Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(10):1186-1195. doi:10.7150/ijbs.24898 This issue Cite
Research Paper
Hepatic Ischemic Preconditioning Alleviates Ischemia-Reperfusion Injury by Decreasing TIM4 Expression
Yu Zhang1 ![]() , Qiang Shen1, Yuanxing Liu1, Hui Chen1, Xiaoxiao Zheng2, Shangzhi Xie2, Haofeng Ji3, Shusen Zheng 1
, Qiang Shen1, Yuanxing Liu1, Hui Chen1, Xiaoxiao Zheng2, Shangzhi Xie2, Haofeng Ji3, Shusen Zheng 1 ![]()
1. Department of Surgery, Division of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, China.
2. Department of Surgery, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, China.
3. Dumont-UCLA Transplant Center, Division of Liver and Pancreas Transplantation, Department of Surgery, David Geffen School of Medicine at University of California-Los Angeles, Los Angeles, CA, USA.
Received 2018-1-12; Accepted 2018-6-3; Published 2018-7-1
Abstract

Ischemia-reperfusion injury (IRI) of the liver is a primary cause of post-liver-surgery complications and ischemic preconditioning (IPC) has been verified to protect against ischemia-reperfusion injury. TIM-4 activation plays an important role in macrophage mediated hepatic IRI. This study aimed to determine whether IPC protects against hepatic IRI through inhibiting TIM-4 activation. In this study, a model of warm liver ischemia (90 min) and reperfusion for 6 h was used. Mice were subjected to ischemia-reperfusion injury with or without ischemic preconditioning and TIM4 blocking antibody. Western blot was determined to detect the expression of TIM4 protein and mitochondrial apoptosis-related protein expression. Liver function was evaluated using the level of alanine transaminase (ALT) and aspartate transaminase (AST), cell apoptosis and pathological examination. We found that compared with the control group, ischemic preconditioning reduced IRI by decreasing hepatocyte apoptosis, ALT, AST, CD68 and CD3 positive cells, tissue myeloperoxidase activity(MPO), and downregulating TIM-4 expression. TIM4 blocking could reduce CD68 and CD3 positive cells in liver. Furthermore, activated monocytes transfusion significantly abolished the protect effect of IPC with increased hepatocyte apoptosis, ALT, AST, CD68 and CD3 positive cells while TIM-4 knockdown monocytes lost this effect. These results suggested that IPC protects against hepatic IRI by downregulating TIM-4 and indicated TIM-4 would be a novel therapeutic target to minimize IRI.
Keywords: TIM4, Ischemic preconditioning (IPC), Ischemia-reperfusion injury (IRI)
Introduction
Ischemia-reperfusion injury (IRI) is a pathophysiological process in which hypoxic organ damage is accentuated following the restoration of blood flow and oxygen delivery to the ischemic tissue. IRI of the liver can occur during various hepatic surgeries, including hepatic trauma surgery, and hepatic resection and transplantation[1], and is a major cause of morbidity and mortality following hepatic surgery. Liver IRI after hepatic resection or transplantation can result in elevated liver enzymes, biliary strictures, clinical dysfunction, and even liver failure [1, 2]. Although a number of experiments have been performed to uncover the mechanisms underlying hepatic IRI, efficient treatments to minimize this injury are still unavailable. Therefore, it is essential to explore novel techniques to ameliorate the detrimental physiological effects of IRI.
Ischemic preconditioning (IPC) is a process during which a short period of ischemia is followed by a period of reperfusion prior to prolonged ischemia. IPC was first described in a renal model[3] and then in a cardiovascular model[4]. In experimental models, hepatic IPC was reported to provide promising liver protection [5, 6]. Consistent with this, the first report of IPC in clinical practice concluded that IPC may protect steatotic livers from IRI[7]. Additionally, IPC has been associated with reduced necrosis and the activation of autophagy in hepatocytes already damaged by chemotherapy among patients with colorectal cancer [8]. A prospective randomized study of 100 consecutive patients undergoing major liver resection proved the protective role of IPC in liver IRI in young but not in older patients[9]. However, the precise mechanism by which IPC confers protection against hepatic IRI has not been fully elucidated.
The T-cell immunoglobulin and mucin domain molecule-4 (TIM4) is conserved between mice and humans, and is mainly expressed in macrophages, mature dendritic cells, and some peritoneal B-cells but not in T-cells [10-14]. TIM-4 regulates adaptive immune responses such as initiation of T-helper cell-2 polarization and induction of T-helper cell-1 apoptosis [11, 15]. TIM-4 is essential for the maintenance of the homeostatic state of resident peritoneal macrophages[16], and the aberrant persistence of TIM-4 leads to dysregulation of lymphocyte activation and autoimmunity[17, 18]. In our previous study, we demonstrated that TIM-4 is required for macrophage migration, phagocytosis, and activation during hepatic IRI[19]. However, the detailed function of TIM-4 in hepatic IRI and its role in IPC are unknown.
In the present study, we confirmed the protective effects of IPC during hepatic IRI in a mouse model. We then determined whether TIM-4 underlies this protective effect of IPC. By providing evidence for the potential mechanism via which IPC protects the liver from IRI, this research provides novel insights into potential therapeutic strategies for and molecular targets in hepatic IRI.
