Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(10):1211-1220. doi:10.7150/ijbs.27411 This issue Cite
Research Paper
Adapted HCV JFH1 variant is capable of accommodating a large foreign gene insert and allows lower level HCV replication and viral production
Qi Wang1,5, Curt Hagedorn2,3,5 ![]() , Shuanghu Liu4,5
, Shuanghu Liu4,5 ![]()
1. Center of Liver Diseases, Beijing Ditan Hospital, Capital Medical University, No.8 Jingshun East Street, Chaoyang District, Beijing, China 100015
2. The Central Arkansas Veterans Healthcare System, 4300 West 7 th St. 111/LR, Little Rock, AR USA 72205
3. Departments of Medicine and Genetics, University of Arkansas for Medical Sciences, 4300 West 7 th St. 111/LR, Little Rock, AR USA 72205
4. Department of Medicinal Chemistry, College of Pharmacy, University of Utah, Salt Lake City, UT USA 84112, United States of America
5. Department of Medicine, School of Medicine, University of Utah, Salt Lake City, UT USA 84112, United States of America
Received 2018-5-22; Accepted 2018-5-22; Published 2018-7-13
Abstract

Infectious HCV carrying reporter genes have further applications in understanding the HCV life cycle including replication, viral assembly and release. In this study, a full-length 3039bp LacZ gene was inserted into the derivative of JFH1-AM120 to develop an additional reporter virus. The results showed that the recombinant reporter virus JFH1-AM120-LacZ can replicate and produce lower titers of infectious virus. However, insertion of the LacZ gene in the C-terminal region of the NS5A in HCV JFH1-AM120-LacZ decreased viral replication and dramatically impaired the production of infectious viral particles. Moreover, the JFH1-AM120-LacZ reporter virus lost the LacZ gene after serial passage. Nevertheless, the JFH1-AM120-LacZ reporter virus displayed the entire life cycle of HCV, from replication to production of infectious virus, in Huh7.5 cells. This study demonstrates that the NS5A region of HCV JFH1-AM120 has the capacity to accommodate large foreign genes up to 3,039 bp and suggests that other relatively large gene inserts can be accommodated at this site.
Keywords: HCV, NS5A, LacZ, Huh7.5, X-Gal, β-Galactosidase
Introduction
Hepatitis C virus (HCV) is a member of the Flaviviridae family and is a single-stranded positive-sense RNA. It infects approximately 3% of the worldwide population (1, 2). HCV can cause acute and chronic hepatitis, and lead to fibrosis, cirrhosis and hepatocellular carcinoma (3). Although much progress has been made in developing highly effective HCV antivirals, an effective vaccine has yet to be developed (4, 5). An internal ribosome entry site (IRES) of HCV drives RNA translation to produce a polyprotein of approximately 3,000 amino acids (aa) that encodes both structural and nonstructural proteins (6-8). The development of HCV replicon systems advanced the understanding of HCV replication, viral protein processing and viral-host cell interactions (9, 10). An establishment of an infectious HCV cell culture system using a genotype 2a isolate (JFH1 strain) of HCV and Huh-7 cells was a major achievement (11-13). In this system, infectious HCV particles are secreted in an envelope glycoprotein-dependent manner and enable a variety of questions to be answered regarding HCV biology and cell infection. However, virus titers of JFH1 released from infected cells are relatively low and limit some applications of this system. Studies have identified adaptive or compensatory mutations that enhance infectious virus particle production from either wild-type JFH1 or intergenotypic chimeras (13-19).
Non-structural protein 5A (NS5A) of HCV is a phosphoprotein, which has 1398 nt and a calculated molecular mass of 49 kDa. It migrates as 56- and 58-kDa species in SDS-polyacrylamide gels and is organized into three domains: I (aa28- 213), II (aa250-342), and III (aa356-447). Domain I coordinate a single zinc atom, and domains II and III are less well characterized but are important for RNA replication and/or virion assembly (20-23). Previous studies have demonstrated that the HCV JFH1 NS5A C-terminal is a flexible region which is capable of accommodating foreign gene inserts (such as EGFP, 720bp and Rennilla luciferase[Rluc], 930bp) and still permit HCV replication and viral production (18, 19, 24-29).
Vesicular stomatitis virus (VSV) is negative-stranded RNA virus that has been used as a gene expression vector. It can accommodate a gene insert as large as 5955 bases (30). One of the more common reporter genes, the E. coli LacZ gene, is 3039 bp in length and encodes the protein β-galactosidase (β-gal). The expression of LacZ can be identified by staining with X-gal substrate to produce a blue color as a measure of enzyme activity (31-33). Human hepatoma cells (Huh7) can express β-galactosidase using adenoviral vectors carrying the LacZ gene (34). It is unclear whether the HCV JFH1 NS5A C-terminal is capable of accommodating a large foreign gene such as LacZ, and still permit HCV replication and production of infectious virus.
In a previous study we demonstrated that there was no viral production after insertion of EGFP (720bp) and Rennilla luciferase (Rluc; 930bp) into the C-terminus of NS5A of wild type JFH1(18). However, a higher titer of HCV reporter virus was produced after inserting EGFP and Rluc into NS5A C-terminus of an adaptively mutated JFH1 strain designated JFH1-AM120 (18). In this study, we used the JFH1-AM120 as a vector to explore if infectious reporter virus would be produced following insertion of LacZ gene that was three time larger than Rluc, into the NS5A C-terminus. The result showed that the NS5A region of HCV JFH1-AM120 has the capacity to accommodate large foreign genes up to 3,039 bp and suggests that other relatively large gene inserts can be accommodated at this site.
