Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(10):1153-1162. doi:10.7150/ijbs.25420 This issue Cite
Research Paper
Combined Effects of PVT1 and MiR-146a Single Nucleotide Polymorphism on the Lung Function of Smokers with Chronic Obstructive Pulmonary Disease
Sijing Zhou1*, Yi Liu2*, Min Li3, Peipei Wu2, Gengyun Sun2, Guanghe Fei2, Xuan Xu4, Xuexin Zhou5, Luqian Zhou5, Ran Wang2 ![]()
1. Hefei Prevention and Treatment Center for Occupational Diseases, Hefei 230022, China
2. Department of respiratory and critical care medicine, the first affiliated hospital of Anhui medical university, Hefei 230022, China
3. Department of oncology, the first affiliated hospital of Anhui medical university, Hefei 230022, China
4. Division of Pulmonary/Critical Care Medicine, Cedars sinai Medical Center, Los Angeles 90015, USA
5. The first clinical college of Anhui medical university, Hefei 230032, China
*These authors contributed equally to the work.
Received 2018-2-7; Accepted 2018-6-16; Published 2018-6-23
Abstract

Non-coding RNAs play an important role in the pathogenesis of chronic obstructive pulmonary disease (COPD). This study was performed to investigate the role of PVT1 and miR-146a single nucleotide polymorphisms (SNPs) in the lung function of COPD smokers. Real-time PCR and Western blot analyses were performed to measure the expression of miR-146 and PVT1 SNPs and determine the effect of these SNPs on the pathogenesis of COPD. A total of 100 COPD smokers were enrolled in this study and divided into four groups as follows: Rs2910164CC/GC + Rs13281615GG; Rs2910164CC/GC + Rs13281615GA/AA; Rs2910164GG + Rs13281615GG; and Rs2910164GG + Rs13281615GA/AA. No obvious differences in terms of age, sex, and body height and weight were found among the four groups. However, the Rs2910164GG + Rs13281615GA/AA was associated with the highest stage of the Global Initiative for Chronic Obstructive Lung Disease and the highest values of the forced vital capacity, forced expiratory volume, and diffusing capacity of carbon monoxide, while the lowest values of these parameters were observed in the Rs2910164CC/GC + Rs13281615GG group. In addition, the highest and lowest COX2 levels were observed in the Rs2910164GG + Rs13281615GA/AA and Rs2910164CC/GC + Rs13281615GG groups, respectively. PVT1 directly and negatively regulated the miR-146a expression, which in turn directly and negatively regulated COX2 expression. Thus, the combined actions of SNP in PVT1 Rs13281615 and miR-146a Rs2910164 affected the lung function in COPD smokers.
Keywords: PVT1, miR-146a, rs13281615, rs2910164, COX2, chronic obstructive pulmonary disease
Introduction
Smoking-induced chronic obstructive pulmonary disease (COPD) is one of the highly preventable chronic diseases in the United States [1]. However, COPD is the third leading cause of mortality worldwide and can also lead to poor prognosis in many other diseases [2, 3]. Cigarette smoke contains a complex mixture of chemicals, such as reactive oxygen species (ROS) and carcinogens. Thus, this smoke can induce airway inflammation by activating relevant signaling pathways regulated by various systemic and pulmonary inflammatory factors, such as TNF-α and COX-2. COX2 is a protective gene activated by different stimuli and was shown to be upregulated in COPD patients [4]. In vivo, COX2 is responsible for PGE2 synthesis, and, compared with normal controls, the fibroblasts collected from COPD patients tended to synthesize more PGE2 [5].
Although nearly 70% of the entire human genome is transcribed, less than 2% of the genome can encode proteins. Non-coding RNAs (ncRNAs) can be categorized into short (≤200 base pairs) or long (lncRNA; >200 base pairs) ncRNAs. The entire human genome encodes approximately 10,000 lncRNA genes [6]. MicroRNAs belong to a class of small but important ncRNAs that are ∼22 nucleotides in length (in mammals) and are derived from hairpin precursors [7]. MiRNAs can guide the binding of RNA-induced silencing complex to the miRNA response elements on their target transcripts and subsequently mediate post-transcriptionally gene expression by translation inhibition or transcript degradation [7]. The genetic variations in the non-coding and coding regions of a gene sequence may affect the degradation, localization, translation, and splicing of its transcripts. These processes ultimately lead to the onset of diseases [8]. In many types of diseases, the disproportionately large number of single nucleotide polymorphisms (SNPs) mapped to the non-coding regions of the genome confirms the role of post-transcriptional gene regulation in the pathogenesis of diseases [9]. For example, SNP rs2910164 in the precursor of miR-146a is involved in different malignancies, such as ovarian cancer, breast cancer, hepatocellular cancer, papillary thyroid cancer, gastric cancer, prostate cancer, and esophageal squamous cell cancer[10]. Jazdewski et al. showed that, by affecting the processing and maturation of pre-miRNAs, the C allele of rs2910164 can reduce the level of mature miR-146a and magnitude of inhibition on its target genes, such as IL-1 receptor-associated kinase 1 and tumor necrosis receptor-associated factor 6. Furthermore, the GG genotype of SNP rs13281615 in PVT1 is associated with downregulated PVT1 expression and an increased risk of breast cancer [11].
In our previous study, we investigated the effect of SNP rs2910164 on the expression of its host, miR-146a, and its target, COX2 [12]. PVT1 could regulate the miR-146a expression by inducing its methylation. In this study, we studied the combined effect of rs13281615 and rs2910164 on the expression of miR-146a and COX2 and evaluated their role in the lung function of COPD smokers.