Materials and Methods
Animals
Male C57BL/6 mice (5-week-old, 19-21 g) were obtained from the Shanghai Laboratory Animal Center, Chinese Academy Sciences, and housed in a controlled 12-h light/dark cycle environment with access to chow and water ad libitum. All animal experiments were performed according to the Zhejiang University guidelines for animal care and were approved by the Animal Ethics Review Committee of Zhejiang University.
Surgical procedures and experimental groups
A total of 48 mice were randomly divided into four groups (n = 12): sham group, IRI group, IPC+IRI group, and IPC+IRI+monocyte group. Mice were anesthetized with 4% chloral hydrate (400 mg/kg, intraperitoneally injected). The mice were fixed in position. After skin preparation, a 1.5-cm-long abdominal incision was made. The abdominal cavity was opened with a hook, and the xiphoid process was pulled up and fixed. The left and middle portal vein branches in the liver were clamped with 4.5-cm-long vascular clips to induce ischemia in approximately 70% of the liver (the caudate lobes, left lobe, and middle lobe). Liver ischemia was confirmed under direct vision as the liver went from dark red to white in color. At this time, the abdominal wall of the clot with a temporary clip 90 min. At this moment, we used a clip to stop temporary closure of the abdominal wall for 90 min the celiac wall with a temporary clot clamping clip 90 min. After 90 min, the vascular clamps were released, and the occurrence of intra-abdominal bleeding was confirmed. The abdomen was then closed. Afterwards, the mice were placed under a lamp to maintain their body temperature until the scheduled collection of specimens 6 h later. In the IPC+IRI group, the mice were subjected to 10 min of ischemia and 10 min of reperfusion prior to sustained ischemia, as previously described[19]. Additionally, 6 mice each from the IRI and IPC+IRI groups were randomly selected to receive an infusion containing a blocking monoclonal antibody against TIM-4 (RMT4-53; 0.5 mg/mouse i.v.; Bio X Cell, West Lebanon, NH, USA) at 1 h prior to the induction of ischemia[19], and 12 mice from the IPC+IRI+monocyte groups were randomly selected to receive a transfusion of activated monocytes transfected with or without Tim-4 siRNA, at 1 h prior to the induction of ischemia. We used CD11b positivity to separate monocytes from peripheral blood samples and then stimulated the monocytes with 100 ng/ml lipopolysaccharide for 24 h. At the end of reperfusion, blood and liver samples were collected and preserved for subsequent procedures. All mice were sacrificed by CO2 inhalation.
Enzyme-linked immunosorbent assay
In each group, blood samples were collected after the treatments and used to measure alanine transaminase (ALT), aspartate transaminase (AST), and myeloperoxidase levels. Quantitation of the ALT, AST, and myeloperoxidase levels was performed using enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Hematoxylin and eosin staining
After the experiment had ended after 6 h of reperfusion, the hepatic tissue samples harvested from the mice were fixed with 4% paraformaldehyde for 24 h. After conventional dehydration, wax infiltration, and paraffin embedding, the tissues were sectioned at intervals of 4 µm and stained with hematoxylin and eosin for 5 min. Inflammatory cell infiltration in the prepared samples was evaluated under an optical microscope.
Immunohistochemical analysis
Mouse liver tissues were fixed in a 10% formalin solution for 24 h, embedded in paraffin, sliced, dewaxed, and hydrated according to conventional techniques. Then, the samples were incubated with 5% fetal bovine serum for 30 min at room temperature followed by overnight incubation at 4°C with the primary antibodies (CD3, ab16669, Abcam, Cambridge, MA, USA; CD68, ab125212, Abcam, Cambridge, MA, USA). The samples were again incubated for 30 min at 37°C after the addition of a horseradish peroxidase-conjugated secondary antibody (anti-rabbit-HPR, 7074, Cell Signaling Technology, Danvers, MA, USA). The samples were then treated with diaminobenzidine for coloration. The cell nuclei were counterstained with hematoxylin, and the samples were dehydrated in a gradient series, vitrified with dimethylbenzene, and finally, mounted with neutral balsam.
TdT-mediated biotin-16-dUTP nick-end labeling assay
TdT-mediated biotin-16-dUTP nick-end labeling assay was performed using the One-Step TUNEL Apoptosis Assay kit (Roche, Basel, Switzerland) in order to detect apoptotic cells in the mouse livers. In brief, 4-μm-thick paraffin sections were deparaffinized, hydrated, treated with proteinase K for 20 min, and subsequently incubated with a mixture of a fluorescent labeling solution of dUTP and the TdT enzyme at 37°C for 1 h in a humidified atmosphere. As a positive control, sections were incubated with DNaseI for 10 min at room temperature (25°C) before the fluorescent labeling procedure. Negative controls were incubated with dUTP for 10 min at room temperature (25°C). Subsequently, the samples were treated with diaminobenzidine, counterstained with hematoxylin (to identify the cell nuclei), dehydrated in a gradient series, vitrified with dimethylbenzene, and finally, mounted with neutral balsam.