Materials and Methods
Cell culture
Human hepatoma cells, Huh7.5, were generously provided by Charles Rice (35) (Rockefeller University) and maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 100 U/ml of penicillin, 100 µg/ml of streptomycin, nonessential amino acids, and 10% fetal bovine serum (FBS) (Invitrogen) at 37˚C in 5% CO2. All experiments described in this study were performed using these cells.
Antibodies and assay kits
The monoclonal antibody to the NS5A protein (9E10) was a gift from Charles Rice (13). Goat anti-mouse conjugated with horseradish peroxidase (HRP) (Sigma), goat anti-mouse IgG conjugated with Alexa Fluor 594 (Invitrogen), X-gal (Genlantis, Cat#A10300k), mammalian β-gal activity assay kit (ThermoFisher, #75707), Renilla Luciferase Assay System (Promega) were obtained commercially.
Construction of the JFH1-AM120-LacZ plasmid
Plasmids constructs were based on JFH-AM120 that has adaptive mutations previous reported (18) and JFH-AM120 based on the consensus sequence of HCV pJFH1, which was kindly provided by Dr. Wakita (11). JFH-AM120-EGFP and JFH-AM120-Rluc had been described previously (18). The EGFP and Renilla luciferace (Rluc) genes were inserted in frame into a unique RsrII site located in the amino acid 398 codon of JFH1-AM120. JFH-AM120-LacZ was also constructed by inserting the LacZ gene (3039bp) into the RsrII site of the NS5A C-terminal region of JFH-AM120. The LacZ fragment was amplified from vector pSV-β-galactosidase Control (Promega, E1081) using the primers LacZ-Foward (5'-TCTCGGTCCGATCGATCCCGTCGTTTTACA-3'), LacZ-Reverse (5'-CTTACGGACCGCCTTTTTGACACCAGACCAACT-3'). The new construct was sequenced and the correct full-length clone, JFH-AM120-LacZ, was used for the subsequent experiments.
Transfection of HCV RNA
To generate full-length genomic RNA, pJFH1-AM120-LacZ and controls of pJFH1-AM120, pJFH1-AM120-EGFP, pJFH1-AM120-Rluc and were linearized at the 3' end of the HCV cDNA with XbaI. The linearized plasmid DNA was purified and used as a template for T7 in vitro transcription (MEGAscript; Ambion, Austin, TX). In vitro transcribed RNAs were transfected into cells by electroporation as described previously (29, 36). Briefly, trypsinized cells were washed twice and resuspended with serum-free Opti-MEM (Invitrogen) at the concentration of 1x107 cells per ml. Ten micrograms of RNA were mixed with 0.4 ml of the cells in a 4-mm cuvette. A Bio-Rad Gene Pulser system was used to deliver a single pulse at 270V and 960 µF and the cells were plated in T75 Costar flasks (Corning). Transfected cells were cultured in complete DMEM for the indicated times. Cells were passaged every three days and the presence of HCV in the corresponding supernatants was determined by immunofluorescence assays (IFA) for the NS5A protein or X-gal staining.
Immunofluorescence assays
Cells infected by HCV or HCV reporter virus were washed with PBS, fixed with 4% paraformaldehyde, and permeabilized with 0.2% Triton X-100. Fixed cells were blocked with 1% bovine serum albumin and 1% normal goat serum in PBS. NS5A protein was detected in cells with an NS5A-specific monoclonal antibody and visualized with a secondary goat anti-mouse antibody conjugated with Alexa Fluor 594 (Invitrogen, 1: 1,000 dilutions). Cover slips were mounted onto slides with DAPI (Vector Labs) and immunostaining visualized by fluorescence microscopy (Nikon E400).
Titration of infectious HCV
The titer of infectious HCV was determined by the number of cell foci staining for NS5A as previously described in detail (12). Briefly, cell supernatants were serially diluted 10-fold in complete DMEM. The supernatant was used to infect 5 × 104 naive Huh 7.5 cells per well in 24-well plates. The inocula were incubated with cells for three hours at 37oC and then supplemented with fresh complete DMEM. The level of HCV infection was determined three days postinfection by immunofluorescence staining for HCV NS5A and NS5A-Rluc, or X-gal staining for NS5A-LacZ and directly visualized the EGFP for NS5A-EGFP. The viral titer is expressed as focus-forming units per milliliter of supernatant (ffu/ml).
X-gal staining
X-Gal positive cells were identified using an X-gal staining assay Kit (Genlantis, Cat#A10300k) and visualized by bright field microscopy (Nikon) following the manufacturer's instructions. Briefly, 5 × 104 naive Huh 7.5 cells per well were seeded in 24-well plates and were transfected with RNA of JFH-AM120-LacZ or infected with the supernatant collected from transfected cells of RNA of JFH-AM120-LacZ or controls. 72 hours post-transfection or infection, cells were fixed with formaldehyde-glutaraldehyde buffer for 15 minutes and stained the cells with X-Gal staining solution for three hours. Blue stained cells were identified by bright field microscopy (Nikon E400). The viral titer of JFH-AM120-LacZ was expressed as focus-forming units per milliliter of supernatant (ffu/ml). Colocalization of X-gal pictures and Immunofluorescence images were processed by Photoshop CC software.
