Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(11):1521-1534. doi:10.7150/ijbs.25334 This issue Cite
Research Paper
Butein suppresses hepatocellular carcinoma growth via modulating Aurora B kinase activity
Yuanfeng Zhou1,3,*, Ming Li6,7,*, Xinyou Yu4, Ting Liu1,2, Tian Li1,2, Li Zhou8, Wenbin Liu9, Wei Li5, ![]() , Feng Gao1,2,
, Feng Gao1,2, ![]()
1. Department of Ultrasonography, The Third Xiangya Hospital of Central South University, Changsha, Hunan, 410013, P.R. China
2. State Key Laboratory of Powder Metallurgy, Central South University, Changsha, Hunan, 410000, P.R. China
3. Department of Infectious Diseases, Taizhou Hospital, Affiliated Hospital of Wenzhou Medical University, Taizhou, P.R. China
4. Shandong Lvdu Bio-Industry Co., Ltd., Binzhou, Shandong 256600, P.R. China
5. Department of Radiology, The Third Xiangya Hospital of Central South University, Changsha, Hunan 410013, P.R. China
6. Changsha Stomatological Hospital, Changsha, Hunan 410004, P.R. China.
7. School of Stomatology, Hunan University of Chinese Medicine, Changsha, Hunan 410208, P.R. China
8. Department of Pathology, Xiangya Hospital of Central South University, Changsha, Hunan 410008, P.R. China
9. Hunan Cancer Hospital and The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, Hunan 410013, P.R. China
* These authors contributed equally to this work.
Received 2018-2-3; Accepted 2018-7-31; Published 2018-9-7
Abstract

Aurora B is aberrantly expressed in various tumors and shown to be a promising target for cancer therapy. Butein, a chalcone isolated from Rhus cerniciflua, has demonstrated antitumor activities in different cancers. In this study, we aimed to validate whether Aurora B kinase was the direct target of butein to exhibit its potency in hepatocellular carcinoma (HCC). Comparing with the normal cell line and tissue, Aurora B was overexpressed in all tested HCC cells and the majority of tumor tissue. Knocking down of Aurora B with shRNA substantially inhibited HCC cell proliferation, colony formation and delayed tumor growth in nude mice. Except computer docking, a series of kinase assays revealed butein directly interacted with Aurora B and inhibited its kinase activity. Along with the decrease of Aurora B and histone H3 phosphorylation, HCC cells were induced G2/M cell cycle arrest and subjected to cell apoptosis. Butein-mediated antitumor activities were substantially impaired in Aurora B knockdown cells, suggesting Aurora B was an important target of butein in HCC. Oral administration of butein substantially restrained HCC xenograft growth and the expressions of Ki67 and phosphor-histone H3 were significantly decreased in butein-treated tissue. To the best of our knowledge, our studies revealed that Aurora B was the direct target of butein in HCC.
Keywords: Apoptosis, Aurora B, Butein, cell cycle arrest, Hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is a great public health problem in worldwide, especially in China. In 2015, it was estimated that 466,100 people were diagnosed with HCC and about 422,100 died from this disease, the incidence and mortality rate ranked the fourth and the third respectively [1]. Owing to most HCC patients were found in the advanced stage, curative treatments such as surgery or local ablation are not feasible and the systemic therapies are considered. Sorafenib is recommended as the first line therapy for advanced HCC patients and has demonstrated to promote the overall survival, however, the efficacy is limited and the median survival is still less than 1year (10.7 month)[2]. Moreover, the recurrence rate is as high as 74% for patients with intermediate and advanced HCC after resection [3]. The reasons of the slow progress are the high heterogeneity of HCC and the shortage of knowledge of critical drivers in HCC progression [4].
The Aurora kinase family is involved in the regulation of multiple steps during cell cycle progression and is indispensable for mitotic and meiotic chromosome assembly and segregation [5]. In mammalian cells, three different kinases are identified, which are named Aurora A, B, C respectively. In mitosis, Aurora A mainly mediates premetaphase events, such as centrosome maturation and separation, bipolar spindle assembly and mitotic entry, while Aurora B is engaged in directing metaphase and post-metaphase, including chromosome alignment, accurate segregation and cytokinesis [6]. Aurora C is less characterized and considered to have redundant functions and substrates as Aurora B [7]. Owing to its importance, the aberrations of Aurora kinase family, such as overexpression or increased gene copy number, have contributed to uncontrolled cell proliferation and defects in chromosome segregation. Overexpression of Aurora B was was detected in various cancers including acute myeloid leukemia (AML) [8], colorectal cancer [9], non-small cell lung cancer (NSCLC) [10] and closely related to poor prognosis. Overexpression of Aurora A plays a crucial role in HCC tumorigenesis [11-13], however, the function of Aurora B in HCC is still elusive. Aurora B overexpression was found to be associated with Aurora A overexpression and p53 mutation. However, despite its interaction with Aurora A and p53, Aurora B overexpression was validated to be correlated with worse clinicopathologic characteristics and was an independent poor prognostic factor [14]. Moreover, Tanaka S et al identified that overexpression of Aurora B kinase was the only independent factor to predict the aggressive recurrence [15]. All these findings made Aurora B as as an attractive target for HCC therapy.
Due to the diversity of chemical structure, the natural products, especially those exists in our daily diet or traditional Chinese medicine, are important resources for novel drug discovery. In our preliminary experiments, through the screening in a series of natural products, we identified that butein had a direct binding affinity to Aurora B. Butein is a chalcone derived from Rhus vericiflua, which is of great medicinal value and is used as traditional medicine for the treatment of various diseases, such as hepatic disorders, gastritis and atherosclerosis in Eastern Asia for a long time [16-18]. Butein has been proved to possess a wide range of biological functions including antioxidant, anti-inflammatory, antidiabetic, hypotensive and neuroprotective effects [19-21]. Beyond that, increasing evidences also demonstrated that butein exerted antitumor activities against various cancers [22-25].
In this study, we aimed to clarify the activities of butein on Aurora B as well as the biological effects caused by Aurora B inhibition. We demonstrated that butein directly interacted with Aurora B in HCC cells and substantially inhibited its activity in selective way (Not targeting Aurora A and Aurora C). With Aurora B inhibition, HCC cells were induced G2/M arrest and subjected to cell apoptosis.
Material and Methods
Cell lines and reagents. HepG2 and Hep3B cells were purchased from the American Type Culture Collection (Manassas, VA, USA). LO2, Huh7, Bel-7402, and HCC-LM3 cells were obtained from Cell Bank of Chinese Academy of Sciences (shanghai, China). All cells were cultured at 37˚C in a 5% CO2 incubator according to the supplier's protocol. The antibodies including p-Aurora B (#2914), Aurora B (#3094), Aurora A (#14475), p-Histone H3 (#53348, #9701 for IHC staining), Histone H3 (#4499), PARP (#9532), and cleaved-caspase 3 (#9664) antibodies were purchased from Cell Signaling Technology(Beverly, MA, USA). β-actin (A5316) antibody was product of Sigma-Aldrich(St. Louis, MO, USA). The Aurora C (ab198705) and Ki67 (ab16667) antibodies were obtained from Abcam (Cambridge, UK). Lentivirus plasmids of Aurora B (TRCN 0000000777, 0000000779) were purchased from Thermo Scientific (Huntsville, AL, USA). The lentiviral packaging plasmid psPAX2 and the envelope plasmid pMD2.G were products of Addgene (Cambridge, MA). LipofectamineTM 2000 was obtained from Invitrogen (Carlsbad CA, USA).
