Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Marker proteins and origin of...
The mechanisms of cardiac...
The pro-fibrotic growth factors...
Signaling pathway involved in...
Transcription factors activated...
Nuclear factor of activated...
Potential therapeutic targets...
Future challenges
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(12):1645-1657. doi:10.7150/ijbs.28103 This issue Cite
Review
Cardiac fibrosis: new insights into the pathogenesis
Zhen-Guo Ma, Yu-Pei Yuan, Hai-Ming Wu, Xin Zhang, Qi-Zhu Tang ![]()
Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan 430060, RP China
Cardiovascular Research Institute of Wuhan University, Wuhan 430060, RP China
Hubei Key Laboratory of Cardiology, Wuhan 430060, RP China
Received 2018-6-25; Accepted 2018-8-2; Published 2018-9-7
Abstract

Cardiac fibrosis is defined as the imbalance of extracellular matrix (ECM) production and degradation, thus contributing to cardiac dysfunction in many cardiac pathophysiologic conditions. This review discusses specific markers and origin of cardiac fibroblasts (CFs), and the underlying mechanism involved in the development of cardiac fibrosis. Currently, there are no CFs-specific molecular markers. Most studies use co-labelling with panels of antibodies that can recognize CFs. Origin of fibroblasts is heterogeneous. After fibrotic stimuli, the levels of myocardial pro-fibrotic growth factors and cytokines are increased. These pro-fibrotic growth factors and cytokines bind to its receptors and then trigger the activation of signaling pathway and transcriptional factors via Smad-dependent or Smad independent-manners. These fibrosis-related transcriptional factors regulate gene expression that are involved in the fibrosis to amplify the fibrotic response. Understanding the mechanisms responsible for initiation, progression, and amplification of cardiac fibrosis are of great clinical significance to find drugs that can prevent the progression of cardiac fibrosis.
Keywords: Cardiac fibrosis, Cardiac fibroblast, TGF-β, Smad
Introduction
Cardiac fibrosis, characterized by the imbalance of extracellular matrix (ECM) production and degradation, which results in the accumulation of scar tissue, is closely associated with cardiac and endocrine diseases [1]. Irrespective of the initial cause, injury to the heart evokes sustained fibrotic response that results in distorted heart architecture and cardiac dysfunction. Cardiac fibrosis increases stiffness of left ventricle, and impedes contraction and relaxation of hearts. Cardiac fibrosis also impairs mechano-electric coupling, thus leading to arrhythmias [2,3]. Growth factors secreted by myofibroblasts directly induce hypertrophy of cardiomyocytes via a paracrine dependent-manner [4], which is another landmark of heart failure. The deposition of collagen and hypertrophy of cardiomyocytes lead to a relatively decreased capillary density, further promoting cardiac remodeling [5]. Currently, no evidence-based therapies show significant efficacy in cardiac fibrosis. The main reason is the underlying basis of fibrosis is still unclear. In this article, we aimed to provide a concise review of advances in pathogenesis of cardiac fibrosis.
Marker proteins and origin of fibroblasts
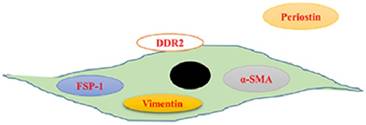
So far, there are no cardiac fibroblasts (CFs)-specific molecular markers. Most previous findings are obtained using co-labelling with panels of antibodies that can recognize CFs. We summarized the markers of CFs in Figure 1. Vimentin, a constituent of the intermediate-filament family of proteins, has been extensively studied as a marker of CFs. Vimentin has been used to identify the purity of isolated CFs in vitro. It can label CFs with high sensitivity, however, it also is expressed by endothelial cells and smooth muscle cells [6]. Upon stimuli, interstitial fibroblasts would express α-smooth muscle actin (α-SMA), a sign of fibroblasts activation [7]. Therefore, α-SMA is used to identify CFs. As α-SMA is also expressed in vascular smooth muscle cells [8], antibody-based strategy to detect this protein is difficult to interpret. S100A4, which is also known as fibroblast-specific protein 1 (FSP1), is specific for fibroblasts in the heart [9]. However, a study reported that FSP1-positive cells were just a small subset of CFs, most of FSP1-postive cells in interstitial fibrotic areas were CD45+ leukocytes [10]. Similar to FSP1, immunostaining of discoidin domain receptor (DDR) 2, a tyrosine kinase receptor, can only label a small part (about 27% as reported) of fibroblasts in the heart [11,12]. Periostin is a secreted protein and closely involved in cell adhesion. It has been reported that periostin specifically was expressed by all newly activated fibroblasts (myofibroblasts) upon heart injury without ectopic expression in other cardiac cell types [13]. However, the majority of periostin is secreted and deposited extracellularly, only a small proportion can be observed in intracellular (Figure 1) [13]. This fact restricts its utility in study fibroblasts [14]. Identification specific markers of CFs are of great importance to understand the mechanisms of cardiac fibrosis.
Schematic displaying the serviceable fibroblast markers. Vimentin is a constituent of the intermediate-filament family of proteins and used to identify the purity of isolated cardiac fibroblasts in vitro. Upon stimuli, interstitial fibroblasts would express α-smooth muscle actin (α-SMA), a sign of fibroblasts activation. Fibroblast-specific protein 1 (FSP1) is also specific for fibroblasts in the heart. Domain receptor (DDR) 2, a membrane tyrosine kinase receptor, can label a part of fibroblasts in the heart. Periostin is a secreted protein and specifically expressed by all newly activated fibroblasts (myofibroblasts) upon heart injury without ectopic expression in other cardiac cell types.