β-Galactosidase assay
The β-Galactosidase activity was measured using a Mammalian β-Galactosidase Assay Kit (ThermoFisher, #75707) and microplate reader (Bio-Tek) following the manufacturer's instructions. Briefly, Huh7.5 cells were transfected with RNA of JFH-AM120-LacZ and controls of JFH-AM120, JFH-AM120-EGFP and JFH-AM120-Rluc and the cells were incubated for 72 hours. Then cells were washed once, 100μL of β-Galactosidase Assay Reagent was added to each well, and cells were incubated for 30 minutes at 37°C. Reactions were stopped by adding 100 μl of β-Galactosidase assay stop solution to each well and absorbance at 405nm was measured.
Western blot analysis
The HCV-transfected Huh 7.5 cells were lysed in a radioimmuno-precipitation assay buffer (50 mM Tris-HCl, pH 7.5, 150 mM sodium chloride, 1% Nonidet P40, 0.5% sodium deoxycholate) containing a cocktail of proteinase inhibitors (Roche). The total protein for each sample was measured with a standard protein assay (Bio-Rad). Twenty-five micrograms of total protein for each sample was analyzed by 10% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked by incubating them with 5% skim milk. HCV proteins were detected with monoclonal antibodies specific to NS5A, horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (Bio-Rad) and a chemiluminescence substrate (Pierce). β-Actin was used as a control and was detected with an anti-β-actin monoclonal antibody (Sigma).
Quantification of HCV RNA by qPCR
Total RNA was extracted with Trizol (Invitrogen) and HCV RNA was measured by qPCR analysis as described previously (37). The relative quantity of HCV RNA in control and HCV samples was calculated with the comparative Ct (cycling threshold) method. A reference gene (β-actin) was used as the control.
RT-PCR
Huh7.5 cells were transfected with the RNA of JFH1-AM120-LacZ and controls of JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc and the cells were passaged at every three days for a total of 15 days. The total RNA in the Huh7.5 cells were extracted with Trizol reagent (Invitrogen). The RNA was reverse transcribed using Superscript III Reverse Transcriptase (Invitrogen) and random primers. The resulting cDNA was used as a template for subsequent PCR with Platinum® Pfx DNA polymerase (Invitrogen) and the following primers: JFH-6815-FOR, 5'-TTAATTCCTATGCTGTCGGGTCCCAGCT-3'; JFH-7765-Rev, 5'-GTGCGTTGTACAGTACACCTTGTTATGG-3'. The PCR amplicons (950bp for NS5A wild type) were analyzed by 1% agarose gel electrophoresis and sequenced using standard methods.
Statistical analysis
The mean and standard deviation of the mean for data were determined and the results for different experimental conditions were compared using the Students t -test. The P value<0.05 was considered to be significant.
Results
Generation of HCV reporter virus construct of JFH1-AM120-LacZ
A previous study (14) demonstrated HCV JFH1 wild type is capable of producing infectious virus but the titer is very low. It also cannot bear the foreign genes of EGFP and Rluc to produce viable reporter viruses (18). Therefore, we used the adaptively mutated JFH1 strain, JFH1-AM120, that produces higher titers of infectious virus as a vector for these studies (18). On this strain, EGFP and Renilla luciferace (Rluc) genes were inserted in frame into a unique RsrII site located in the amino acid 398 codon of NS5A and reporter viruses of JFH1-AM120-EGFP and JFH-AM120-Rluc were produced in Huh7.5 (18). The JFH1-AM120-LacZ was constructed by in frame inserting full LacZ gene fragment (3039bp) to the same RsrII site of JFH-AM120 NS5A C-terminal region and the LacZ fragment was amplified from vector pSV-β-galactosidase control (Promega, E1081) using primers LacZ-F(5'-TCTCGGTCCGATAGATCCCGTCGTTTTACA-3') and LacZ-R(5'-CTTACGGACCGCCTTTTTGACACCAGACCAACT-3'). The new clone was sequenced and the correct full-length clone was chosen for the subsequent experiments. All the constructs used in this study are shown in Fig. 1.
Schematic representation of HCV constructs used in this study. JFH1-AM120, JFH1-AM120-Rluc and JFH1-AM120-EGFP were generated in our previous study (18). EGFP and Renilla luciferace (Rluc) genes were inserted in frame into a unique RsrII site located in the amino acid 398 codon of NS5A of JFH1-AM120. JFH-AM120-LacZ was generated by in frame insertion of full LacZ gene, which was amplified from vector pSV-β-galactosidase Control (Promega, E1081) to the same RsrII site. The detailed protocol was described in Material and Method.