MTS assay. The cells were seeded (3×103/well/100 mL) into 96-well plates, after treated with different concentrations of butein for different time point, cell proliferation was assessed by MTS assay (Promega, Madison, WI) according to the instructions provided.
Anchorage-independent cell growth assay
Cells (8×103 per well) were seeded into 6-well plates with 0.3% Basal Medium Eagle agar containing 10% FBS and cultured. The cultures were maintained at 37°C in a 5% CO2 incubator for 2 or 3 weeks and colonies were counted under a microscope as previously described [26].
Virus infection. For generation of Aurora B knocking down cells, pLKO.1-sh-GFP or pLKO.1-sh-Aurora B lentivirus plasmid was co-transfected into 293T cells with psPAX2 and pMD2.G. Viral supernatant fractions were collected at 72 hours after transfection and filtered through a 0.45 μm filter followed by infection into HCC cells together with 8µg/mL polybrene. The medium was replaced with fresh medium containing 2μg/mL puromycin the next day, and cells were incubated in puromycin containing medium for another 6 days.
Natural products screening. To identify Aurora B inhibitors from natural products, we selected the compounds from the Natural Product Library (Cat. No. L1400-01/02) of Selleckchem Company. Meanwhile, we also minimized the numbers of compounds by searching on Pubmed with the key words of “Compound name + Cancer”, the compounds with publications less than 100 were selected for study. The list of the tested compounds was shown in Figure S2. Firstly, the tested compounds were conjugated to Sepharose 4B beads (GE Healthcare Biosciences) by following manufacturer's instructions. Then the compound-Sepharose 4B beads were incubated with HepG2 cell lysates (400μg) at 4°C overnight. The beads were washed with binding buffer and subjected to western blotting, and then probed with anti-Aurora B antibody.
Molecular modeling. To predict the binding mode of butein targeting Aurora B, the crystal structure of the kinase domain (PDB ID: 4C2V) was obtained from the Protein Data Bank. This structure was then prepared using the default parameters of Protein Preparation Wizard in Schrödinger Suite 2013. Hydrogen atoms were added consistent with a pH of 7, and all water molecules were removed. Finally, an ATP-binding site-based receptor grid was generated at the centroid of the ligand, barasertib, from the crystal structure, with default settings in Receptor Grid Generation in Schrödinger Suite 2013. The 3D structures of butein was generated and prepared in the module of LigPrep in Schrödinger Suite 2013, with other parameters kept default. Docking was performed using the program of Glide in Schrödinger Suite 2013 with default parameters under the standard precision mode. Three poses of each prepared structure of butein were output to analyze the scores and binding modes.
In vitro ATP competitive binding and ex vivo pull-down assays. The in vitro ATP competitive binding and ex vivo pull-down assays were performed as described previously [27]. The butein-conjugated Sepharose 4B beads were prepared according to the manufacturer's protocol (GE Healthcare Biosciences). Hep3B or HepG2 cell lysates (400 μg) was incubated with butein-Sepharose 4B beads or Sepharose 4B beads only overnight at 4°C. The beads were washed with binding buffer for 3 times and boiled with 5×SDS loading buffer for western blotting analysis. For ATP competition assays, the active Aurora B kinase was incubated with different doses of ATP at 4°C overnight. Then the butein-conjugated Sepharose 4B or Sepharose 4B beads only were added into the reaction and followed by incubation at 4°C for another 4 h. The binding activity was analyzed by Western blotting.
In vitro Aurora B kinase assay. The active Aurora A/B kinases were purchased from Millipore (Cat. 14-835, 14-511). The in vitro kinase assay was performed as described previously [28]. 1 µg of Histone H3 and 100 ng of active Aurora B/A/C kinase were incubated with various concentrations of butein or barasertib (Aurora B inhibitor)/hesperadin (Aurora A/B inhibitor)/danusertib (pan Aurora A/B/C inhibitor) in a 20 µL reaction [29]. The mixture was conducted at 30°C for 30 min in a 100 µM ATP and 1× kinase buffer (Cell Signaling Technology). Reactions were stopped by boiling samples in 5×SDS loading buffer, and proteins were analyzed by Western blot. The results were analyzed and quantified with Image-Pro Plus software (version 6.2) program (Media Cybernetics).
Western blotting. Proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore), the membranes were blocked with 5% non-fat milk and incubated with primary antibodies overnight at 4°C, after washing with PBST, the membranes were hybridized with horseradish peroxidase (HRP)-conjugated secondary antibody and then the protein bands on the membrane were visualized with ECL chemiluminescence reagents (Pierce Chemical Co., Rockford, lllinois, USA).
Cell cycle and apoptosis assay. Flow cytometry analysis was performed as described previously [30]. After the treatment of butein for 24h, HCC cells were harvested. For cell cycle analysis, HCC cells were fixed with cold 70% ethanol solution at 4°C for 24h, cells were stained with 50 μg/ml Propidium Iodide and 100 μg ribonuclease A and then analyzed with flow cytometry. For apoptosis assay, the cells harvested were centrifugated at 800 g for 5 min and suspended with binding buffer, Annexin V-FITC and Propidium Iodide were added as manufacturer 's instruction and incubated for 15 mins avoiding light, and the stained cells were subjected to FACS analysis. All results were analyzed with the FlowJo software (Version 7.6).
Immunofluorescence staining. Hep3B Cells were fixed in 4% paraformaldehyde and permeabilized in 0.5% Triton X-100 for 30 minutes. Fixed cells were blocked with 5% BSA in PBS and incubated with a p-Histone H3 rabbit antibody (ab5176, Abcam) overnight at 4°C followed by incubation with green fluorescent Alexa Fluor 488 dye-labeled anti-rabbit IgG (ab150077, Abcam,). Nuclei were stained with DAPI. Samples were viewed with a fluorescence microscopy system.
In vivo experiment. The in vivo animal study was performed following guidelines approved by the Animal Ethics Committee of Central South University. HCC cell suspension were inoculated s.c. into the right flank of athymic nude mice. After the xenografts were formed, the mice were randomly grouped. The control and the treatment group were orally administrated with the vehicle (5% dimethyl sulfoxide, 5% polyethylene glycol in PBS) or 10mg/kg butein respectively once per day. The weight of mice and the tumor volume were recorded twice per week.
Immunohistochemistry. Immunohistochemistry was performed as described previously [31]. The HCC tissue microarray (LivH150CS03) was product of Shanghai Outdo Biotech Co., Itd. including 75 cases of hepatocellular carcinoma and matched adjacent normal tissue. Briefly, tumor tissue was dewaxed in xylene and hydrated in ethanol respectively. The endogenous peroxidase was blocked with 3% H2O2 solution. The antigen was retrieved in boiling citric acid solution (pH=6.0) and the non-specific binding sites were blocked with serum from the host of the secondary antibody. The tissues were incubated with anti-Aurora B (1:100), anti-phospho-histone H3 (1:50), anti-Ki-67 (1:500) primary antibodies at 4°C overnight. After washing, the tissues were hybridized with biotinylated secondary antibody (1:100) and then incubated with HRP conjugated streptavidin. After developing with with 3,3-diaminobenzidine solution, the tissues were counterstained with hematoxylin, dehydrated and mounted. The results were analyzed with Image-Pro Plus software (version 6.2) program (Media Cybernetics).