Results
Patient characteristics
A total of 100 COPD smokers were enrolled in this study. Patient information, including age, sex, body height, body weight, smoking history, stage of the Global Initiative for Chronic Obstructive Lung Disease (GOLD), forced vital capacity (FVC), forced expiratory volume (FEV), and diffusion capacity for carbon monoxide (DLCO), was collected and listed in Table 1. All subjects were assigned to one of the four groups according to their genotypes of PVT1 Rs13281615 and miR-146a Rs2910164, namely, Rs2910164CC/GC + Rs13281615GG (n=35), Rs2910164CC/GC + Rs13281615GA/AA (n=23), Rs2910164GG + Rs13281615GG (n=25), and Rs2910164GG + Rs13281615GA/AA (n=17). Student t-tests were performed for statistical evaluation. No obvious differences in terms of age, sex, body height, weight, and smoking history were found among the different groups. However, the highest GOLD stage and FVC, FEV, and DLCO values were observed in the Rs2910164GG + Rs13281615GA/AA group, while the lowest values of these parameters were observed in the Rs2910164CC/GC + Rs13281615GG group. All genotype frequencies were in accordance with the Hardy-Weinberg equilibrium.
Demographic and clinicopathological characteristics of the participants in this study
| Characteristics | Rs2910164 CC/GC | Rs2910164 GG | P value | ||||
|---|---|---|---|---|---|---|---|
| Rs13281615 GG (N=35) | Rs13281615 GA/AA (N=23) | Rs13281615 GG (N=25) | Rs13281615 GA/AA (N=17) | ||||
| Sex (Male/Female) | 33/2 | 22/1 | 23/2 | 16/1 | 0.8421 | ||
| Age (years) | 62.4 ± 5.1 | 61.6 ± 4.3 | 60.4 ± 5.3 | 60.2 ± 5.5 | 0.4215 | ||
| Height (cm) | 169.5 ± 7.7 | 172.4 ± 8.2 | 170.4 ± 6.4 | 170.4 ± 6.8 | 0.6251 | ||
| Weight (kg) | 70.4 ± 5.5 | 68.4 ± 7.4 | 69.1 ± 5.9 | 68.4 ± 7.5 | 0.5412 | ||
| Smoking (pack years) | 41.4 ± 10.4 | 40.2 ± 9.8 | 38.5 ± 8.8 | 39.4 ± 9.4 | 0.7216 | ||
| Smoking status | |||||||
| Current smoker | 20 | 13 | 14 | 9 | |||
| Ex-smoker | 15 | 10 | 11 | 8 | 0.9934 | ||
| GOLD stage | |||||||
| I | 2 | 3 | 3 | 5 | |||
| II | 10 | 6 | 15 | 10 | |||
| III | 17 | 12 | 15 | 2 | |||
| IV | 6 | 2 | 2 | 0 | 0.0076 | ||
| FVC (% of predicted value) | 86.1 ± 7.4 | 89.3 ± 7.9 | 91.4 ± 7.4 | 93.5 ± 9.3 | <0.01 | ||
| FEV1 (% of predicted value) | 43.8 ± 5.4 | 49.2 ± 6.7 | 51.4 ± 7.1 | 52.6 ± 7.8 | <0.01 | ||
| DLCO (% of predicted value) | 58.4 ± 6.2 | 60.7 ± 7.2 | 64.8 ± 7.6 | 66.8 ± 8.1 | <0.01 | ||
Expression of miR-146a and COX2 differed among the four groups
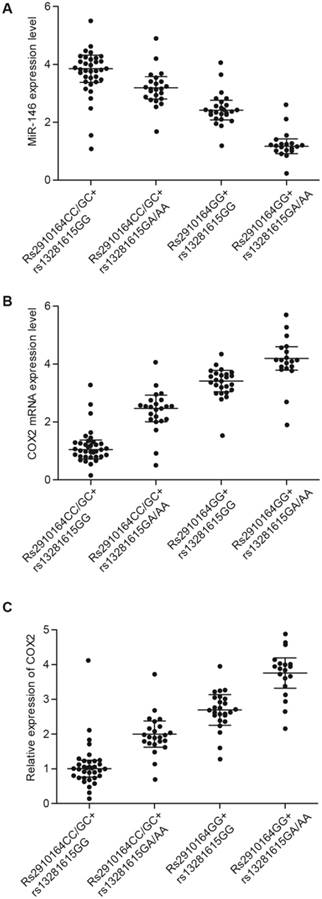
Lung tissue samples were collected from the 100 subjects, and the samples were analyzed by real-time PCR and Western blot to measure the expression of miR-146a and COX2 in the four patient groups. The highest and lowest levels of miR-146a were found in the Rs2910164CC/GC + Rs13281615GG and Rs2910164GG + Rs13281615GA/AA groups, respectively (Fig. 1A). In addition, the highest and lowest mRNA (Fig. 1B) and protein (Fig. 1C) levels of COX2 were observed in the Rs2910164GG + Rs13281615GA/AA and Rs2910164CC/GC + Rs13281615GG groups, respectively.