Real-time polymerase chain reaction
Total RNA was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA, USA), and the RNA concentration was measured using spectrophotometry. Single-stranded cDNA was synthesized using a cDNA synthesis kit (Takara, Kyoto, Japan) according to the procedures. Reverse transcription-polymerase chain reaction assays were performed using Applied Biosystems SYBR Green Mix kits (Applied Biosystems, CA, USA). The following primers were used: IL-1β, AAATCTCGCAGCAGCACAT (forward) and CACACACCAGCAGGTTATCA (reverse); Cxcl-1, CTGGGATTCACCTCAAGAACATC (forward) and CAGGGTCAAGGCAAGCCTC (reverse); Cxcl-2, CCAACCACCAGGCTACAGG (forward) and GCGTCACACTCAAGCTCTG (reverse); Tim-4, ACACATTTTCCCTGCCTCGT (forward) and GCTGTGGCAAGGATTTCACC (reverse); IL-6, CCACTTCACAAGTCGGAGGCTTA (forward) and CCAGTTTGGTAGCATCCATCATTTC (reverse); TNF, TATGGCCCAGACCCTCACA (forward) and GGAGTAGACAAGGTACAACCCATC (reverse); and IFNγ, CATCAGCAACAACATAAGTGTCATC (forward) and CATTGACAGCTTTGTGCTGGA (reverse). GAPDH was used as a housekeeping gene. The results were presented as the ratio of the gene to GAPDH mRNA (sense and antisense).
Western blot analysis
Total proteins were extracted and quantified using the bicinchoninic acid method. Equivalent weights of proteins (40 μg/lane) were separated on 10% SDS-PAGE gels and then transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat milk in TBST buffer for 1 h and then incubated overnight with the following primary antibodies: Bax, 2772 (Cell Signaling Technology, Danvers, MA, USA); Bcl-2, ab32124 (Cell Signaling Technology, Danvers, MA, USA); Bcl-xl, 2764 (Cell Signaling Technology, Danvers, MA, USA); cleaved caspase-3, 9664 (Cell Signaling Technology, Danvers, MA, USA); GAPDH, 2118 (Cell Signaling Technology, Danvers, MA, USA); Tim-4, ab47637 (Abcam, Cambridge, MA, USA). After being washed twice with TBST, the membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (anti-rabbit-HPR, 7074, Cell Signaling Technology, Danvers, MA, USA) at 1:2000 dilution. Specific bands were visualized using an enhanced chemiluminescence detection kit.
Flow cytometric analysis
Peripheral blood mononuclear cells were isolated by centrifugation on Ficoll (Lymphoprep, PAA, Nycomed, Oslo, Norway) gradients from buffy coat preparations obtained from mice, and then washed twice with PBS. The cells were then incubated with a primary anti-TIM4 antibody (ab47637, Abcam, Cambridge, MA, USA) for 30 min at 4°C, washed twice with PBS, and incubated with a secondary FITC-conjugated antibody (ab6717, Abcam, Cambridge, MA, USA) for 30 min at 4°C. After this, the cells were washed twice with PBS, incubated with 600 µl PBS at room temperature, and examined using flow cytometry.
Statistical analysis
All values are expressed as the mean ± SD. Data were analyzed with an unpaired, two-tailed Student t-test and checked using analysis of variance. P < 0.05 was considered statistically significant.
Results
IPC ameliorates hepatic IRI and relieves hepatic inflammation during IRI
To determine whether IPC ameliorated hepatic IRI, we assessed the serum ALT and AST levels, liver histology, cellular apoptosis, and mitochondrial apoptosis-related protein expression in the sham, IRI, and IPC+IRI groups. After 6 h of reperfusion, serum transaminase levels were significantly higher in the IRI group than in the sham group (P < 0.05). IPC prior to IRI reduced the serum transaminase levels, though the difference was not statistically significant (Fig 1A). Histological examination of the liver tissue revealed that IRI was followed by significant periportal congestion, severe hepatocyte apoptosis, and diffuse vacuolation within hepatocytes, as compared to the sham group. The mice in the IPC+IRI group showed less apoptosis and damage than did those in the IRI group. The sham-operated animals exhibited minimal changes (Fig 1B-C). Moreover, the expressions of the BCL-2 and BCL-XL proteins, which inhibit apoptosis, were higher in the IPC+IRI group than in the IRI group, while those of the apoptosis promoter BAX protein and the apoptosis marker cleaved caspase-3 were lower in the IPC+IRI group than in the IRI group (Fig 1D).
Changes in serum transaminase levels, liver histology, cell apoptosis, and apoptosis-related proteins after IRI with or without IPC. (A) Serum alanine transaminase (ALT) and aspartate transaminase (AST) levels determined using enzyme-linked immunosorbent assay (*P < 0.05, **P < 0.01, ***P < 0.001, #P < 0.05). (B-C) Liver histology examined using hematoxylin and eosin (HE) staining, and cell apoptosis examined using TdT-mediated biotin-16-dUTP nick-end labeling (TUNEL) assay (magnification, ×400; *P < 0.05, **P < 0.01 vs. control; ##P < 0.01 vs. IRI; n = 3 for each experiment). (D) Expression of apoptosis-related proteins measured using western blot analysis (*P < 0.05, ***P < 0.001 vs. control; ##P < 0.01 vs. IRI; n = 3 for each experiment). (E-F) CD68- and CD3-positive cells on immunohistochemical analysis (**P < 0.01, ***P < 0.001 vs. control; n = 3 for each experiment). (G) Detection of IL-6, CXCL-1, CXCL-2, TNF-a, IL-1β, and IFN-γ using quantitative polymerase chain reaction assays (*P < 0.05, **P < 0.01, ***P < 0.001 vs Control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs IR). (H) Myeloperoxidase (MPO) concentration determined using enzyme-linked immunosorbent assay (*P < 0.05 vs Control; #P < 0.05 vs IR). (I) Western blot analysis of TIM4 expression (***P < 0.001 vs. control; n = 3 for each experiment). (J) TIM4 mRNA levels determined using by quantitative polymerase chain reaction assay (*P < 0.05, ***P < 0.001 vs Control; #P < 0.05 vs IR). (K) Flow cytometric analysis of TIM4-positive cells in serum samples.