Statistical Analysis. Data was expressed as means ± SD and statistical analysis was determined by a Student t test or one-way ANOVA with SPSS 16.0 (SPSS, Inc, Chicago, IL). p˂ 0.05 indicated significant difference.
Results
Aurora B was overexpressed in HCC cell lines and tumor tissue
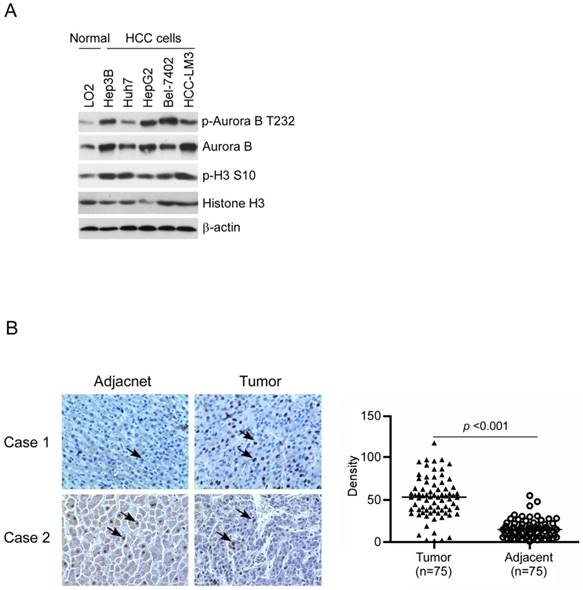
To confirm Aurora B expression in HCC, firstly we examined the difference between normal liver cell and HCC cells by western blotting. Compared with the normal liver cell LO2, Aurora B in all tested HCC cell lines was significantly elevated and had the highest expression in Hep3B, HepG2, and HCC-LM3 (Figure 1A). We further examined the phosphorylation of Aurora B and downstream target protein H3. Our data showed that Aurora B and Histone H3 were highly phosphorylated in HCC cell lines, which suggested that the Aurora B signaling pathway was hyperactivated in HCC cells (Figure 1A). Additionally, the expression of Aurora B in the tissue of HCC patients was also detected by immunohistochemistry. As the results shown in Figure 1B, in paired adjacent normal tissue, Aurora B expression was relatively low, while overexpression of Aurora B was found in the majority of HCC tissues (90.6%, 68/75, p˂0.001). All these results suggested that the abnormalities of Aurora B signaling pathway was involved in HCC development and progression.
Aurora B was overexpressed in HCC cell lines and tumor tissue. A, Aurora B signaling related proteins in normal liver cell and HCC cell lines were examined as indicated. B, Aurora B expression in HCC tissues and paired adjacent normal tissue (n=75) was detected by immunohistochemistry. left: the representative images of IHC staining; right: the statistical results. The asterisk (***, p˂0.001) indicated a significant difference.
Aurora B knockdown inhibited HCC cell proliferation and tumor growth
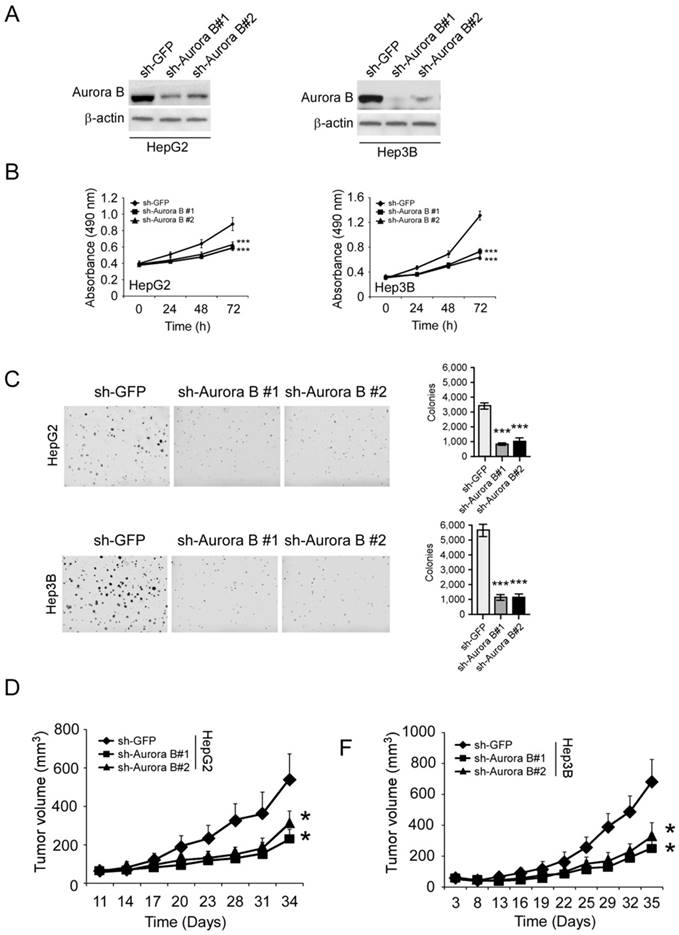
To validate the importance of Aurora B, we used shRNA to knockdown Aurora B expression in HCC cells. The protein level of Aurora B was significantly reduced in lentivirus infected stable cell lines (Figure 2A). With the knockdown of Aurora B, the proliferative abilities of HepG2 and Hep3B cells were significantly decreased (35.1% and 53.8% at 72 hours respectively, Figure 2B). In addition to the cell proliferation inhibition, the colony formation in the sh-Aurora B groups was also substantially suppressed compared with the control group (Figure 2C). To further investigate the function of Aurora B, we tested the tumor growth rate of sh-Aurora B HCC cells in nude mice. The results showed that silencing Aurora B expression in HepG2 and Hep3B significantly restrained the xenograft tumor growth (Figure 2D-F, Figure S1). All these results demonstrated that Aurora B had a critical role to sustain HCC cell growth both in vitro and in vivo.
Aurora B knockdown substantially inhibited HCC cell proliferation and tumor growth. A, HepG2 (left) or Hep3B (right) cells were transfected with Aurora B shRNA and Aurora B expression was determined by western blotting. B, The proliferative ability of HepG2 (left) or Hep3B (right) were examined by MTS assay after Aurora B was knockdown with shRNA. C, Anchorage independent assays were performed to assess the clone formation ability of HepG2 (upper) or Hep3B (below) cells transfected with Aurora B shRNA. Representative photographs were shown (left), and the graphs (right) were the results of three independent experiments. D, HepG2 (left) or Hep3B (right) cells transfected with Aurora B shRNA were injected into nude mice and the tumor growth rate was examined, the asterisks (*, p<0.05, Student's t test) indicated a significant difference.