MiR-146a and COX2 were differentially expressed among the four different groups. A. The highest and lowest miR-146a levels were found in the Rs2910164CC/GC + Rs13281615GG and Rs2910164GG + Rs13281615GA/AA groups, respectively. In addition, miR-146a expression was markedly higher in the Rs2910164CC/GC + Rs13281615GA/AA group than in the Rs2910164GG + Rs13281615GG group. B. The highest and lowest COX2 mRNA levels were observed in the Rs2910164GG + Rs13281615GA/AA and Rs2910164CC/GC + Rs13281615GG groups, respectively. In addition, COX2 mRNA level was considerably higher in the Rs2910164GG + Rs13281615GG group than in the Rs2910164CC/GC + Rs13281615GA/AA group. C. The highest and lowest COX2 protein levels were observed in the Rs2910164GG + Rs13281615GA/AA and Rs2910164CC/GC + Rs13281615GG groups, respectively. In addition, COX2 protein level was markedly higher in the Rs2910164GG + Rs13281615GG group than in the Rs2910164CC/GC + Rs13281615GA/AA group. D. The highest and lowest COX2 protein levels were observed in the Rs2910164GG + rs13281615GA/AA and Rs2910164CC/GC + Rs13281615GG groups, respectively. In addition, the COX2 protein level was markedly higher in the Rs2910164GG + Rs13281615GG group than in the Rs2910164CC/GC + Rs13281615GA/AA group.
MiR-146a directly targeted PVT1 and COX2
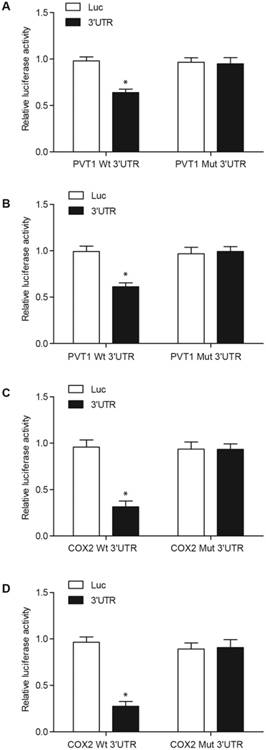
The results from the computational analyses showed a putative binding site for miR-146a in PVT1 and 3′-UTR of COX2. In the luciferase assay of fibroblasts (Figs. 2A and 2C) and pulmonary artery smooth muscle cells (PASMCs) (Figs. 2B and 2D), miR-146a substantially reduced the luciferase activity of wild-type PVT1 and COX2 3′-UTR but showed no effect on the luciferase activity of mutant PVT1 and mutant COX2 3′-UTR. This result suggested that miR-146a directly targeted and repressed the expression of PVT1 and COX2.
PVT1 directly targeted miR-146a, while COX2 was a virtual target gene of miR-146a. A. MiR-146a inhibited the luciferase activity of wild-type PVT1 but did not affect the luciferase activity of mutant PVT1 in the fibroblasts. B. MiR-146a inhibited the luciferase activity of wild-type PVT1 but did not affect the luciferase activity of mutant PVT1 in the PASMCs. C. MiR-146a inhibited the luciferase activity of wild-type COX2 3′-UTR but did not affect the luciferase activity of the mutant COX2 3′-UTR in the fibroblasts. D. MiR-146a inhibited the luciferase activity of the wild-type COX2 3′-UTR but did not affect the luciferase activity of the mutant COX2 3′-UTRin the PASMCs.
COX2 was differently expressed among the four groups of patients
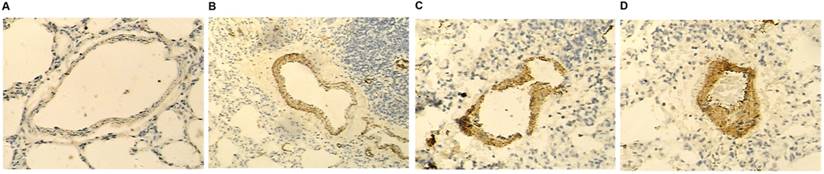
Immunohistochemical (IHC) results (Fig. 3) showed that the highest and lowest levels of COX2 were in the Rs2910164GG + Rs13281615GA/AA and Rs2910164CC/GC + Rs13281615GG groups, respectively.
COX2 was differentially expressed among the four different groups. The highest and lowest COX2 protein levels were observed in the Rs2910164GG + Rs13281615GA/AA (D) and Rs2910164CC/GC + Rs13281615GG (A) groups, respectively. In addition, COX2 protein was considerably higher in the Rs2910164GG + Rs13281615GG group (C) than in the Rs2910164CC/GC + Rs13281615GA/AA group (B).