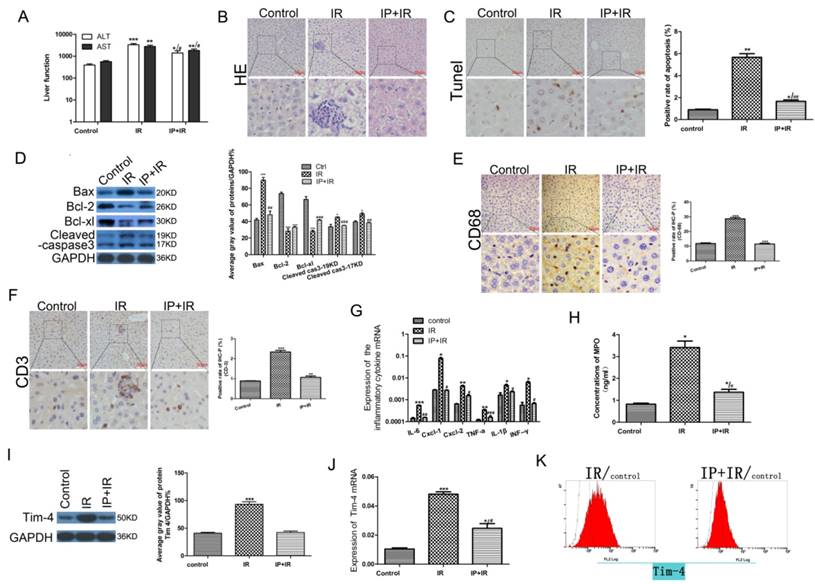
To assess the inflammatory response after hepatic IRI[20], we next examined the levels of inflammatory cells and cytokines in each study group. Immunohistochemical analysis showed that the levels of the macrophage marker CD68 and T-cell marker CD3 were lower in the IPC+IRI group than in the IRI group, which indicated less macrophage and T-cell infiltration in the IPC+IRI group (Fig 1E-F). Quantitative polymerase chain reaction assays showed that the expressions of the inflammatory cytokines IL-6, CXCL-1, CXCL-2, TNF-a, IL-1β, and IFN-γ were lower in the IPC+IRI group than in the IRI group (Fig 1G). Additionally, the myeloperoxidase concentration was lower in the IPC+IRI group than in the IRI group, indicating lower polymorphonuclear leukocyte activation in the former group (Fig 1H). The above results collectively indicated that IPC significantly inhibited inflammatory responses during IRI.
In our previous study, we proved that TIM-4 is required for macrophage migration, phagocytosis, and activation leading to hepatic IRI, and that the blocking of TIM4 signaling relieves hepatic IRI[19]. Thus, we hypothesized that IPC may protect against hepatic IRI by decreasing TIM4 signaling. To validate this hypothesis, we examined the expression of TIM4 in the sham, IRI, and IPC+IRI groups. The results showed that IRI obviously increased the protein expression and transcription of TIM4, while IPC reduced this IRI-induced TIM4 elevation (Fig 1 I-K).
TIM-4 participates in IPC-mediated hepatic protection from IRI
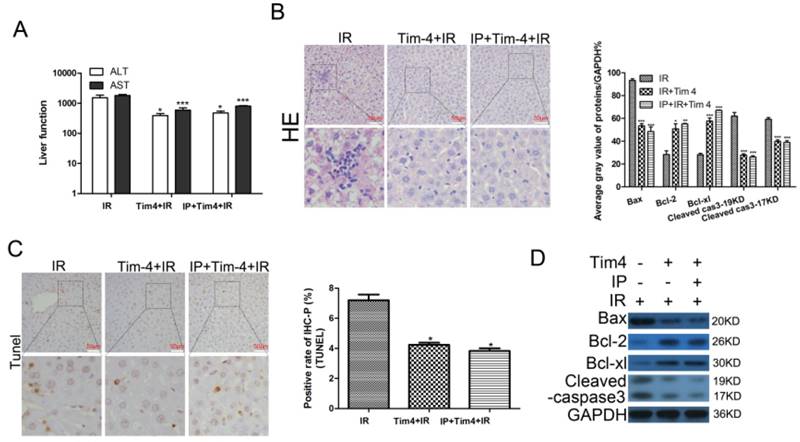
To further explore the role of TIM4 in the liver-protective effects of IPC, we examined the serum ALT and AST levels, liver histology, cell apoptosis, and mitochondrial apoptosis-related protein expression in mice subjected to IRI, IRI+TIM4 antibody treatment, or IPC+IRI+TIM4 antibody treatment. The results showed that TIM4 blocking significantly decreased the ALT and AST levels (Fig 2A). Liver tissue histology and TUNEL analysis revealed that TIM4 blocking could minimize hepatic IRI and hepatocyte apoptosis (Fig 2B-C). Moreover, TIM4 blocking promoted BCL2 and BCL-XL expression, but inhibited BAX and cleaved caspase-3 levels (Fig 2D). However, IPC combined with TIM4 blocking did not further enhance IPC-mediated protection against IRI, possibly because TIM4 downregulation is involved in the liver-protective effects of IPC.