Butein inhibited Aurora B kinase activity in vitro and ex vivo
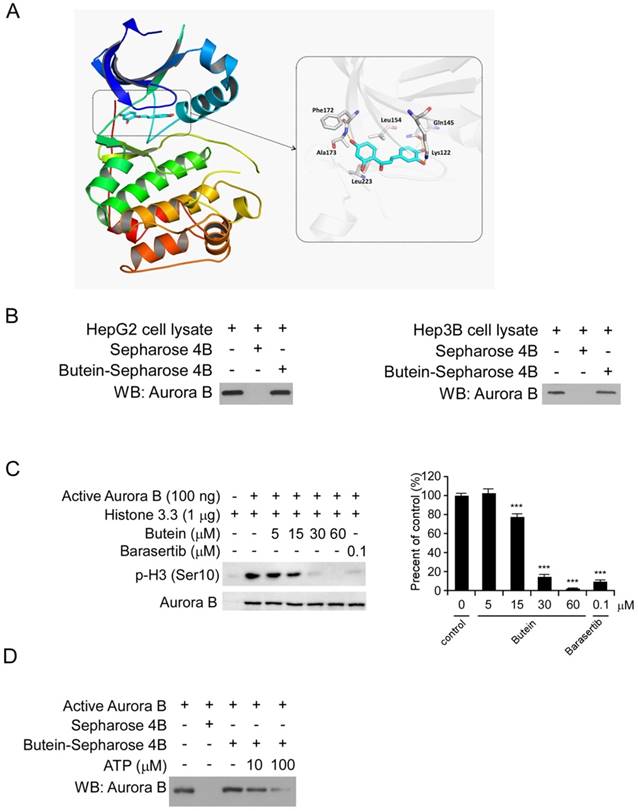
To discover natural compounds (listed in Figure S2A) which can specifically inhibit HCC cells via targeting Aurora B signaling pathway, we firstly examined the binding activities between Aurora B and the candidate compounds using the ex vivo pull-down assay. The results showed that several candidate compounds including sinomenine, limonin, butein and bergenin can directly pull-down Aurora B protein in HepG2 cell lysates (Figure S2B). However, among these four compounds, cell proliferation experiments demonstrated that only butein significantly inhibited HepG2 cell proliferation at the concentration of 10 μM (Figure S2C). Based on these screening data, we speculated that Aurora B might be a target of butein to exert its potency in HCC cells. Through molecular modeling, we found that butein may bind with human Aurora B kinase. The docked pose suggested that flat scaffold of butein fit well with the pocket by forming hydrophobic interactions with Phe172, Leu223, Leu154, etc. Three of the oxygen atoms in butein formed hydrogen bonds with Ala173, Lys122, and Gln145, respectively (Figure 3A). After incubation with HepG2 and Hep3B cell lysates, the Aurora B kinase in the cell lysates was demonstrated to be interacted with butein-Sepharose 4B beads (Figure 3B), however, the Aurora A/C kinase didn't bind with butein-Sepharose 4B significantly (Figure S3). All of these data illustrated that there is a direct interaction between butein and Aurora B. Histone H3 is one of the most important downstream targets of Aurora B. To examine the inhibitory effect of butein on Aurora B kinase activity, in vitro aurora kinase assays were conducted and the results demonstrated that butein dose dependently inhibited Aurora B activity, histone H3 phosphorylation was completely blocked by 60μM butein (Figure 3C). In contrast with the Aurora B activity inhibition, butein had no significant inhibitory effects on Aurora A/C kinase (Figure S4). Furthermore, with the increase of ATP concentration, the binding between butein and Aurora B was decreased, indicating there is a competition (Figure 3D). To better verify the specificity of butein against Aurora B, kinase profiling was performed to evaluate the activities of butein in 25 different kinases. As the results shown, in all tested kinases, buten had demonstrated the strongest inhibition against Aurora B. After treated with butein, the activity of Aurora B was decreased by nearly 70% (Table S1).
butein inhibited the Aurora B kinase in vitro and ex vivo. A, Butein was docked into the ATP binding pocket of Aurora B and the interactions between butein and Aurora B was identified. B, HepG2 (left) and Hep3B (right) cell lysates (400 μg) was incubated with butein-Sepharose 4B beads (Sepharose 4B beads only as control) overnight at 4°C, and the beads were subjected to western blotting. C, Inactive Histone H3.3 protein was incubated with different concentrations of butein, active Aurora B kinase and 100 μM ATP reaction buffer at 30°C for 30 min, then the phosphorylation was examined by western blotting, barasertib was used as positive control. D, active Aurora B was incubated with different concentrations of ATP overnight, then with butein-Sepharose 4B beads for 4h, Auroro B expression on the beads was detected by western blotting.
Butein inhibited HCC cell proliferation and colony formation in vitro
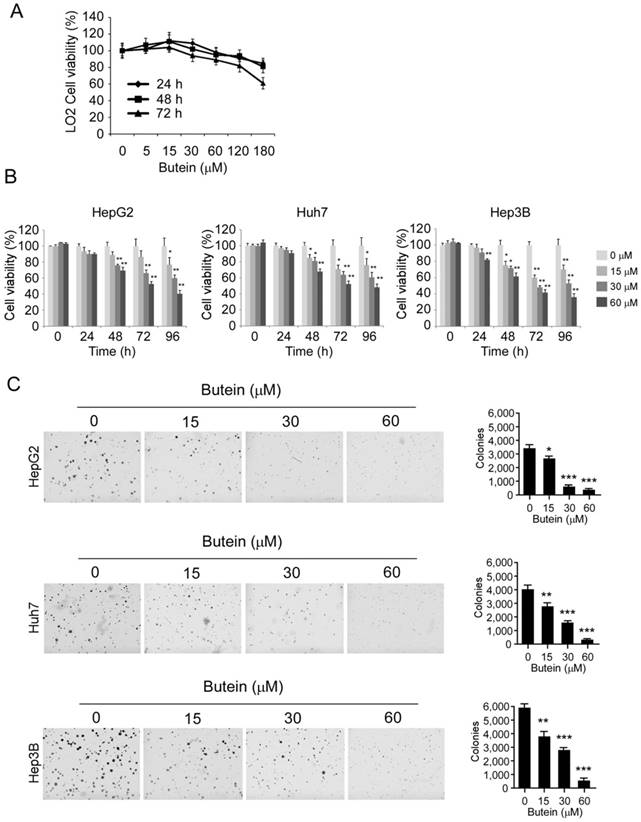
The effects of butein on cell proliferation were further assessed by the MTS assay. As the results shown, butein had no significant cytotoxicity on normal liver cell LO2 at the concentrations ˂60 μM (Figure 4A), whereas the proliferation of HepG2 (left), Huh-7 (middle), and Hep3B (right) cells were substantially inhibited in a dose-dependent manner after the treatment of butein (Figure 4B). The growth inhibition rate of 60 μM butein on HCC cells was about 77.9%, 67.8%, and 77.2% respectively in HepG2, Huh-7 and Hep3B cells after 96 hour treatment. Moreover, in anchorage-independent assay, the colony formation of HCC cells in soft agar was inhibited by butein dose-dependently (Figure 4C). The number of clones decreased 90.6%, 93.4%, and 92.7% respectively in HepG2, Huh-7 and Hep3B cells by 60 μM butein.
butein inhibited HCC cell proliferation and anchorage independent growth in vitro. A and B, LO2 cells (A) or HCC cells (B) were treated with indicated concentrations of butein for different times, and cell viability was accessed by MTS assay. C, butein significantly suppressed the clone formation of HepG2 (upper), Huh-7 (middle) and Hep3B (below). The asterisks (*, p<0.05, **, p<0.01, ***p<0.001,) indicated a significant difference versus the control.