Insertion of the JFH1 AM120 NS5A C-terminal region with full LacZ gene did not completely disrupt HCV viral replication
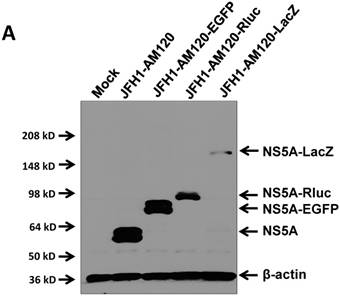
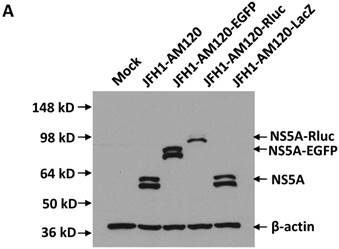
In this study the LacZ gene (3039 bp) was inserted into the NS5A C-terminal region of JFH1-AM120, designated JFH1-AM120-LacZ. JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc were used as controls. RNAs were transcribed from plasmids of JFH1-AM120, JFH1-AM120-EGFP, JFH1-AM120-Rluc and JFH1-AM120-LacZ and were transfected to Huh7.5 cells to determine whether cells transfected with the RNAs from each plasmid expressed HCV proteins and HCV RNA. Three days after RNA transfection of cells, wild-type NS5A (58kd) and the NS5A-EGFP (84kd), NS5A-Rluc (92kd) and NS5A-LacZ (169kd) fusion protein with the predicted molecular mass were detected in cell lysate by western blotting (Fig. 2A). However, the band of NS5A-LacZ protein was obviously weaker than NS5A and other fusion proteins. The HCV RNAs were also quantified by qPCR. The results showed that HCV RNA amounts (Fig. 2B) correlated with changes in protein levels by western blotting. These results indicated that the replication level of JFH1-AM120-LacZ was significantly decreased after insertion of LacZ gene into the NS5A C-terminal of JFH1-AM120. However, JFH1-AM120-LacZ can still replicate in Huh7.5 cells even though the LacZ gene is 3039bp in length.
Replication analysis of HCV JFH1-AM120-LacZ followed RNA transfection. (A) Huh7.5 cells were transfected with the RNA of JFH1-AM120-LacZ and controls of JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc. Three days post-transfection, the cells were lysed for Western blotting using anti-NS5A and anti-β-actin antibodies as described in Methods. Western blot analyses were carried out two times and representative examples are shown. (B) Three days after transfection of Huh7.5 cells in six-well plates with RNAs of JFH-AM120-LacZ and JFH-AM120, JFH-AM120-EGFP and JFH-AM120-Rluc, the total RNA was extracted from cells and qPCR analysis of HCV RNA was performed. Experiments were performed three times and the data presented as the mean ± SD (* P< 0.05, * * P< 0.01).
NS5A-LacZ fusion protein can both be visualized by X-gal staining and NS5A staining and measured the enzyme activity of β-galactosidase
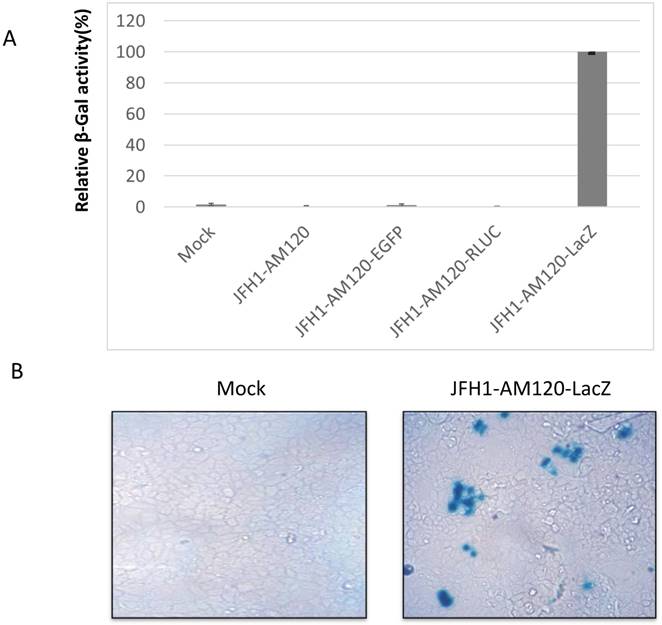
The LacZ gene encodes β-galactosidase (β-gal) and the protein levels of this gene can be determined via staining with X-gal substrate and measuring the activity of the β-galactosidase enzyme. The protein levels and enzyme activity of β-galactosidase should be detected in JFH1-AM120-LacZ infected Huh7.5 cells. To verify this statement, the RNA of JFH1-AM120-LacZ was transfected into Huh7.5 cells. RNAs of JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc were used as controls. Three days post transfection, β-Galactosidase activity was measured using a Mammalian β-Galactosidase Assay Kit (ThermoFisher). The results showed only the sample of JFH1-AM120-LacZ had detectable β-Galactosidase activity. All other transfected RNAs were negative (Fig. 3A). Using the same method, additional cells were transfected with JFH-AM120-LacZ RNA or control RNAs and fixed with paraformaldehyde, stained using an X-gal staining assay Kit (Genlantis) and visualized the cell with blue color under bright field microscope. Blue cells were only seen in cells transfected with JFH1-AM120-LacZ RNA (Fig.3B) and there was not any on the other three transfected RNAs (data not shown). These results confirmed the JFH1-AM120-LacZ could replicate in Huh7.5 cells.