Changes in serum transaminase levels, liver histology, cell apoptosis, and apoptosis-related proteins in mice subjected to y IRI with or without IPC and TIM4 blocking. (A) Serum alanine transaminase (ALT) and aspartate transaminase (AST) levels determined using enzyme-linked immunosorbent assay. (*P < 0.05, ***P < 0.001) (B-C) Liver histology examined using hematoxylin and eosin (HE) staining, and cell apoptosis examined using TUNEL assay (magnification, ×400; *P < 0.05 vs. IRI; n = 3 for each experiment). (D) Apoptosis-related protein expression determined using western blot analysis (*P < 0.05, ***P < 0.001 vs. IRI; n = 3 for each experiment).
TIM4 blocking underlies IPC-mediated alleviation of hepatic inflammatory responses during IRI
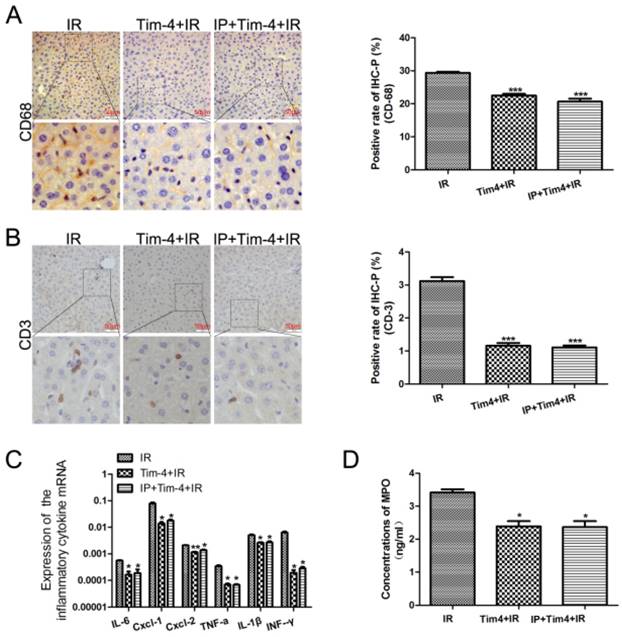
To further explore the potential contribution of TIM4 blocking in IPC-mediated liver protection, we examined the levels of inflammatory cells and cytokines after TIM4 blocking. We found that TIM4 blocking obviously reduced CD68 and CD3 expression (Fig 3A-B) as well as IL-6, CXCL-1, CXCL-2, TNF-a, IL-1β, and IFN-γ levels (Fig 3C). Moreover, TIM4 blocking decreased serum myeloperoxidase concentrations (Fig 3D). However, IPC combined with TIM4 blocking did not further inhibit the IRI-induced inflammatory responses. These results indicated that IPC may inhibit IRI-induced inflammatory responses by downregulating TIM4 expression.
Changes in inflammatory cells and cytokines in in mice subjected to IRI with or without IPC and TIM4 blocking. (A-B) CD68- and CD3-positive cells on immunohistochemical analysis (***P < 0.001 vs. IRI; n = 3 for each experiment). (C) Detection of IL-6, CXCL-1, CXCL-2, TNF-a, IL-1β, and IFN-γ using quantitative polymerase chain reaction assays. (*P < 0.05, **P < 0.01 vs IR). (D) MPO concentration determined using enzyme-linked immunosorbent assay (*P < 0.05 vs IR).
The liver-protective effects of IPC are mediated through Tim-4 inhibition
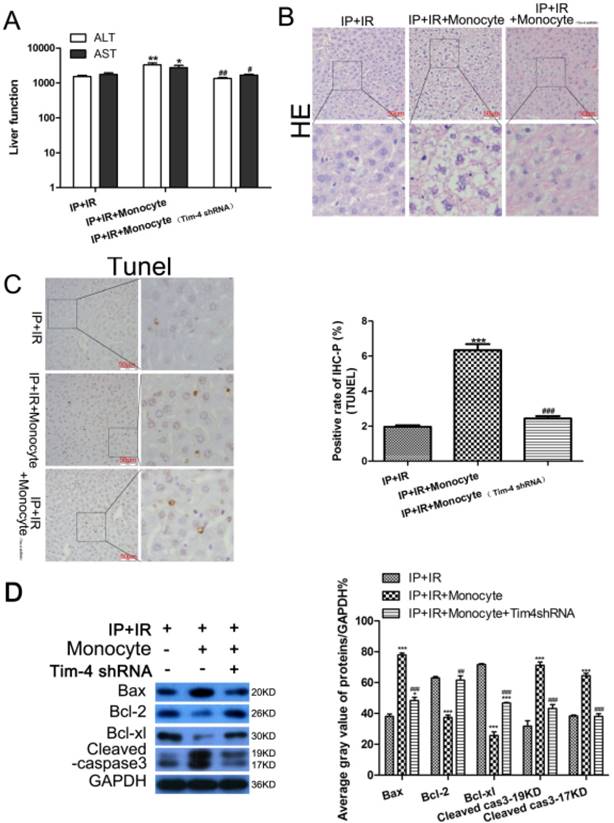
To determine whether the liver-protective effects of IPC depend on TIM-4 expression, we used CD14 to separate mononuclear cells from peripheral blood, stimulated the cells with 100 ng/ml lipopolysaccharide for 24 h, and then transfused the mice with activated monocytes. We found that the liver-protective effects of IPC disappeared in the mice that received the monocyte transfusion compared with those in the IR+IP group. Activated monocytes significantly increased the ALT and AST levels compared to the levels in the IR+IP group (Fig 4A). Liver tissue histology and TUNEL analysis revealed that activated monocytes could minimize hepatic IRI and hepatocyte apoptosis (Fig 4B, 4C), but interestingly, TIM-4 knockdown could reverse these effects. Almost immediately, activated monocyte treatment promoted BCL2 and BCL-XL expression and inhibited BAX and cleaved caspase-3 levels (Fig 4D). However, TIM-4 inhibition also decreased BCL2 and BCL-XL expression and increased BAX and cleaved caspase-3 levels. These findings indicate that the liver-protective effects of IPC were related to TIM-4 expression.