Butein induced G2/M arrest and apoptosis in HCC cells
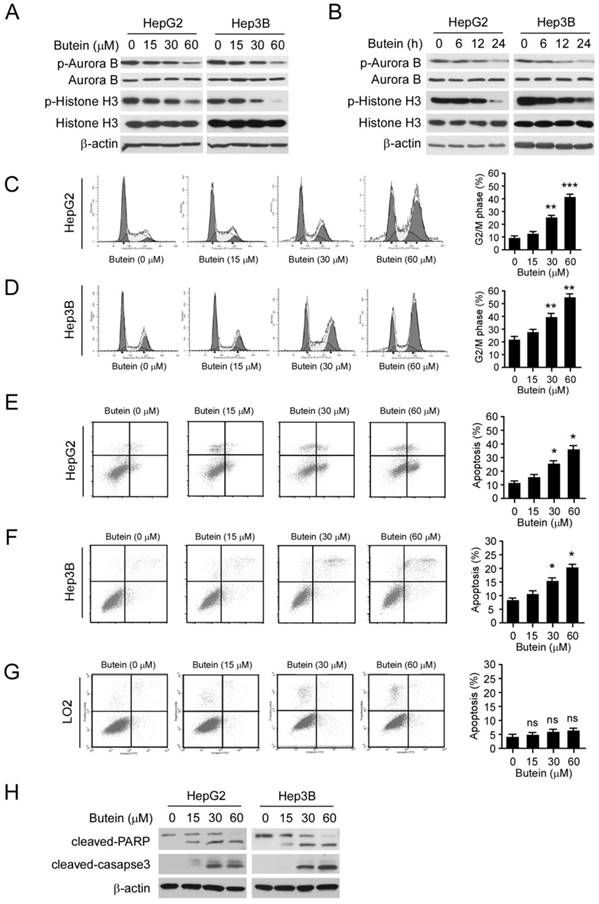
To further confirm the effect of butein on Aurora B in HCC cells, we examined the phosphorylation of Aurora B and histone H3 after butein treatment. As the results shown, the phosphorylation of both Aurora B and histone H3 in HepG2 and Hep3B cells were inhibited by butein in a dose- and time-dependent manner (Figure 5A-B). However, the down-regulation of p-Aurora B and p-Histone H3 were not observed in LO2 cells (Figure S5A). With the suppression of Aurora B activity, butein treatment for 24 h caused G2/M phase arrest in HepG2 and Hep3B cells and the proportion of cells occupying the G2/M phase increased significantly (31% in HepG2 and 23% in Hep3B at 60 μM respectively, Figure 5C-D). Moreover, as the results shown in Figure 5E-F, exposure of HCC cells to butein gave rise to apoptosis induction as measured by Annexin V/propidium iodide (PI) double staining. In contrast with the effects on HCC cells, no apoptosis induction (Figure 5G) or cell cycle arrest (Figure S5B) was detected in the normal liver cell LO2. Importantly, butein didn't promote the protein level of cleaved-caspase 3/-PARP (Figure S5A). The results of western blotting also demonstrated that the expression of cleaved caspase 3 and PARP were dramatically increased after butein treatment, further confirming that HepG2 and Hep3B cells were induced apoptosis (Figure 5H).
butein induced G2/M arrest and apoptosis in HCC cells. A-B, butein inhibited Aurora B kinase activity in a dose (A) or time (B) dependent manner in HCC cells. HepG2 and Hep3B cells were treated with different concentrations of butein for 24 h (A) or with 30μM butein for indicated times (B), cell lysates were harvested and subjected to western blotting. C-D, HepG2(C) or Hep3B (D) cells were treated with butein for 24h and subjected to cell cycle analysis by flow cytometry. left; the representative FACS results, right; statistical analysis of three independent experiments. The asterisks (**, p<0.01; ***, p<0.001) indicated a significant difference versus the control. E-H, HepG2 (E), Hep3B (F) or LO2 (G) cells were incubated with butein for 24h, the cells were collected and cell apoptosis were analyzed by Annexin V-PI double staining. left; the representative FACS results, right; statistical analysis of three independent experiments. The asterisks (*, p<0.05) indicated a significant difference versus the control. H, cleaved caspase-3, and PARP was analyzed by western blotting in butein-treated HepG2 (left) or Hep3B (right) cells.
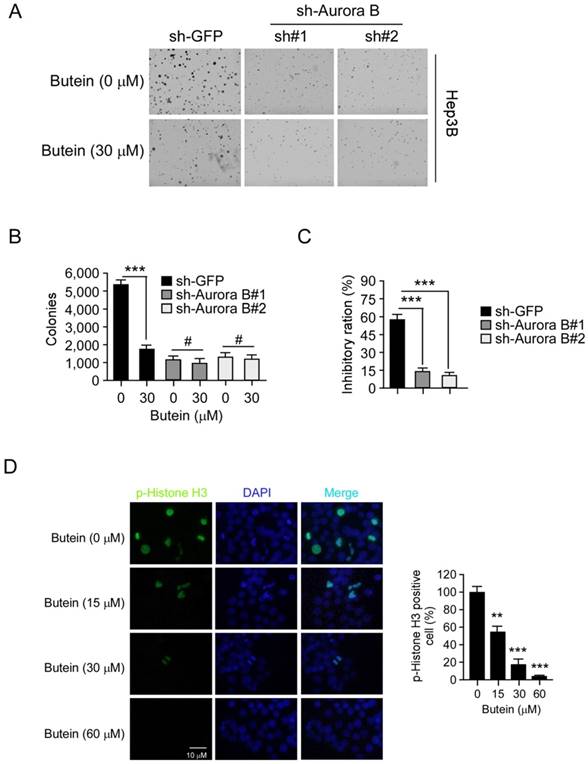
Aurora B knockdown impaired the sensitivity of HCC cell to butein
To validate the importance of Aurora B, we also examined the activity of butein in Aurora B knockdown Hep3B cells. As shown in the results (Figure 6A-C), the clone formation of HCC cells transfected with Aurora B shRNA was decreased compared with the mock group (GFP shRNA). 30 μM butein inhibited the anchorage-independent growth of Hep3B cells transfected with GFP shRNA by about 57.2%, while the inhibition was decreased to about 14.3% in Hep3B cells transfected with Aurora B shRNA. These results demonstrated that with the silence of Aurora B, the sensitivities of HCC cells to butein was significantly decreased, meanwhile, also suggested the inhibition of Aurora B played a critical role for butein to exert its activity. The immunofluorescence staining was conducted to further determine the inhibitory effect of butein on Aurora B kinase activity. We found that butein dose-dependently inhibited the phosphorylation of histone H3. Treatment of Hep3B cells with 60 μM almost completely suppressed histone H3 phosphorylation (Figure 6D). These results further demonstrated that butein decreased Aurora B signaling.