Analysis of NS5A-LacZ activity followed RNA transfection of JFH1-AM120-LacZ. (A) Huh7.5 cells were transfected with the RNA of JFH1-AM120-LacZ and controls of JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc as described in Methods. Three days post-transfection, the absorption peaks of β-Galactosidase activity were measured using a Bio-Tec plate reader at 405 nm. The values were relative to JFH1-AM120-LacZ. Experiments were performed three times and the data presented as the mean ± SD. (B) Three days post-transfection of Huh7.5 cells in 24-well plates with RNA of JFH1-AM120-LacZ, cells were fixed with 4% paraformaldehyde followed X-gal staining. Cover slips were visualized and images were taken (100x) by bright field microscopy. Experiments were performed two times and representative results are shown. (C) Co-localization of NS5A and β-galactosidase. Three days post-transfection of Huh7.5 cells in 24-well plates with RNA of JFH1-AM120-LacZ, Cells were fixed with 4% paraformaldehyde and were immunostained with anti-NS5A antibody (red) and Nuclei were counterstained using DAPI (blue). Images were taken by immunofluorescence microscopy (200x). Then the coverslip was washed and were processed with X-gal staining followed visualizing and imaging by bright field microscopy (200x). Colocalization assay was performed by Photoshop CC software. Experiments were performed three times and representative results are shown.
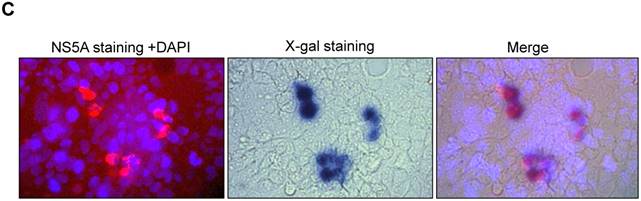
To examine the concurrent expression of HCV NS5A and the β-galactosidase fusion protein in Huh7.5 cells after transfection of JFH1-AM120-LacZ RNA, the colocalization of the NS5A protein and β-galactosidase protein were determined by staining with NS5A immunofluorescence assays and X-gal three days after transfection. Cells were fixed with 4% paraformaldehyde and immunostained with anti-NS5A antibody (red) and nuclei counterstained with DAPI (blue). Images were taken by immunofluorescence microscopy (200x). The coverslip was then washed and X-gal staining was performed, followed by visualizing and imaging by bright field microscopy (200x). Colocalization assay was performed with Photoshop CC software (Fig.3C). Images showed that NS5A positive cells (red) were also with the blue color of X-gal staining. This result provided evidence that fusion protein of NS5A and β-galactosidase can be co-expressed in Huh7.5 cells after transfection of JFH1-AM120-LacZ RNA.
Insertion of the JFH1 AM120 NS5A C-terminal region with LacZ gene significantly impaired HCV viral production
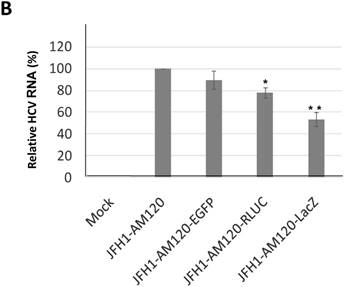
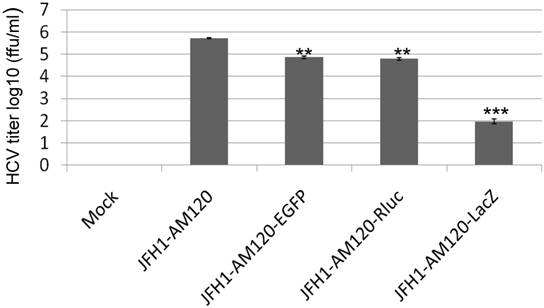
To determine if transfection of Huh 7.5 cells with JFH1-AM120-LacZ RNA resulted in production of infectious viral particles, we inoculated naïve Huh 7.5 cells with supernatants collected at 6th days post-transfection with JFH1-AM120-LacZ RNA. Supernatants collected from the cells transfected with RNAs from JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc were used as controls. At three days post-infection, the cells infected with the supernatant of JFH-AM120-LacZ were stained by X-gal and blue color cells were detected, while all of the others were negative. EGFP positive cells were only found in JFH-AM120-EGFP infected cells. JFH1-AM120 and JFH1-AM120-Rluc infected cells expressed NS5A and NS5A-Rluc, as determined by the immunofluorescence assay (IFA). The viral titers are expressed as focus-forming units per milliliter of supernatant (ffu/ml). The titer of JFH1-AM120, JFH1-AM120-EGFP, JFH1-AM120-Rluc and JFH1- AM120- LacZ were 5.5x105 ffu/ml, 4.5x104 ffu/ml, 4.2x104 ffu/ml and 0.98x102 ffu/ml, respectively. The viral titers of JFH1-AM120-EGFP and JFH1-AM120-Rluc were about 10 times lower than JFH-AM120, however, the titer of JFH1-AM120-LacZ was dramatically lower (more than 5000 times) compared to JFH-AM120 (Fig.4). These results demonstrated that the insertion of LacZ in NS5A C-terminal region of JFH1-AM120 significantly impaired the production of infectious viral particles. This result provided direct evidence that long exogenous genes can influence the capability of infectious HCV virus production at difference levels with a gene length dependent manner.