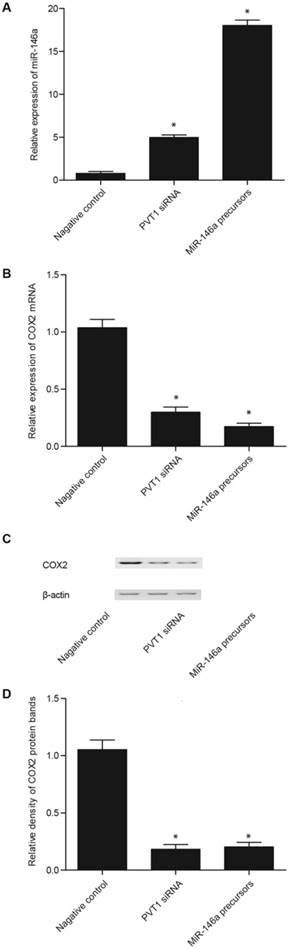
Effect of PVT1 siRNA and miR-146a precursor on the level of miR-146a and COX2 in the fibroblasts and PASMCs
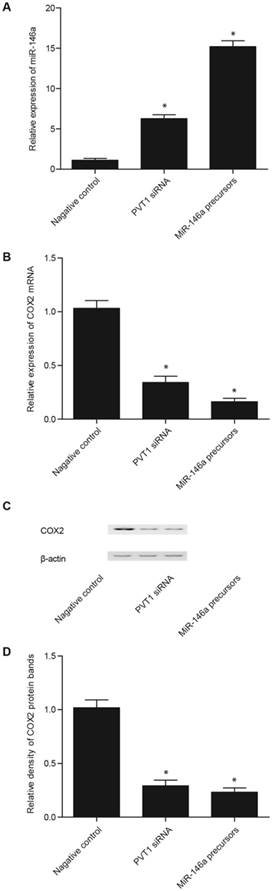
Real-time PCR and Western blot analyses were performed to measure the levels of miR-146a and COX2 in the fibroblasts and PASMCs transfected with PVT1 siRNA, miR-146a precursor, pcDNA 3.1-PVT1, or anti-miR-146a. The transfection of PVT1 siRNA and miR-146a precursor significantly enhanced miR-146a expression, although the effect of miR-146a precursor was considerably stronger than that of PVT1 siRNA (Figs. 4A and 5A). In addition, the transfection of PVT1 siRNA and miR-146a precursor significantly downregulated the expression of COX2 mRNA (Figs. 4B and 5B) and proteins (Figs. 4C, 4D, 5C, and 5D), although the effect of the miR-146a precursor was remarkably stronger than that of PVT1 siRNA.
Effect of PVT1 siRNA and miR-146a precursor on the level of miR-146a and COX2 in the fibroblasts. A. The PVT1 siRNA and miR-146a precursor significantly upregulated the miR-146a expression, but the effect of miR-146a precursor was markedly stronger than that of PVT1 siRNA. B. The PVT1 siRNA and miR-146a precursor significantly downregulated the expression of COX2 mRNA, but the effect of miR-146a precursor was markedly stronger than that of PVT1 siRNA. C. The PVT1 siRNA and miR-146a precursor significantly downregulated the expression of COX2 protein, but the effect of miR-146a precursor was markedly stronger than that of PVT1 siRNA. D. PVT1 siRNA and miR-146a precursor significantly downregulated the expression of COX2 protein, but the effect of miR-146a precursor was markedly stronger than that of PVT1 siRNA.
Effect of PVT1 siRNA and miR-146a precursor on the miR-146a and COX2 levels in the PASMCs. A. The PVT1 siRNA and miR-146a precursor significantly upregulated miR-146a expression, but the effect of miR-146a precursor was markedly stronger than that of PVT1 siRNA. B. The PVT1 siRNA and miR-146a precursor significantly downregulated the expression of COX2 mRNA, but the effect of miR-146a precursor was markedly stronger than that of PVT1 siRNA. C. The PVT1 siRNA and miR-146a precursor significantly downregulated the expression of COX2 protein, but the effect of miR-146a precursor was markedly stronger than that of PVT1 siRNA. D. The PVT1 siRNA and miR-146a precursor significantly downregulated the expression of COX2 protein, but the effect of miR-146a precursor was markedly stronger than that of PVT1 siRNA.
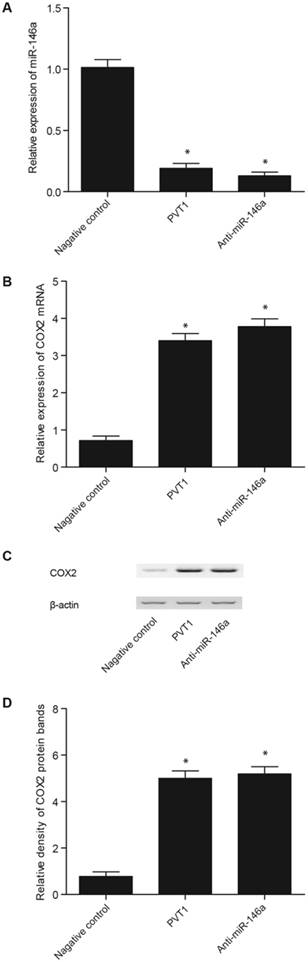
Effect of pcDNA-PVT1 and anti-miR-146a on the levels of miR-146a and COX2 in the fibroblasts and PASMCs
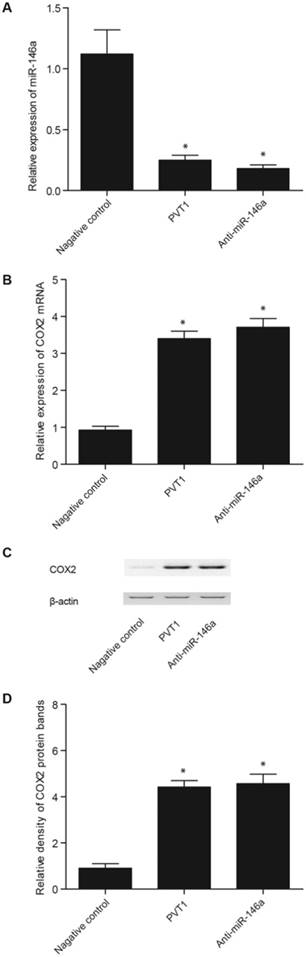
The transfection of pcDNA 3.1-PVT1 and anti-miR-146a in the fibroblasts (Fig. 5A) and PASMCs (Fig. 6A) significantly inhibited miR-146a expression, although the effect of anti-miR-146a was considerably stronger than that of pcDNA 3.1-PVT1 (Figs. 6 and 7). In addition, the expression of COX2 mRNA (Figs. 6B and 7B) and proteins (Figs. 6C, 6D, 7C, and 7D) in the fibroblasts (Fig. 6) and PASMCs (Fig. 7) was apparently upregulated by the transfection of pcDNA 3.1-PVT1 and anti-miR-146a, although the effect of anti-miR-146a was markedly stronger.
