Feng and Jie Ding carried out all the major experiments, collected the data, analyzed and interpreted the data, and wrote the manuscript. Xinhua Zhu and Wenxian Guan obtained funding, provided technical and material support, and participated in drafting the manuscript. Min Wang and Jie Zhang involved in research design, analysis and interpretation of the data, and critical review.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Campana L, Iredale JP. Regression of Liver Fibrosis. Semin Liver Dis. 2017;37:1-10
2. Ellis EL, Mann DA. Clinical evidence for the regression of liver fibrosis. J Hepatol. 2012;56:1171-80
3. Terai S, Tsuchiya A. Status of and candidates for cell therapy in liver cirrhosis: overcoming the "point of no return" in advanced liver cirrhosis. J Gastroenterol. 2017;52:129-40
4. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. 2017;127:55-64
5. Tacke F, Trautwein C. Mechanisms of liver fibrosis resolution. J Hepatol. 2015;63:1038-9
6. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A. et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(E):3186-95
7. Gao J, Jiang Z, Wang S, Zhou Y, Shi X, Feng M. Endoplasmic reticulum stress of Kupffer cells involved in the conversion of natural regulatory T cells to Th17 cells in liver ischemia-reperfusion injury. J Gastroenterol Hepatol. 2016;31:883-9
8. Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS. et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288-95
9. Spleen-derived lipocalin-2 in the portal vein regulates Kupffer cells activation and attenuates the development of liver fibrosis in mice. Lab Invest. 2017; 97:890-902.
10. Feng M, Wang Q, Zhang F, Lu L. Ex vivo induced regulatory T cells regulate inflammatory response of Kupffer cells by TGF-beta and attenuate liver ischemia reperfusion injury. Int Immunopharmacol. 2012;12:189-96
11. Lu L, Feng M, Gu J, Xia Z, Zhang H, Zheng S. et al. Restoration of intrahepatic regulatory T cells through MMP-9/13-dependent activation of TGF-beta is critical for immune homeostasis following acute liver injury. J Mol Cell Biol. 2013;5:369-79
12. Feng M, Wang H, Wang Q, Guan W. Matrix metalloprotease 9 promotes liver recovery from ischemia and reperfusion injury. J Surg Res. 2013;180:156-61
13. Qin L, Han YP. Epigenetic repression of matrix metalloproteinases in myofibroblastic hepatic stellate cells through histone deacetylases 4: implication in tissue fibrosis. Am J Pathol. 2010;177:1915-28
14. Zhang X, Feng M, Liu X, Bai L, Kong M, Chen Y. et al. Persistence of cirrhosis is maintained by intrahepatic regulatory T cells that inhibit fibrosis resolution by regulating the balance of tissue inhibitors of metalloproteinases and matrix metalloproteinases. Transl Res. 2016;169(e1-2):67-79
15. Lo RC, Kim H. Histopathological evaluation of liver fibrosis and cirrhosis regression. Clin Mol Hepatol. 2017;23:302-7
16. Han YP. Matrix metalloproteinases, the pros and cons, in liver fibrosis. J Gastroenterol Hepatol. 2006;21(Suppl 3):88-91
17. Ma ZY, Qian JM, Rui XH, Wang FR, Wang QW, Cui YY. et al. Inhibition of matrix metalloproteinase-9 attenuates acute small-for-size liver graft injury in rats. Am J Transplant. 2010;10:784-95
18. Veidal SS, Vassiliadis E, Barascuk N, Zhang C, Segovia-Silvestre T, Klickstein L. et al. Matrix metalloproteinase-9-mediated type III collagen degradation as a novel serological biochemical marker for liver fibrogenesis. Liver Int. 2010;30:1293-304
19. Yan C, Zhou L, Han YP. Contribution of hepatic stellate cells and matrix metalloproteinase 9 in acute liver failure. Liver Int. 2008;28:959-71
20. Wen G, Zhang C, Chen Q, Luong le A, Mustafa A, Ye S. et al. A Novel Role of Matrix Metalloproteinase-8 in Macrophage Differentiation and Polarization. J Biol Chem. 2015;290:19158-72
21. Westergren-Thorsson G, Bagher M, Andersson-Sjoland A, Thiman L, Lofdahl CG, Hallgren O. et al. VEGF synthesis is induced by prostacyclin and TGF-beta in distal lung fibroblasts from COPD patients and control subjects: Implications for pulmonary vascular remodelling. Respirology. 2018;23:68-75
22. Ramachandran P, Iredale JP, Fallowfield JA. Resolution of liver fibrosis: basic mechanisms and clinical relevance. Semin Liver Dis. 2015;35:119-31
23. Atta H, El-Rehany M, Hammam O, Abdel-Ghany H, Ramzy M, Roderfeld M. et al. Mutant MMP-9 and HGF gene transfer enhance resolution of CCl4-induced liver fibrosis in rats: role of ASH1 and EZH2 methyltransferases repression. PLoS One. 2014;9(e):112384
Author contact
![]() Corresponding authors: Address: Department of General Surgery, Nanjing Drum Tower Hospital, the Affiliated Hospital of Nanjing University Medical School, Nanjing 210008, China. Fax: +86 25 83304616. E-mail address: drzhuxhcom (X. Zhu) or guan-wxcom (W. Guan).
Corresponding authors: Address: Department of General Surgery, Nanjing Drum Tower Hospital, the Affiliated Hospital of Nanjing University Medical School, Nanjing 210008, China. Fax: +86 25 83304616. E-mail address: drzhuxhcom (X. Zhu) or guan-wxcom (W. Guan).