Changes in serum transaminase levels, liver histology, and apoptosis-related proteins in mice subjected to IRI and ischemic preconditioning (IPC) with or without transfusion of activated monocytes and TIM4 blocking. (A) ALT and AST levels determined using enzyme-linked immunosorbent assay (*P < 0.05, **P < 0.01 vs. IPC+IRI; #P < 0.05, ##P < 0.01 vs. IPC+IRI+monocyte). (B and C) Liver histology examined using hematoxylin and eosin (HE) staining, and cell apoptosis examined using TUNEL assay (magnification, ×400; ***P < 0.001 vs. IPC+IRI; ###P < 0.001 vs. IPC+IRI+monocyte; n = 3 for each experiment). (D) Western blot analysis of apoptosis proteins expression (***P < 0.001 vs. IPC+IRI; ##P < 0.01 vs. IPC+IRI+monocyte; n = 3 for each experiment).
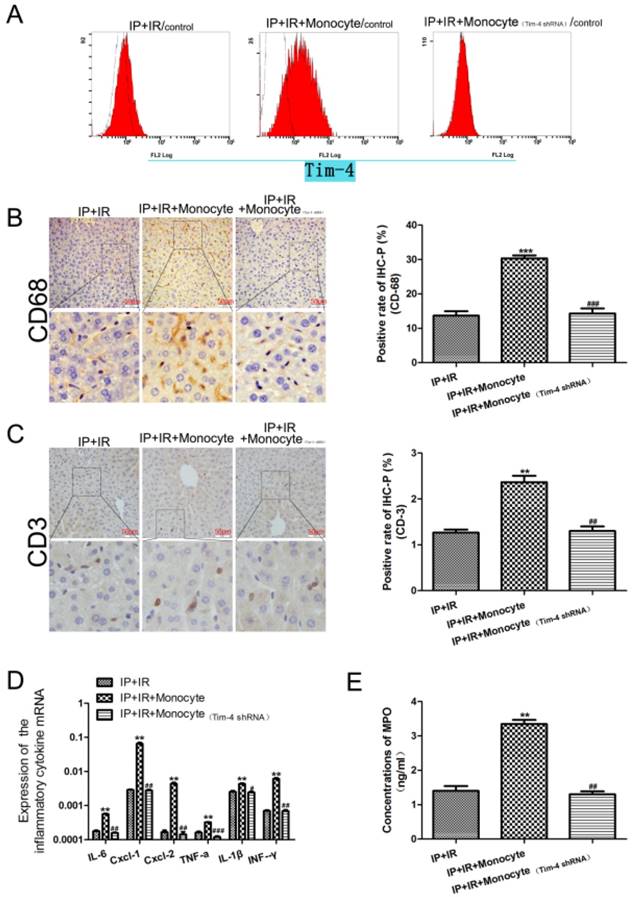
IPC-mediated alleviation of hepatic inflammation disappeared after treatment with activated monocytes during IRI
In this study, we demonstrated that TIM-4 blocking inhibited the IRI-induced inflammatory responses, and that IPC did not affect this finding. We hypothesized that IPC may inhibit IRI-induced inflammatory responses by downregulating TIM4 expression. To confirm this, we transfused TIM-4-positive monocytes; we found that this increased cell apoptosis compared to the IR+IPC group, and that this effect was reversed after TIM-4 knockdown (Fig 5A). IPC did not inhibit IRI-induced inflammation in mice treated with activated monocytes. In addition, CD68 and CD3 expression (Fig 5B, 5C) as well as IL-6, CXCL-1, CXCL-2, TNF-a, IL-1β, and IFN-γ levels (Fig 5D) were increased in these mice compared with the IR+IP group. However, these levels were decreased after TIM-4 inhibition. Furthermore, TIM-4 knockdown decreased MPO expression, while activated monocytes increased MPO expression in the IR+IP group (Fig 5E).
Changes in inflammatory cells and cytokines in mice subjected to IRI and IPC with or without transfusion of activated monocytes and TIM4 knockdown. (A) Flow cytometry analysis was used to detect cell apoptosis. (B and C) CD68- and CD3-positive cells on immunohistochemical analysis (**P < 0.01, ***P < 0.001 vs. IPC+IRI; ##P < 0.01, ###P < 0.001 vs. IPC+IRI+monocyte; n = 3 for each experiment). (D) Detection of IL-6, CXCL-1, CXCL-2, TNF-a, IL-1β, and IFN-γ using quantitative polymerase chain reaction assays (**P < 0.01 vs. IPC+IRI; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. IPC+IRI+monocyte). (E) MPO concentration determined using enzyme-linked immunosorbent assay (**P < 0.01 vs. IPC+IRI; ##P < 0.01 vs. IPC+IRI+monocyte).