Effect of pcDNA-PVT1 and anti-miR-146a on the levels of miR-146a and COX2 in the fibroblasts. A. The pcDNA-PVT1 and anti-miR-146a significantly downregulated miR-146a expression, but the effect of anti-miR-146a was markedly stronger than that of pcDNA-PVT1. B. The pcDNA-PVT1 and anti-miR-146a significantly downregulated the expression of COX2 mRNA, but the effect of anti-miR-146a was markedly stronger than that of pcDNA-PVT1. C. The pcDNA-PVT1 and anti-miR-146a significantly downregulated the expression of COX2 protein, but the effect of anti-miR-146a was markedly stronger than that of pcDNA-PVT1. D. The pcDNA-PVT1 and anti-miR-146a significantly downregulated the expression of COX2 protein, but the effect of anti-miR-146a was markedly stronger than that of pcDNA-PVT1.
Effect of pcDNA-PVT1 and anti-miR-146a on the levels of miR-146a and COX2 in the PASMCs. A. The pcDNA-PVT1 and anti-miR-146a significantly downregulated miR-146a expression, but the effect of anti-miR-146a was markedly stronger than that of pcDNA-PVT1. B. The pcDNA-PVT1 and anti-miR-146a significantly downregulated the expression of COX2 mRNA, but the effect of anti-miR-146a was markedly stronger than that of pcDNA-PVT1. C. The pcDNA-PVT1 and anti-miR-146a significantly downregulated the expression of COX2 protein, but the effect of anti-miR-146a was markedly stronger than that of pcDNA-PVT1. D. The pcDNA-PVT1 and anti-miR-146a significantly downregulated the expression of COX2 protein, but the effect of anti-miR-146a was markedly stronger than that of pcDNA-PVT1.
Discussion
The important role of lncRNAs in COPD [13] has been demonstrated by the altered expression of PVT1, LINC00883, and LINC00882 in the lung tissues of COPD patients [14]. In addition, Sato et al. showed that the tumor necrosis factor (TNF)-α and interleukin (IL)-1β, which are the two potent activators of the NF-кB signaling pathway, upregulated the miR-146a expression in the fibroblasts of the COPD patients, but to a lesser extent compared with that in the fibroblasts from the control subjects [15]. Similarly, miR-146a was downregulated in the sputum of COPD patients and smokers [16] but with higher downregulation in smokers [17]. Furthermore, PVT1 downregulates the miR-146a expression by inducing the methylation in the promoter of miR-146a [18]. Prostate cancer cells transfected with pcDNA3-PVT1 showed significantly downregulated miR-146a expression, because PVT1 acted as an antagonist of miR-146a [18]. In our previous and current studies, we showed that COX2 was a direct target of miR-146a, whereas lncRNAPVT1 was an upstream regulator of miR-146a. COX2 is a cyclooxygenase isoenzyme and helps catalyze the synthesis of thromboxane and prostaglandins. In addition to its constitutive expression in the brain and kidneys, COX2 primarily acts as an inducible enzyme activated in a number of different cells upon stimulation by mitogens, cytokines, and endotoxins [19]. COX2 primarily helps in the conversion of prostaglandin-E2 (PGE2) from arachidonic acid (AA).
Thus, PGE2 level often indicates the local activity of cyclooxygenase [20]. PGE2 is a pro-inflammatory factor synthesized by stromal cells, cancer cells, and infiltrating myeloid cells, and this molecule can bind to G-protein-coupled receptors. COX2 is a key factor controlling the synthesis of PGE2 during an immune response. Lovgren et al. demonstrated that, when responding to environmental stimuli, COX2-/- mice showed increased fibrosis and severely hindered pulmonary functions [21]. The beneficial function of PGE2 in the lungs has also been confirmed by the results from other experimental models of inflammation and pulmonary fibrosis. For example, PGE2 inhibits the growth of fibroblasts and the synthesis of collagen in the airway epithelium. In addition, PGE2 can also inhibit the transition from the fibroblast to the myofibroblast upon TGF-/- induction [22]. Furthermore, PGE2 suppresses the activation of NF-κB in LPS-induced monocytes. This process decreases the production of TNF-α while promoting the synthesis of IL-10, which is an anti-inflammatory cytokine [23]. Several studies have consistently shown that PGE2 participates in the regulation of lymphocyte differentiation and trafficking [24]. In this context, Sato et al. demonstrated that miRNA-146a expression was downregulated in the fibroblasts of COPD smokers, and this phenomenon led to upregulated COX2 and PGE2 in these patients [15]. Another report confirmed that miR-146a expression was inhibited by the activated inflammatory signaling in smokers [25].