Aurora B knockdown in Hep3B cells reduced the sensitivity to butein. A-B, the anchorage-independent growth of Hep3B cells transfected with Aurora B shRNA, with or not with butein treatment. C, The inhibitory ratio of butein on colony formation ability of Hep3B cells transfected with sh-GFP or sh-Aurora B. D, representative images of Hep3B cells treated with butein at the indicated concentrations for 24 h. Cells were fixed and stained with p-histone H3 antibody (green) and DAPI (blue). The asterisks **p<0.01, ***p<0.001) indicated a significant difference.
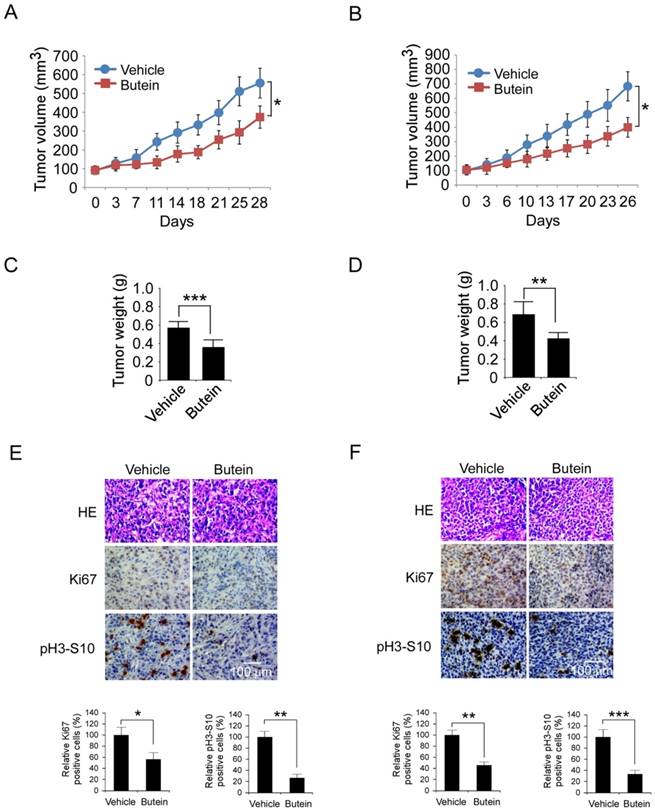
Buein suppressed HepG2 and Hep3B xenograft growth
The antitumor activities of butein against HCC cells were further validated in HepG2 and Hep3B xenograft models. As the results shown in Figure 7A-D and Figure S6, after butein treatment, the growth of HepG2 and Hep3B xenografts in nude mice was significantly inhibited. In HepG2 model, the average of tumor volume in butein-treated group was 374±59 mm3, whereas the vehicle group reached about 556±78 mm3 (33.8% inhibition). In Hep3B model, the tumor growth inhibition rate was 41.6% (683±101 mm3 versus 399±68 mm3, p˂0.05). The tumor weight of butein treated group was also substantially decreased (0.582±0.061g versus 0.366±0.044g in HepG2, 0.688±0.129g versus 0.426±0.062g in Hep3B, p˂0.05). The body weight of tumor bearing mice had no significant change during the treatment, indicating that butein was well tolerant (Figure S6). Furthermore, the effects of butein on phosphor-histone H3 and Ki67 a biomarker of proliferative potential were investigated by immunohistochemistry. As shown in Figure 7E-F, the expressions of both were dramatically decreased after butein treatment. These results demonstrated that butein inhibited HCC xenografts growth by inhibiting Aurora B activity.
Butein inhibited the HCC xenografts growth in vivo by targeting Aurora B. A-D, butein suppressed HepG2 and Hep3B xenograft growth in nude mices. The mice with HepG2 or Hep3B xenograft (n=5) were treated with butein, the tumor growth curve of HepG2 (A) or Hep3B (B) xenograft was examined. After the experiments, the xenografts of HepG2 (C) or Hep3B (D) model were isolated and weighed. The asterisks (*, p<0.05,**,p<0.01, ***p<0.001) indicated a significant difference versus the vehicle. E-F, butein decreased the expression of Ki67 and phosphor-Histone H3 in tumor tissue. HepG2 (E) or Hep3B (F) tumor tissue treated with butein was stained with Ki67 or phosphor-Histone H3 antibody. Upper, the representative images of IHC staining. Below, the expression of indicated marker were quantified and the statistical results expressed as Mean±SD, the asterisks (*, p<0.05, **, p<0.01, ***p<0.001) indicated a significant difference.
Discussion
Due to the complexity of the pathogenesis of HCC, the progress of its clinical treatment is still limited. With the advance of basic research, the dysregulation of signaling pathways including the EGFR [32], FGFR [33], HGF/c-Met [34], mTOR pathway [35] are proved to be involved in the development of HCC. So far, different inhibitors targeting above pathways have been tested in clinical trials. However, except sorafenib, which is an inhibitor of multiple angiogenic tyrosine kinases, was approved as first line therapy, the other signaling pathway inhibitors have shown limited clinical efficacy, so there is a great demand for additional or alternative strategies. Abnormal proliferation is an important characteristic of cancer cells, the success of CDK4/6 inhibitors, such as palbociclib, ribociclib, and abemaciclib which mainly blocks the transition of G1 to S phase, proved that it would be of great value to modulate cell cycle regulation [36-38]. Beyond that, the clinical benefits of antimitotic inhibitors like taxanes also validated that it's an effective intervention strategy to block mitosis in cancer cells. As a critical kinase responsible for the transition from G2 to M phase, Aurora B is essential for the chromosome biorientation, function of the spindle assembly checkpoint and cytokinesis [39]. Aberrant Aurora B expression was detected in various tumors and closely related to the chemotherapy sensitivity and poor prognosis [10, 40-43]. In our study, Aurora B overexpression was observed in all tested cell lines and the majority of HCC tumor tissue, meanwhile, the results of shRNA experiments demonstrated that both in vitro proliferation and in vivo tumor growth were significantly impaired after Aurora B was knockdown in overexpressed HCC cells. All these data suggested that Aurora B was of great importance in HCC development and it would be an effective approach by interfering with its function.