Discussion
IRI is a multifactorial process that plays a major role in liver damage during hepatic resection or transplantation [1, 21]. Strategies to relieve IRI can be broadly classified into biochemical, genetic, and surgical. However, no efficient therapeutic strategies to inhibit IRI are as yet available[22]. Considering the harmful clinical effects of IRI, it is essential to develop more efficient strategies to overcome IRI.
One strategy that has been used in clinical practice to confer protection against subsequent IRI is IPC. IPC was first proposed by Zager et al. to limit damage to the kidney[3]. In animal experiments, the protective role of IPC has been proved in several organs, such as the brain[23], lung[24], and kidney [25]. IPC of the liver was first reported by Lloris-Carsi et al. in 1993[26]. Accumulating evidence has proved that IPC can limit the damage caused during hepatic IRI[27]. However, a few studies have reported that IPC does not prevent hepatic IRI [28, 29]. Hence, more studies are needed to confirm the benefit of IPC during hepatic IRI. In the present study, we employed a mouse model to verify the role of IPC in hepatic IRI. The results showed that the AST and ALT levels and apoptosis rate were significantly lower after IPC treatment than after IRI alone. IPC could reduce macrophage and T-cell infiltration and cytokine secretion during hepatic IRI. Our results provide a theoretical foundation for the use of IPC to minimize hepatic IRI and show that IPC significantly inhibited inflammatory responses during IRI and protected the mouse liver against IRI.
To further explore the underlying mechanism, we focused on the TIM4 protein, as our previous study had shown that TIM-4 signaling was required for innate immunity-driven hepatic inflammatory damage during IRI[19]. Tim-4, a member of the TIM family, has attracted much research attention as a potential regulator of immune responses[15, 30, 31]. Tim-4 overexpression has been reported t o decrease nitric oxide production and inducible nitric oxide synthase expression in lipopolysaccharide- or IFN-γ-stimulated macrophages[32]. These findings indicate that TIM4 may participate in the IPC-mediated protection of the liver from IRI. In the present study, we for the first time found that IPC could reduce IRI-induced TIM4 expression. Furthermore, we proved that IPC combined with TIM4 blocking did not further enhance IPC-mediated protection against IRI and TIM4 blocking produced the same protective effects as IPC by inhibiting inflammatory responses. Moreover, the liver-protective effects of IPC disappeared after monocyte transfusion in mice subjected to IRI; however, these effects were restored after TIM-4 inhibition. Thus, our results indicate that TIM4 mediates the liver-protective effect of IPC during IRI.
Taken together, our study demonstrated that IPC could protect the mouse liver against IRI by reducing inflammatory reaction and that TIM4 was a potential mediator of these protective effects of IPC. Although further basic and clinical experiments are needed to confirm our results, our study suggests that IPC is an efficient strategy to minimize hepatic IRI, and that Tim-4 may be a novel target for treating IRI.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No.81302547 and No.81570591). We would like to thank the native English speaking scientists of Elixigen Company (Huntington Beach, California) for editing our manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schemmer P, Lemasters JJ, Clavien PA. Ischemia/Reperfusion injury in liver surgery and transplantation. HPB Surg. 2012;2012:453295
2. Weigand K, Brost S, Steinebrunner N, Buchler M, Schemmer P, Muller M. Ischemia/Reperfusion injury in liver surgery and transplantation: pathophysiology. HPB Surg. 2012;2012:176723
3. Zager RA, Baltes LA, Sharma HM, Jurkowitz MS. Responses of the ischemic acute renal failure kidney to additional ischemic events. Kidney Int. 1984;26:689-700
4. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124-36
5. Hu GH, Lu XS. Effect of normothermic liver ischemic preconditioning on the expression of apoptosis-regulating genes C-jun and Bcl-XL in rats. World J Gastroenterol. 2005;11:2579-82
6. Gomez D, Homer-Vanniasinkam S, Graham AM, Prasad KR. Role of ischaemic preconditioning in liver regeneration following major liver resection and transplantation. World J Gastroenterol. 2007;13:657-70
7. Clavien PA, Yadav S, Sindram D, Bentley RC. Protective effects of ischemic preconditioning for liver resection performed under inflow occlusion in humans. Ann Surg. 2000;232:155-62
8. Domart MC, Esposti DD, Sebagh M, Olaya N, Harper F, Pierron G. et al. Concurrent induction of necrosis, apoptosis, and autophagy in ischemic preconditioned human livers formerly treated by chemotherapy. J Hepatol. 2009;51:881-9
9. Clavien PA, Selzner M, Rudiger HA, Graf R, Kadry Z, Rousson V. et al. A prospective randomized study in 100 consecutive patients undergoing major liver resection with versus without ischemic preconditioning. Ann Surg. 2003;238:843-50 discussion 51-2
10. Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE. et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927-40