SNP rs13281615 is located in the PVT1 gene on chromosome 8q24. The GG genotype of rs13281615 has been linked to an increased risk of breast cancer. Several SNPs on 8q24 have been identified as risk alleles and demonstrated to participate in the altered expression of PVT1 or MYC in human cancers [26]. The GG genotype of SNP rs13281615 may play similar roles in breast cancer by affecting the PVT1 expression [26]. Rs2910164 is a C/G polymorphism in the precursor stem region of the pre-miR146a on chromosome 5q33.[27] Jazdzewski et al. first showed that rs2910164 could regulate the miR-146a expression and was associated with the genetic predisposition to PTC [27]. In addition, the minor allele of rs2910164 polymorphism decreased the miR-146a level but increased the COX2 level and its catalytic product PGE2. This phenomenon improved lung functions and alleviated the severity of COPD [12]. Hence, rs2910164 polymorphism may become a new marker for COPD prognosis, whereas miR-146a may become a novel therapeutic target for COPD treatment [12]. In our previous study, we showed that rs2910164 polymorphism could affect COX2 expression by regulating the miR-146a expression. Given that PVT1 is a regulator of COX2 and rs13281615 can affect the PVT1 expression, we speculated that COX2 expression may be affected by a synergistic effect of the two polymorphisms. In this study, a total of 100 COPD smokers were enrolled and genotyped for rs2910164 and rs13281615. The results showed that highest and lowest GOLD stage and FVC, FEV, and DLCO values were observed in the Rs2910164GG + Rs13281615GA/AA and Rs2910164CC/GC + Rs13281615GG groups, respectively.
This study has several limitations. First, the sample size was relatively small, and a study with big sample size is needed to validate the conclusions from this study. Second, only two polymorphisms were included in this study, and further comprehensive study with more variants is also warranted.
In summary, we demonstrated that rs2910164 and rs13281615 in PVT1 and miR-146a, respectively, exerted a combined effect on the lung function of COPD smokers. We found that the rs13281615 polymorphism in PVT1 affected PVT1 expression, while the minor allele of rs2910164 downregulated miR-146a expression. Both SNPs upregulated COX2 expression, which plays an important role in the lung function and severity of COPD.
Materials and methods
Sample collection
This study was approved by the Ethical Committee of Anhui Medical University and conducted according to the Declaration of Helsinki. A total of 100 COPD smokers were enrolled in this study, and 66 lung tumor samples were obtained from the subjects after surgery. The COPD stage of the patients was determined according to the following criteria in GOLD: FEV1 in 1 s <30%, GOLD4 and “very severe”; 30% ≤FEV1<50%, GOLD3 and “severe”; 50%≤FEV1<80%, GOLD2 and “moderate”; and FEV1>80%, GOLD1 and “mild”. Peripheral blood samples were also collected from all participants. Participants or their first-degree relatives signed informed consents before the start of the study.
Genotyping by sequencing
The experiments were performed as previously described [28]. The genomic DNA from the tissue samples was isolated using a DNA extraction kit (Shunhua Bioengineering Co. Ltd., China) and subsequently amplified using PCR. In the next step, an ExoSAP-IT purification kit (USB, London, UK) was utilized to purify the PCR products before the sequencing of two SNPs (i.e., rs2910164 and rs13281615).
RNA isolation and real-time PCR
The experiments were performed as previously described [29, 30]. An RNeasy Mini kit (Qiagen, Germany) was utilized to extract the total RNA from the fibroblasts, PASMCs, and tissue samples. The absorbance of the extracted RNA was measured on a spectrophotometer (Thermo Scientific, Waltham, MA, US), and its value was used to determine the RNA quality. Subsequently, the RNA samples were treated with DNase I (Thermo Fisher Scientific, Waltham, MA, US) and converted to cDNA using M-MLV Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA, US). The mRNA levels of PVT1, miR-146, and COX2 in the samples were quantified on an AB7500 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, US) using TaqMan Gene Expression Assays (Thermo Fisher Scientific, Waltham, MA, US). The RT-qPCR reaction was conducted under the following conditions: 95 °C for 10 min, followed by 45 two-step cycles of 95 °C for 15 s and 60 °C for 1 min. The products of RT-qPCR were analyzed on a 2% agarose gel, and their nucleotide sequences were confirmed by Sanger sequencing on an ABI Prism 3100 Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, US). The relative expression of PVT1, miR-146, and COX2 was computed using the ΔΔCT method and the Relative Quantitation (RQ) software (Thermo Fisher Scientific, Waltham, MA, US). All experiments were repeated thrice.
Vector construction and mutagenesis
The 3′-UTR segment of the PVT1 gene was amplified by PCR, followed by the insertion of the PCR product into a pcDNA3.1 (+) (Invitrogen, CA, USA) vector immediately upstream of the firefly luciferase reporter gene. This construct was named pcDNA3.1 (+)-COX2-3′UTR. Subsequently, the Site-Directed Mutagenesis Kit (SBS Genetech, Beijing, China) was utilized to mutate the 3′-UTR of PVT1, and the mutant was then inserted into the same location of another pcDNA3.1 (+) vector. This mutant construct was named pcDNA3.1(+)-mut- PVT1-3′UTR. Similarly, the 3′-UTR of the wild-type COX2 was amplified using PCR, and the PCR product was inserted into a pcDNA3.1 (+) vector immediately downstream of the firefly luciferase reporter gene. This construct was named pcDNA3.1 (+)-COX2-3′UTR. Subsequently, the Site-Directed Mutagenesis Kit was utilized to mutate the miR-146a binding site in the 3′-UTR of COX2, and the mutant was then inserted into the same location of another pcDNA3.1 (+) vector. This mutant construct was named pcDNA3.1(+)-mut-COX2-3′UTR. In addition, the full sequence of PVT1 was amplified and inserted into a pcDNA3.1 (Invitrogen, CA, USA) vector to create pcDNA 3.1-PVT1. After these constructs were created, direct Sanger sequencing was performed to confirm the correctness of their sequences. All experiments were repeated thrice.