Infectivity assay of virus particles followed RNAs transfection of JFH1-AM120-LacZ and controls. The RNA of JFH1-AM120-LacZ and controls of JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc were electroporated into Huh-7.5 and the infectivity titers in the cultured supernatants at the 6th day were measured (Described in Material and Method). The viral titer is expressed as focus-forming units per ml of supernatant (ffu/ml) as determined by the average number of NS5A-positive foci detected by immunofluorescence for NS5A (Mock, JFH-AM120 and JFH-AM120-Rluc), or directly visualized EGFP positive cells(JFH1-AM120-EGFP) and detected blue color cells after X-gal staining (JFH1-AM120-LacZ). Assays were performed three times and the data are presented as mean ± standard deviation. (** P< 0.01, *** P< 0.001)
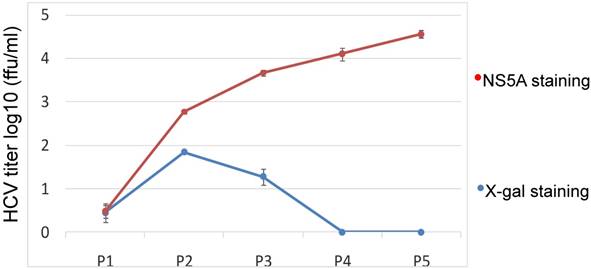
Kinetics and stability of the JFH1-AM120-LacZ after multiple passages
Our data show that JFH1-AM120-LacZ can replicate and produce a low titer of JFH1-AM120-LacZ HCV reporter virus in Huh7.5 cells. However, the genetic stability of a reporter virus is essential for its further application. To explore the durability of the JFH1-AM120-LacZ reporter in Huh7.5 cells subculture, a kinetics assay was performed by passaging the cells every three days for a total of 15 days. Supernatants were collected and designated P1 to P5. Double titrations were carried out by X-gal staining for β-galactosidase and immunofluorescence staining for NS5A protein (see Materials and Methods). The titer measured by these two methods was different. The titer after NS5A antibody staining was increased from P1 to P5. The titer after X-gal staining was increase from P1 to P2 then decreased from P2 to P3 and reached zero at P4 and P5 (Fig.5). The discrepancy of these result indicated the LacZ gene may be deleted and one strain lost LacZ gene became a dominant and produced a higher titer virus.
Kinetics assay of the JFH1-AM120-LacZ HCV reporter after multiple passages. The cells transfected with the RNA of JFH1-AM120-LacZ were passaged at every three days for a total of 15 days. Supernatants collected were designated P1 to P5. The double titrations were carried out by X-gal staining for β-galactosidase and immunofluoresence staining for NS5A protein (see Materials and Methods). Experiments were performed three times and the data presented as the mean ± SD.
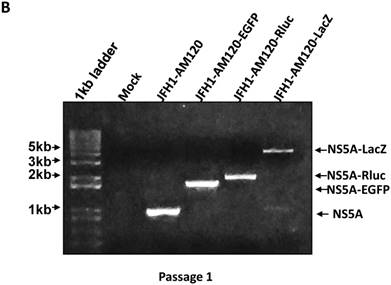
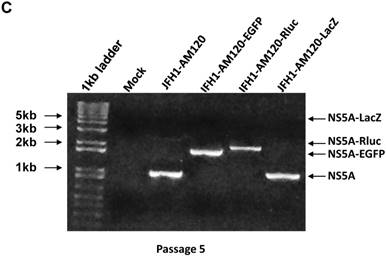
To confirm these results, Western blotting was carried out for cells transfected with JFH1-AM120-LacZ RNA and control RNAs at P1 and P5. The results show no band of NS5A-LacZ (163Kd) in P5 cells and the NS5A fragment reverting to the size of wild-type NS5A (56 and 58Kd) (Fig. 6A) compared to the much larger NS5A-LacZ fusion protein in P1 cells (Fig.2A). NS5A-EGFP and NS5A-Rluc did not change after the same number of passages (Fig.2A & 6A). To verify these results, RT-PCR was performed on P1 and P5 cells and agarose gel analysis showed the predicted size PCR products (Fig. 6B and 6C). The size of the NS5A-WT, NS5A-EGFP and NS5A-Rluc PCR products did not change at P1 and P5. However, the size of NS5A-LacZ PCR product was reverted to the same size of the NS5A-WT product at P5. This PCR product was sequenced and only six nucletitides of the LacZ gene remained, while the other nucleotides of the LacZ gene were absent, providing direct evidence for the loss of the LacZ gene with multiple passaging of cells. On the Fig. 2A and 6B, a weak band of similar size to wild-type NS5A were seen in the LacZ lane. This suggests that the deletion of the LacZ gene may occur early in the first passage. These results indicated the JFH1-AM120-LacZ reporter virus was unstable after progressive cell culture.
Stability assay of JFH1-AM120-LacZ and controls followed the RNA transfection. (A) Huh7.5 cells were transfected with the RNA of JFH1-AM120-LacZ and controls of JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc and the cells were passaged at every three days for a total of 15 days. The cells of passage 1 (Fig.2A) and passage 5 were lysed for Western blotting using anti-NS5A and anti-β-actin bodies as described in Methods.The experiment was performed twice and representative example are shown. (B & C) Detection of HCV RNA. Huh7.5 cells were transfected with the RNA of JFH1-AM120-LacZ and controls of JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc and the cells were passaged at every three days for a total of 15 days. The total RNA was extracted and RT-PCR was performed described as in methods. The PCR products were analyzed by 1% agarose gel electrophoresis. Experiments were performed two times and a representative experiment is shown (B showed passage 1st result and C showed passage 5th result).