In HCC cells, the inhibition of EGFR activity [44], suppression of protein kinase C(PKC) and cAMP-dependent protein kinase [45], blockade of JNK pathway [46], inhibition of signal transducer and activator of transcription 3 (STAT3) [47] were reported to be the underlying molecular mechanisms for butein to exert its antitumor activities. Apoptosis induction is an important biological effect after butein treatment and different molecular mechanisms including PI3K/Akt/mTOR pathway inhibition [22], suppression of STAT3 activation [47], reactive oxygen species generation [48], inhibition of cyclooxygenase-2 expression [49] were reported to be engaged in the apoptosis induction by butein. Consistent with the concentrations used in other studies [23, 24, 50], in our study, at the concentration rang of 10-60μM, butein dose-dependently inhibited HCC cell proliferation, colony formation and induced G2/M cell cycle arrest and apoptosis. Differently, for the first time, we clarified that Aurora B was a potential target of butein in HCC. The molecular docking results showed that butein interacted with the kinase domain of Aurora B and formed hydrophobic interactions (Figure 3A). In vitro biochemical assays revealed that butein directly bond to the Aurora B in HCC cells and suppressed its kinase activity (Figure 3B-D). The decreased phosphorylation of histone H3 in HCC cells and tumor tissue after butein treatment further confirmed that butein inhibited Aurora B kinase (Figure 5A-B, 7E-F). In contrast with the potency in HCC cells, the effect of butein on proliferation and apoptosis of LO2 cell had no statistically significant difference (Figure 4A and 5G), which implied that Aurora B expression was closely related to butein's efficacy. Moreover, the activities of butein in Aurora B knockdown cells were substantially attenuated (Figure 6), which further suggested that Aurora B was an important target of butein in HCC cells. Beyond that, at 60μM, no inhibition was observed in the in vitro Aurora A/C kinase assay (Figure S3), suggesting that Aurora A/C was not the direct target of butein in HCC.
Since the importance of Aurora B in cancer progression, it has become an attractive target for antitumor drug development [24]. So far, many inhibitors of Aurora B including pan Aurora kinase inhibitors and selective inhibitors have entered into clinical trials and demonstrated promising efficacy [51]. Such as barasertib (AZD1152), the best studied selective Aurora B inhibitor, have shown superior efficacy in AML. In contrast with the standard therapy (arabinoside), after barasertib treatment, about 35.4% of patients received complete remission (CR), while in the arabinoside group, the ratio was only 11.5%. However, barasertib had dose-limiting toxicity to neutropenia patients [52, 53]. Therefore, discovery of Aurora B kinase inhibitor with fewer side effects are in need. In present study, none of mice treated with butein had shown significant signs of adverse effect, suggesting butein was well tolerated. Given butein is used as food additive and traditional herb for a long time, the lack of systemic toxicities and the potential to reduce toxic doses make butein a good candidate hit targeting Aurora B kinase.
In summary, we revealed a novel mechanism by which butein induced cell cycle arrest and apoptosis in HCC cells. For the first time, Aurora B was validated to be the direct target of butein. By suppressing Aurora B, butein had substantial antitumor activities against HCC in vitro and in vivo.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was supported by the New Xiangya Talent Project of the Third Xiangya Hospital of Central South University (No.JY2015011), We also thank the technical support of the State Key Laboratory of Powder Metallurgy of Central South University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
2. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
3. Zhong JH, Ke Y, Gong WF, Xiang BD, Ma L, Ye XP. et al. Hepatic resection associated with good survival for selected patients with intermediate and advanced-stage hepatocellular carcinoma. Ann Surg. 2014;260:329-40
4. Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014;20:2072-9
5. Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene. 2015;34:537-45
6. D'Assoro AB, Haddad T, Galanis E. Aurora-A Kinase as a Promising Therapeutic Target in Cancer. Front Oncol. 2015;5:295
7. Yan X, Cao L, Li Q, Wu Y, Zhang H, Saiyin H. et al. Aurora C is directly associated with Survivin and required for cytokinesis. Genes Cells. 2005;10:617-26
8. Lucena-Araujo AR, de Oliveira FM, Leite-Cueva SD, dos Santos GA, Falcao RP, Rego EM. High expression of AURKA and AURKB is associated with unfavorable cytogenetic abnormalities and high white blood cell count in patients with acute myeloid leukemia. Leuk Res. 2011;35:260-4
9. Zhang T, Fields JZ, Opdenaker L, Otevrel T, Masuda E, Palazzo JP. et al. Survivin-induced Aurora-B kinase activation: A mechanism by which APC mutations contribute to increased mitoses during colon cancer development. Am J Pathol. 2010;177:2816-26
10. Takeshita M, Koga T, Takayama K, Ijichi K, Yano T, Maehara Y. et al. Aurora-B overexpression is correlated with aneuploidy and poor prognosis in non-small cell lung cancer. Lung Cancer. 2013;80:85-90
11. Wang B, Hsu CJ, Chou CH, Lee HL, Chiang WL, Su CM. et al. Variations in the AURKA Gene: Biomarkers for the Development and Progression of Hepatocellular Carcinoma. Int J Med Sci. 2018;15:170-5
12. Zhao Y, Hu X, Wei L, Song D, Wang J, You L. et al. PARP10 suppresses tumor metastasis through regulation of Aurora A activity. Oncogene. 2018
13. Wang LL, Jin XH, Cai MY, Li HG, Chen JW, Wang FW. et al. AGBL2 promotes cancer cell growth through IRGM-regulated autophagy and enhanced Aurora A activity in hepatocellular carcinoma. Cancer Lett. 2018;414:71-80
14. Lin ZZ, Jeng YM, Hu FC, Pan HW, Tsao HW, Lai PL. et al. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer. 2010;10:461
15. Tanaka S, Arii S, Yasen M, Mogushi K, Su NT, Zhao C. et al. Aurora kinase B is a predictive factor for the aggressive recurrence of hepatocellular carcinoma after curative hepatectomy. Br J Surg. 2008;95:611-9
16. Kim JH, Shin YC, Ko SG. Integrating traditional medicine into modern inflammatory diseases care: multitargeting by Rhus verniciflua Stokes. Mediators Inflamm. 2014;2014:154561
17. Moon JE, Shin JH, Kwon O, Kim JY. A Standardized Extract of Rhus verniciflua Stokes Protects Wistar Rats Against Lipopolysaccharide-Induced Acute Inflammation. J Med Food. 2015;18:1223-30
18. Kim JH, Jung CH, Jang BH, Go HY, Park JH, Choi YK. et al. Selective cytotoxic effects on human cancer cell lines of phenolic-rich ethyl-acetate fraction from Rhus verniciflua Stokes. Am J Chin Med. 2009;37:609-20
19. Padmavathi G, Roy NK, Bordoloi D, Arfuso F, Mishra S, Sethi G. et al. Butein in health and disease: A comprehensive review. Phytomedicine. 2017;25:118-27
20. Song Z, Shanmugam MK, Yu H, Sethi G. Butein and Its Role in Chronic Diseases. Adv Exp Med Biol. 2016;928:419-33