Cell culture and transfection
Human fibroblasts were harvested as described previously [31], while PASMCs were obtained from ATCC. Both cells were cultured at 5% CO2 and 37 °C in DMEM (Gibco, Thermo Fisher Scientific, Massachusetts, USA) supplemented with 10% fetal bovine serum, 100 U streptomycin, and 100 mg/mL penicillin. When the cells reached 80% confluence, they were seeded into 96-well plates and co-transfected with 40 ng of luciferase constructs (wild-type or mutant PVT1 3′-UTR or COX2 3′-UTR), 400 ng of miR-146a, a rellina luciferase control vector (Promega, Fitchburg, WI, USA), and 4 ng of pRL-TK. For the inhibition experiments, the cells were transfected with pcDNA3.1-PVT1, miR-146a precursor, PVT1 siRNA, or anti-miR-146a. All transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and repeated thrice.
Luciferase assay
The experiments were performed as previously described [32]. The luciferase activity of transfected cells was measured on a TD-20/20 luminometer (Turner Biosystems, Sunnyvale, CA, USA) after 48 h of transfection. All experiments were repeated thrice.
Western blot analysis
The experiments were performed as previously described [33, 34]. Prior to the analysis, the cultured cells and tissue samples were washed thrice in ice-cold phosphate buffered saline (PBS) and then lysed in a RIPA buffer (Pierce, Waltham, MA, USA). Subsequently, the lysate was incubated on ice for 30 min to extract the total protein. A BCA kit (Thermo Fisher, Waltham, MA, USA) was utilized to measure the concentration of the extracted protein. Subsequently, 30 μg of the extracted protein were separated by 6% or 10% SDS-PAGE and transferred onto a PVDF membrane (Millipore, Billerica, MA). Then, the membrane was blocked at room temperature for 60 min in a TBS-Tween 20 (TBST) buffer supplemented with 5% bovine serum albumin and incubated at 4 °C overnight with primary anti-COX2 antibodies (1:6,000 dilution, Abcam, Cambridge, UK) or anti-β-actin antibodies (1:10,000 dilution, Cell Signaling Technology, Beverly, MA). After being washed thrice with TSBT, the membrane was incubated for 120 min at room temperature with horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibodies (1:12,000 dilution, Santa Cruz Biotechnology Inc., California, US). Finally, ECL detection reagents (Millipore, Billerica, MA) were utilized to visualize the membrane and the intensity of protein bands was measured by a Syngene GeneGenius gel imaging system (Syngene, Frederick, MD, UK). All tests were repeated thrice.
Immunohistochemistry assay
The experiments were performed as previously described [35, 36]. All tissue sample sections were deparaffinized in xylene and rehydrated in gradient alcohol. Subsequently, the samples were incubated with 5% goat serum to eliminate the non-specific binding. In the next step, the sample sections were incubated at 4 °C overnight with mouse anti-COX2 primary antibodies (1:600 dilution, Abcam, Cambridge, UK), followed by 60 min of incubation at room temperature with mouse anti-HRP secondary antibodies (1:2000 dilution, Santa Cruz Biotechnology Inc., California, US). Finally, the sections were developed at room temperature for 5 min in PBS containing 3,3′-di-aminobenzidine (Boster Biological Technology, Ltd.). All IHC staining results were independently reviewed by two pathologists. All tests were repeated thrice.
Statistical analysis
All data were expressed as mean ± standard deviation. The significance of differences between two groups was examined using Student's t-tests, while the significance of differences among multiple groups was evaluated using ANOVA in conjunction with Bonferroni post hoc tests. A P value less than 0.05 was considered as statistically significant. Statistical analysis was performed using SPSS 15.0 (IBM Inc., Chicago, IL). The genotype frequency was evaluated for Hardy-Weinberg equilibrium.
Acknowledgements
This research was supported by the fund from the Natural Science Foundation of China (No.81300041), the fund for the academic backbone of the excellent young and middle-age people of Anhui medical university (2013), the fund from the first affiliated hospital of Anhui medical university for reserve talents(2014), the fund for Excellent top talent cultivation project of Anhui Higher Education Institutions (gxyqZD2017030).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A. et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347-65
2. Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2010. Natl Vital Stat Rep. 2013;61:1-117
3. Hansell AL, Walk JA, Soriano JB. What do chronic obstructive pulmonary disease patients die from? A multiple cause coding analysis. Eur Respir J. 2003;22:809-14
4. Chen Y, Chen P, Hanaoka M, Droma Y, Kubo K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology. 2008;13:1014-21
5. Togo S, Holz O, Liu X, Sugiura H, Kamio K, Wang X. et al. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med. 2008;178:248-60
6. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775-89
7. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
8. Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet. 2005;42:737-48
9. Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166-76
10. Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci U S A. 2008;105:7269-74