Discussion
Some RNA viruses can tolerate the insertion of relatively large exogenous genes without disrupting replication and the production of functional recombinant viruses (30, 38). Vesicular stomatitis virus (VSV), has been used as a gene expression vector and can accommodate the insertion of a foreign gene as large as 5955 bp (30). Previous reports have shown that the HCV adapted JFH-1 strain can tolerate the insertion of exogenous genes less than 1000 bp, such as EGFP (720bp) and Rluc(930bp), into the C terminus of NS5A without disrupting viral replication and the production of infectious virions (18, 19, 25, 26, 28).
A reporter gene as large as the LacZ gene (3039bp), has not been successfully inserted into the HCV genome with production of infectious virions. Our results demonstrate that the LacZ reporter gene can be inserted into the NS5A C-terminus of HCV JFH1-AM120 and will express the predicted NS5A-LacZ fusion protein, which can be detected by western blotting three days after RNA transfection of cells. However, compared to the control vectors of JFH1-AM120, JFH1-AM120-EGFP and JFH1-AM120-Rluc, there was an obvious decrease in the expression of the NS5A fusion protein (Fig. 2A). β-galactosidase was also detected in these cells by several measures (Fig.3) and the supernatants of these cells produced infectious virus of JFH1-AM120-LacZ (Fig.4). However, the infectious virus titer was much lower, 0.98 x102 ffu/ml compared to 5.5x105 ffu/ml of parent JFH1-AM120 virus. In addition, the titer of infectious HCV JFH1-AM120-LacZ progressively decreased with serial passaging of cells, with the eventual loss of X-gal positive cells (Fig.5). This observation is consistent with Western blot and NS5A PCR analysis indicating the loss of the LacZ reporter gene (Fig.6). The reason for the loss of the inserted LacZ reporter gene is unclear. There may be three possible mechanisms on it. First, the HCV genome RNA might have the capability of repairing itself and removing exogenous genes. Second, the HCV NS5A gene has a limit to it's carrying capacity for exogenous genes, and LacZ is too large for stable expression. Third, the combined effects of viral RNA repair, expression stability and replication might result in LacZ depletion in some strains, which could become the dominant strains during subsequent replication process, and this is consistent with the basic principle of natural adaptive mutation selection.
Although HCV-EGFP and HCV-Rluc reporter viruses we have described previously were quite stabile with repeated passages, resulted in relatively high titers of infectious virions and have been used in a number of studies (19, 39-41), it is still unclear the capacity of HCV to accommodate large foreign genes. The production of infectious JFH1-AM120-LacZ virions reported here demonstrate that much larger gene inserts can be engineered into the C terminus of NS5A of JFH1-AM120 for other applications and approaches. However, the instability of this reporter virus and the loss of LacZ gene are likely to limit its use. Nevertheless, the fact that a reporter gene up to 3039bp in size can be inserted into the C terminus of NS5A indicates that additional studies can be performed to guide further engineering of JFH1-AM120 in the research of HCV.
Acknowledgements
We thank Dr. T. Wakita for providing the plasmid containing the HCV JFH1 plasmid. We also thank Dr. C.M. Rice for providing Huh7.5 cells and the anti-NS5A monoclonal antibody. This work was supported in part by NIH Grant CA176130 to C.H.H.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55(Suppl 1):S10-5
2. Rosen HR. Clinical practice. Chronic hepatitis C infection. N Engl J Med. 2011;364:2429-38
3. Jahan S, Ashfaq UA, Qasim M, Khaliq S, Saleem MJ, Afzal N. Hepatitis C virus to hepatocellular carcinoma. Infect Agent Cancer. 2012;7:2
4. Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, Abergel A, Mangia A, Lai CL, Chan HL, Mazzotta F, Moreno C, Yoshida E, Shafran SD, Towner WJ, Tran TT, McNally J, Osinusi A, Svarovskaia E, Zhu Y, Brainard DM, McHutchison JG, Agarwal K, Zeuzem S, Investigators A-. Sofosbuvir and Velpatasvir for HCV Genotype 1, 2, 4, 5, and 6 Infection. N Engl J Med. 2015;373:2599-607
5. Falade-Nwulia O, Suarez-Cuervo C, Nelson DR, Fried MW, Segal JB, Sulkowski MS. Oral Direct-Acting Agent Therapy for Hepatitis C Virus Infection: A Systematic Review. Ann Intern Med. 2017;166:637-648
6. Murray CL, Rice CM. Turning hepatitis C into a real virus. Annu Rev Microbiol. 2011;65:307-27
7. Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology. 2004;39:5-19
8. Reed KE, Rice CM. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol. 2000;242:55-84
9. Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972-4
10. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110-3
11. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791-6
12. Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294-9
13. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623-6