11. Meyers JH, Chakravarti S, Schlesinger D, Illes Z, Waldner H, Umetsu SE. et al. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol. 2005;6:455-64
12. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435-9
13. Mizui M, Shikina T, Arase H, Suzuki K, Yasui T, Rennert PD. et al. Bimodal regulation of T cell-mediated immune responses by TIM-4. Int Immunol. 2008;20:695-708
14. Rodriguez-Manzanet R, Sanjuan MA, Wu HY, Quintana FJ, Xiao S, Anderson AC. et al. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci U S A. 2010;107:8706-11
15. Ge RT, Zeng L, Mo LH, Xu LZ, Zhang HP, Yu HQ. et al. Interaction of TIM4 and TIM3 induces T helper 1 cell apoptosis. Immunol Res. 2016;64:470-5
16. Wong K, Valdez PA, Tan C, Yeh S, Hongo JA, Ouyang W. Phosphatidylserine receptor Tim-4 is essential for the maintenance of the homeostatic state of resident peritoneal macrophages. Proc Natl Acad Sci U S A. 2010;107:8712-7
17. Feng BS, Chen X, He SH, Zheng PY, Foster J, Xing Z. et al. Disruption of T-cell immunoglobulin and mucin domain molecule (TIM)-1/TIM4 interaction as a therapeutic strategy in a dendritic cell-induced peanut allergy model. J Allergy Clin Immunol. 2008;122:55-61 e1-7
18. Yang PC, Xing Z, Berin CM, Soderholm JD, Feng BS, Wu L. et al. TIM-4 expressed by mucosal dendritic cells plays a critical role in food antigen-specific Th2 differentiation and intestinal allergy. Gastroenterology. 2007;133:1522-33
19. Ji H, Liu Y, Zhang Y, Shen XD, Gao F, Busuttil RW. et al. T-cell immunoglobulin and mucin domain 4 (TIM-4) signaling in innate immune-mediated liver ischemia-reperfusion injury. Hepatology. 2014;60:2052-64
20. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229-317
21. Casillas-Ramirez A, Mosbah IB, Franco-Gou R, Rimola A, Rosello-Catafau J, Peralta C. [Ischemia-reperfusion syndrome associated with liver transplantation: an update]. Gastroenterol Hepatol. 2006;29:306-13
22. Clavien PA, Petrowsky H, DeOliveira ML, Graf R. Strategies for safer liver surgery and partial liver transplantation. N Engl J Med. 2007;356:1545-59
23. Glazier SS, O'Rourke DM, Graham DI, Welsh FA. Induction of ischemic tolerance following brief focal ischemia in rat brain. J Cereb Blood Flow Metab. 1994;14:545-53
24. Du ZY, Hicks M, Winlaw D, Spratt P, Macdonald P. Ischemic preconditioning enhances donor lung preservation in the rat. J Heart Lung Transplant. 1996;15:1258-67
25. Turman MA, Bates CM. Susceptibility of human proximal tubular cells to hypoxia: effect of hypoxic preconditioning and comparison to glomerular cells. Ren Fail. 1997;19:47-60
26. Lloris-Carsi JM, Cejalvo D, Toledo-Pereyra LH, Calvo MA, Suzuki S. Preconditioning: effect upon lesion modulation in warm liver ischemia. Transplant Proc. 1993;25:3303-4
27. Chu MJ, Vather R, Hickey AJ, Phillips AR, Bartlett AS. Impact of ischaemic preconditioning on experimental steatotic livers following hepatic ischaemia-reperfusion injury: a systematic review. HPB (Oxford). 2015;17:1-10
28. Zapata-Chavira HA, Cordero-Perez P, Casillas-Ramirez A, Escobedo-Villarreal MM, Perez-Rodriguez E, Torres-Gonzalez L. et al. Is Ischemic Preconditioning a Useful Therapeutic Strategy in Liver Transplantation? Results from the First Pilot Study in Mexico. Arch Med Res. 2015;46:296-302
29. Chu MJ, Vather R, Hickey AJ, Phillips AR, Bartlett AS. Impact of ischemic preconditioning on outcome in clinical liver surgery: a systematic review. Biomed Res Int. 2015;2015:370451
30. Yang B, Li LJ, Xu LZ, Liu JQ, Zhang HP, Geng XR. et al. Histone acetyltransferease p300 modulates TIM4 expression in dendritic cells. Sci Rep. 2016;6:21336
31. Albacker LA, Karisola P, Chang YJ, Umetsu SE, Zhou M, Akbari O. et al. TIM-4, a receptor for phosphatidylserine, controls adaptive immunity by regulating the removal of antigen-specific T cells. J Immunol. 2010;185:6839-49
32. Xu LY, Qi JN, Liu X, Ma HX, Yuan W, Zhao PQ. et al. Tim-4 Inhibits NO Generation by Murine Macrophages. Plos One. 2015:10
Author contact
![]() Corresponding authors: Dr. Shusen Zheng, Department of Surgery, Division of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital, School of Medicine, Zhejiang University, NO.79, Qingchun Road, Hangzhou 310003, Zhejiang, China. Tel: 86- 571-87236765; Fax: 86- 571-87236736 E-mail: shusenzhengedu.cn and Yu Zhang, Department of Surgery, Division of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital, School of Medicine, Zhejiang University, NO.79, Qingchun Road, Hangzhou 310003, Zhejiang, China. Tel: 86- 571-87236765; Fax: 86- 571-87236736 E-mail: dr_zhangyuedu.cn
Corresponding authors: Dr. Shusen Zheng, Department of Surgery, Division of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital, School of Medicine, Zhejiang University, NO.79, Qingchun Road, Hangzhou 310003, Zhejiang, China. Tel: 86- 571-87236765; Fax: 86- 571-87236736 E-mail: shusenzhengedu.cn and Yu Zhang, Department of Surgery, Division of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital, School of Medicine, Zhejiang University, NO.79, Qingchun Road, Hangzhou 310003, Zhejiang, China. Tel: 86- 571-87236765; Fax: 86- 571-87236736 E-mail: dr_zhangyuedu.cn