11. Zhang Z, Zhu Z, Zhang B, Li W, Li X, Wu X. et al. Frequent mutation of rs13281615 and its association with PVT1 expression and cell proliferation in breast cancer. J Genet Genomics. 2014;41:187-95
12. Wang R, Li M, Zhou S, Zeng D, Xu X, Xu R. et al. Effect of a single nucleotide polymorphism in miR-146a on COX-2 protein expression and lung function in smokers with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2015;10:463-73
13. De Smet EG, Mestdagh P, Vandesompele J, Brusselle GG, Bracke KR. Non-coding RNAs in the pathogenesis of COPD. Thorax. 2015;70:782-91
14. Perry MM, Tsitsiou E, Austin PJ, Lindsay MA, Gibeon DS, Adcock IM. et al. Role of non-coding RNAs in maintaining primary airway smooth muscle cells. Respir Res. 2014;15:58
15. Sato T, Liu X, Nelson A, Nakanishi M, Kanaji N, Wang X. et al. Reduced miR-146a increases prostaglandin E(2)in chronic obstructive pulmonary disease fibroblasts. Am J Respir Crit Care Med. 2010;182:1020-9
16. Van Pottelberge GR, Mestdagh P, Bracke KR, Thas O, van Durme YM, Joos GF. et al. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183:898-906
17. Ezzie ME, Crawford M, Cho JH, Orellana R, Zhang S, Gelinas R. et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67:122-31
18. Liu HT, Fang L, Cheng YX, Sun Q. LncRNA PVT1 regulates prostate cancer cell growth by inducing the methylation of miR-146a. Cancer medicine. 2016;5:3512-3519
19. Kurumbail RG, Kiefer JR, Marnett LJ. Cyclooxygenase enzymes: catalysis and inhibition. Curr Opin Struct Biol. 2001;11:752-60
20. Giuliano F, Warner TD. Origins of prostaglandin E2: involvements of cyclooxygenase (COX)-1 and COX-2 in human and rat systems. J Pharmacol Exp Ther. 2002;303:1001-6
21. Bozinovski S, Anthony D, Vlahos R. Targeting pro-resolution pathways to combat chronic inflammation in COPD. Journal of thoracic disease. 2014;6:1548-56
22. Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. 2003;29:537-44
23. Conte E, Bonaiuto C, Nesci C, Crimi N, Vancheri C, Messina A. Nuclear factor-kappaB activation in human monocytes stimulated with lipopolysaccharide is inhibited by fibroblast conditioned medium and exogenous PGE2. FEBS Lett. 1997;400:315-8
24. Okamoto T, Hino O. Expression of cyclooxygenase-1 and -2 mRNA in rat tissues: tissue-specific difference in the expression of the basal level of mRNA. Int J Mol Med. 2000;6:455-7
25. Zago M, Rico de Souza A, Hecht E, Rousseau S, Hamid Q, Eidelman DH. et al. The NF-kappaB family member RelB regulates microRNA miR-146a to suppress cigarette smoke-induced COX-2 protein expression in lung fibroblasts. Toxicol Lett. 2014;226:107-16
26. Zhang Z, Zhu Z, Zhang B, Li W, Li X, Wu X. et al. Frequent mutation of rs13281615 and its association with PVT1 expression and cell proliferation in breast cancer. J Genet Genomics. 2014;41:187-95
27. Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci U S A. 2008;105:7269-74
28. Zhou S, Li M, Zeng D, Xu X, Fei L, Zhu Q. et al. A Single Nucleotide Polymorphism in 3' Untranslated Region of Epithelial Growth Factor Receptor Confers Risk for Pulmonary Hypertension in Chronic Obstructive Pulmonary Disease. Cell Physiol Biochem. 2015;36:166-178
29. Zhou S, Sun L, Cao C, Wu P, Li M, Sun G. et al. Hypoxia-induced microRNA-26b inhibition contributes to hypoxic pulmonary hypertension via CTGF. J Cell Biochem. 2018;119:1942-1952
30. Ding X, Zhou S, Li M, Cao C, Wu P, Sun L. et al. Upregulation of SRF Is Associated With Hypoxic Pulmonary Hypertension by Promoting Viability of Smooth Muscle Cells via Increasing Expression of Bcl-2. J Cell Biochem. 2017;118:2731-2738
31. Wang R, Ding X, Zhou S, Li M, Sun L, Xu X. et al. Microrna-26b attenuates monocrotaline-induced pulmonary vascular remodeling via targeting connective tissue growth factor (CTGF) and cyclin D1 (CCND1). Oncotarget. 2016;7:72746-72757
32. Zhou SJ, Li M, Zeng DX, Zhu ZM, Hu XW, Li YH. et al. Expression variations of connective tissue growth factor in pulmonary arteries from smokers with and without chronic obstructive pulmonary disease. Scientific reports. 2015;5:8564
33. Zhou S, Li M, Zeng D, Sun G, Zhou J, Wang R. Effects of basic fibroblast growth factor and cyclin D1 on cigarette smoke-induced pulmonary vascular remodeling in rats. Experimental and therapeutic medicine. 2015;9:33-38
34. Wang R, Zhou SJ, Zeng DX, Xu R, Fei LM, Zhu QQ. et al. Plasmid-based short hairpin RNA against connective tissue growth factor attenuated monocrotaline-induced pulmonary vascular remodeling in rats. Gene Ther. 2014;21:931-7
35. Wang R, Xu YJ, Liu XS, Zeng DX, Xiang M. CCN2 promotes cigarette smoke-induced proliferation of rat pulmonary artery smooth muscle cells through upregulating cyclin D1 expression. J Cell Biochem. 2012;113:349-59
36. Wang R, Xu YJ, Liu XS, Zeng DX, Xiang M. Knockdown of connective tissue growth factor by plasmid-based short hairpin RNA prevented pulmonary vascular remodeling in cigarette smoke-exposed rats. Arch Biochem Biophys. 2011;508:93-100
Author contact
![]() Corresponding author: Ran Wang Ph.D., Department of respiratory and critical care medicine, The first affiliated hospital of Anhui medical university, Hefei, 230022, China. Phone: 86-551-62922913; Fax: 86-551-62922913; E-mail: ranwangtjmucom
Corresponding author: Ran Wang Ph.D., Department of respiratory and critical care medicine, The first affiliated hospital of Anhui medical university, Hefei, 230022, China. Phone: 86-551-62922913; Fax: 86-551-62922913; E-mail: ranwangtjmucom