14. Kaul A, Woerz I, Meuleman P, Leroux-Roels G, Bartenschlager R. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J Virol. 2007;81:13168-79
15. Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J Virol. 2006;80:11082-93
16. Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouille Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol. 2007;88:2495-503
17. Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc Natl Acad Sci U S A. 2008;105:4370-5
18. Liu S, Xiao L, Nelson C, Hagedorn CH. A cell culture adapted HCV JFH1 variant that increases viral titers and permits the production of high titer infectious chimeric reporter viruses. PLoS One. 2012;7:e44965
19. Liu S, Chen R, Hagedorn CH. Direct visualization of hepatitis C virus-infected Huh7.5 cells with a high titre of infectious chimeric JFH1-EGFP reporter virus in three-dimensional Matrigel cell cultures. J Gen Virol. 2014;95:423-33
20. Qiu D, Lemm JA, O'Boyle DR 2nd, Sun JH, Nower PT, Nguyen V, Hamann LG, Snyder LB, Deon DH, Ruediger E, Meanwell NA, Belema M, Gao M, Fridell RA. The effects of NS5A inhibitors on NS5A phosphorylation, polyprotein processing and localization. J Gen Virol. 2011;92:2502-11
21. Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J Virol. 2008;82:1073-83
22. Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J Biol Chem. 2004;279:48576-87
23. Tellinghuisen TL, Marcotrigiano J, Rice CM. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature. 2005;435:374-9
24. Moradpour D, Evans MJ, Gosert R, Yuan Z, Blum HE, Goff SP, Lindenbach BD, Rice CM. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J Virol. 2004;78:7400-9
25. Han Q, Xu C, Wu C, Zhu W, Yang R, Chen X. Compensatory mutations in NS3 and NS5A proteins enhance the virus production capability of hepatitis C reporter virus. Virus Res. 2009;145:63-73
26. Kim CS, Jung JH, Wakita T, Yoon SK, Jang SK. Monitoring the antiviral effect of alpha interferon on individual cells. J Virol. 2007;81:8814-20
27. Liu S, Ansari IH, Das SC, Pattnaik AK. Insertion and deletion analyses identify regions of non-structural protein 5A of Hepatitis C virus that are dispensable for viral genome replication. J Gen Virol. 2006;87:323-7
28. Schaller T, Appel N, Koutsoudakis G, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J Virol. 2007;81:4591-603
29. Liu S, Nelson CA, Xiao L, Lu L, Seth PP, Davis DR, Hagedorn CH. Measuring antiviral activity of benzimidazole molecules that alter IRES RNA structure with an infectious hepatitis C virus chimera expressing Renilla luciferase. Antiviral Res. 2011;89:54-63
30. An HY, Kim GN, Wu K, Kang CY. Genetically modified VSV(NJ) vector is capable of accommodating a large foreign gene insert and allows high level gene expression. Virus Res. 2013;171:168-77
31. Nielsen DA, Chou J, MacKrell AJ, Casadaban MJ, Steiner DF. Expression of a preproinsulin-beta-galactosidase gene fusion in mammalian cells. Proc Natl Acad Sci U S A. 1983;80:5198-202
32. Ide Y, Zhang L, Chen M, Inchauspe G, Bahl C, Sasaguri Y, Padmanabhan R. Characterization of the nuclear localization signal and subcellular distribution of hepatitis C virus nonstructural protein NS5A. Gene. 1996;182:203-11
33. Takimoto R, Kato J, Terui T, Takada K, Kuroiwa G, Wu J, Ohnuma H, Takahari D, Kobune M, Sato Y, Takayama T, Matsunaga T, Niitsu Y. Augmentation of antitumor effects of p53 gene therapy by combination with HDAC inhibitor. Cancer Biol Ther. 2005;4:421-8
34. Qiao L, Zhang H, Yu J, Francisco R, Dent P, Ebert MP, Rocken C, Farrell G. Constitutive activation of NF-kappaB in human hepatocellular carcinoma: evidence of a cytoprotective role. Hum Gene Ther. 2006;17:280-90
35. Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001-14
36. Krieger N, Lohmann V, Bartenschlager R. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J Virol. 2001;75:4614-24
37. Papic N, Maxwell CI, Delker DA, Liu S, Heale BS, Hagedorn CH. RNA-sequencing analysis of 5' capped RNAs identifies many new differentially expressed genes in acute hepatitis C virus infection. Viruses. 2012;4:581-612
38. de Haan CA, Haijema BJ, Boss D, Heuts FW, Rottier PJ. Coronaviruses as vectors: stability of foreign gene expression. J Virol. 2005;79:12742-51
39. Meyer K, Kwon YC, Liu S, Hagedorn CH, Ray RB, Ray R. Interferon-alpha inducible protein 6 impairs EGFR activation by CD81 and inhibits hepatitis C virus infection. Sci Rep. 2015;5:9012
40. Hong M, Schwerk J, Lim C, Kell A, Jarret A, Pangallo J, Loo YM, Liu S, Hagedorn CH, Gale M Jr, Savan R. Interferon lambda 4 expression is suppressed by the host during viral infection. J Exp Med. 2016;213:2539-2552
41. Aydin Y, Chedid M, Chava S, Danielle Williams D, Liu S, Hagedorn CH, Sumitran-Holgersson S, Reiss K, Moroz K, Lu H, Balart LA, Dash S. Activation of PERK-Nrf2 oncogenic signaling promotes Mdm2-mediated Rb degradation in persistently infected HCV culture. Sci Rep. 2017;7:9223
Author contact
![]() Corresponding authors: Shuanghu Liu, e-mail: shuanghu.liuutah.edu and Curt Hagedorn, e-mail: chhagedornedu
Corresponding authors: Shuanghu Liu, e-mail: shuanghu.liuutah.edu and Curt Hagedorn, e-mail: chhagedornedu