21. Lee DS, Jeong GS. Butein provides neuroprotective and anti-neuroinflammatory effects through Nrf2/ARE-dependent haem oxygenase 1 expression by activating the PI3K/Akt pathway. Br J Pharmacol. 2016;173:2894-909
22. Bai X, Ma Y, Zhang G. Butein suppresses cervical cancer growth through the PI3K/AKT/mTOR pathway. Oncol Rep. 2015;33:3085-92
23. Jung SK, Lee MH, Lim DY, Lee SY, Jeong CH, Kim JE. et al. Butein, a novel dual inhibitor of MET and EGFR, overcomes gefitinib-resistant lung cancer growth. Mol Carcinog. 2015;54:322-31
24. Cho SG, Woo SM, Ko SG. Butein suppresses breast cancer growth by reducing a production of intracellular reactive oxygen species. J Exp Clin Cancer Res. 2014;33:51
25. Lai YW, Wang SW, Chang CH, Liu SC, Chen YJ, Chi CW. et al. Butein inhibits metastatic behavior in mouse melanoma cells through VEGF expression and translation-dependent signaling pathway regulation. BMC Complement Altern Med. 2015;15:445
26. Liu H, Li W, Yu X, Gao F, Duan Z, Ma X. et al. EZH2-mediated Puma gene repression regulates non-small cell lung cancer cell proliferation and cisplatin-induced apoptosis. Oncotarget. 2016;7:56338-54
27. Liu H, Hwang J, Li W, Choi TW, Liu K, Huang Z. et al. A derivative of chrysin suppresses two-stage skin carcinogenesis by inhibiting mitogen- and stress-activated kinase 1. Cancer Prev Res (Phila). 2014;7:74-85
28. Sheng Y, Li W, Zhu F, Liu K, Chen H, Yao K. et al. 3,6,2',4',5'-Pentahydroxyflavone, an orally bioavailable multiple protein kinase inhibitor, overcomes gefitinib resistance in non-small cell lung cancer. J Biol Chem. 2014;289:28192-201
29. Thoresen SB, Campsteijn C, Vietri M, Schink KO, Liestol K, Andersen JS. et al. ANCHR mediates Aurora-B-dependent abscission checkpoint control through retention of VPS4. Nat Cell Biol. 2014;16:550-60
30. Yu X, Li W, Deng Q, You S, Liu H, Peng S. et al. Neoalbaconol inhibits angiogenesis and tumor growth by suppressing EGFR-mediated VEGF production. Mol Carcinog. 2017;56:1414-26
31. Li W, Gao F, Ma X, Wang R, Dong X, Wang W. Deguelin inhibits non-small cell lung cancer via down-regulating Hexokinases II-mediated glycolysis. Oncotarget. 2017;8:32586-99
32. Berasain C, Avila MA. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol. 2014;49:9-23
33. Hagel M, Miduturu C, Sheets M, Rubin N, Weng W, Stransky N. et al. First Selective Small Molecule Inhibitor of FGFR4 for the Treatment of Hepatocellular Carcinomas with an Activated FGFR4 Signaling Pathway. Cancer Discov. 2015;5:424-37
34. Goyal L, Muzumdar MD, Zhu AX. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res. 2013;19:2310-8
35. Bhat M, Sonenberg N, Gores GJ. The mTOR pathway in hepatic malignancies. Hepatology. 2013;58:810-8
36. Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K. et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med. 2016;375:1925-36
37. Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S. et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med. 2016;375:1738-48
38. Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L. et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016;6:740-53
39. Gonzalez-Loyola A, Fernandez-Miranda G, Trakala M, Partida D, Samejima K, Ogawa H. et al. Aurora B Overexpression Causes Aneuploidy and p21Cip1 Repression during Tumor Development. Mol Cell Biol. 2015;35:3566-78
40. Al-Khafaji AS, Davies MP, Risk JM, Marcus MW, Koffa M, Gosney JR. et al. Aurora B expression modulates paclitaxel response in non-small cell lung cancer. Br J Cancer. 2017;116:592-9
41. Bufo P, Sanguedolce F, Tortorella S, Cormio L, Carrieri G, Pannone G. Expression of mitotic kinases phospho-aurora A and aurora B correlates with clinical and pathological parameters in bladder neoplasms. Histol Histopathol. 2010;25:1371-7
42. Yu J, Zhou J, Xu F, Bai W, Zhang W. High expression of Aurora-B is correlated with poor prognosis and drug resistance in non-small cell lung cancer. Int J Biol Markers. 2018. 1724 600817753098
43. Yu X, Liang Q, Liu W, Zhou L, Li W, Liu H. Deguelin, an Aurora B Kinase Inhibitor, Exhibits Potent Anti-Tumor Effect in Human Esophageal Squamous Cell Carcinoma. EBioMedicine. 2017;26:100-11
44. Yang EB, Guo YJ, Zhang K, Chen YZ, Mack P. Inhibition of epidermal growth factor receptor tyrosine kinase by chalcone derivatives. Biochim Biophys Acta. 2001;1550:144-52
45. Yang EB, Zhang K, Cheng LY, Mack P. Butein, a specific protein tyrosine kinase inhibitor. Biochem Biophys Res Commun. 1998;245:435-8
46. Ma CY, Ji WT, Chueh FS, Yang JS, Chen PY, Yu CC. et al. Butein inhibits the migration and invasion of SK-HEP-1 human hepatocarcinoma cells through suppressing the ERK, JNK, p38, and uPA signaling multiple pathways. J Agric Food Chem. 2011;59:9032-8
47. Rajendran P, Ong TH, Chen L, Li F, Shanmugam MK, Vali S. et al. Suppression of signal transducer and activator of transcription 3 activation by butein inhibits growth of human hepatocellular carcinoma in vivo. Clin Cancer Res. 2011;17:1425-39
48. Moon DO, Kim MO, Choi YH, Hyun JW, Chang WY, Kim GY. Butein induces G(2)/M phase arrest and apoptosis in human hepatoma cancer cells through ROS generation. Cancer Lett. 2010;288:204-13
49. Li Y, Ma C, Qian M, Wen Z, Jing H, Qian D. Butein induces cell apoptosis and inhibition of cyclooxygenase2 expression in A549 lung cancer cells. Mol Med Rep. 2014;9:763-7
50. Tang YL, Huang LB, Lin WH, Wang LN, Tian Y, Shi D. et al. Butein inhibits cell proliferation and induces cell cycle arrest in acute lymphoblastic leukemia via FOXO3a/p27kip1 pathway. Oncotarget. 2016;7:18651-64
51. Falchook GS, Bastida CC, Kurzrock R. Aurora Kinase Inhibitors in Oncology Clinical Trials: Current State of the Progress. Semin Oncol. 2015;42:832-48
52. Kantarjian HM, Martinelli G, Jabbour EJ, Quintas-Cardama A, Ando K, Bay JO. et al. Stage I of a phase 2 study assessing the efficacy, safety, and tolerability of barasertib (AZD1152) versus low-dose cytosine arabinoside in elderly patients with acute myeloid leukemia. Cancer. 2013;119:2611-9
53. Collins GP, Eyre TA, Linton KM, Radford J, Vallance GD, Soilleux E. et al. A phase II trial of AZD1152 in relapsed/refractory diffuse large B-cell lymphoma. Br J Haematol. 2015;170:886-90
Author contact
![]() Corresponding authors: Feng Gao, Address: Department of Ultrasonography, The Third Xiangya Hospital of Central South University, Changsha, Hunan, 410013, P.R. China. Tel: +86-0731-88618401; E-mail: gf0731edu.cn and Wei Li, Address: Department of Radiology, The Third Xiangya Hospital of Central South University, Changsha, Hunan 410013, P.R. China. Tel: +86-0731-84805448; E-mail: liw3biocom
Corresponding authors: Feng Gao, Address: Department of Ultrasonography, The Third Xiangya Hospital of Central South University, Changsha, Hunan, 410013, P.R. China. Tel: +86-0731-88618401; E-mail: gf0731edu.cn and Wei Li, Address: Department of Radiology, The Third Xiangya Hospital of Central South University, Changsha, Hunan 410013, P.R. China. Tel: +86-0731-84805448; E-mail: liw3biocom