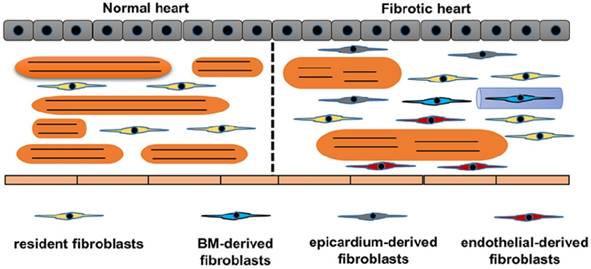
Another significant issue in studying fibroblasts is heterogeneous origins of fibroblasts. We summarized the origin of CFs in Figure 2. The traditional attitude held is that cardiac fibrosis is the result of proliferation and activation of resident fibroblasts, which is supported by the study that transforming growth factor (TGF)-β induced the proliferation and activation of isolated fibroblasts [15]. Further study using a transgenic collagen1α1-GFP reporter mice provided more evidence. Moore-Morris et al found that pressure overload resulted in activation and proliferation of these resident lineages [10]. This notion has been challenged by the following findings: fibroblasts rapidly transformed into myofibroblasts even without stimuli in vitro experiment; and proliferating fibroblast-like cells only existed in the vicinity of the vessels [16,17]. In addition to the activation of resident CFs, epithelial cells also contributed to the total pool of CFs through an endothelial-mesenchymal transition (EndMT)[18,19]. In this study, the authors used Tie1 to identify endothelial cells and used FSP1 to label CFs, and found that 27% to 35% of all (α-SMA+ or FSP1+) fibroblasts were generated from endothelial cells [18,19]. However, Tie1 could label both endothelial and immune lineages. Indeed, other genetic fate mapping studies using different endothelial-specific markers, for example fetal liver kinase 1 (Flk1) or VE-Cadherin, showed limited contribution of endothelial to total pool of CFs in the normal heart [20,21]. The contribution of EndMT to cardiac fibrosis need to be further clarified. The epicardium-derived fibroblasts have been summarized by a review [22]. Zhou et al. further found that epicardium-derived cells migrated into the region of the forming annulus fibrosis using Cre-LoxP technology in the normal mammalian hearts [23]. Epicardium-derived cells occurred in ischemic hearts, but was less apparent in hypertensive heart disease [24]. In the damaged hearts, activation of the epicardium and cardiac fibroblasts by Wnt1/β-catenin promoted cardiac repair [25]. Cardiac fibroblasts also originate from the bone marrow. One study reported that bone marrow-derived cells constituted up to 24% of all myofibroblasts in the infarct hearts [26]. However, this view has been challenged by another study in which the authors used Vav-Cre labeled over 95% of CD45+ immune cells in hearts, and did not show any contribution from bone marrow during pressure overload [27]. Further fate-mapping studies focusing on the contribution of bone marrow-derived CFs would be of interest.
Origins of cardiac fibroblasts. Schematic demonstrates the origins of fibroblasts in the heart. Normal heart only has resident fibroblasts. Upon stimuli, resident fibroblasts proliferates. Endothelial cells, bone marrow cells and epicardium contribute to the total pool of fibroblasts. In response to stimuli, cardiomyocytes become hypertrophy and muscle fibers become disarranged.
The mechanisms of cardiac fibrosis
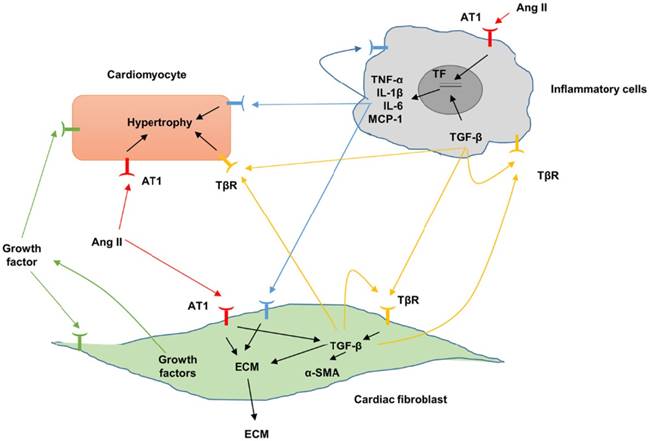
Briefly, the fibrotic response can be divided into three phases: the initiating phase, the effective phase and amplificative phase. After stimuli, the levels of circulating and myocardial pro-fibrotic growth factors and cytokines are increased, triggering the fibrotic response [28]. In the effective phase, this pro-fibrotic growth factors and cytokines bind to their receptors and then trigger the activation of signaling pathway and transcriptional factors including Smad, mitogen-activated protein kinases (MAPKs), protein kinase B (PKB, also called AKT) and nuclear factor kappa B (NF-κB). These pathological activations lead to the transformation of CFs into myofibroblasts, which would express the highly contractile protein α-SMA and produce a number of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs) to regulate homeostasis of ECM. Moreover, these pro-fibrotic transcriptional factors regulate the synthesis and secretion of pro-fibrotic growth factors and cytokines in CFs. The secreted growth factors and cytokines by CFs or other cells, for example, cardiomyocytes, endothelial cells and inflammatory cells, can function on CFs or cardiomyocytes, forming a positive feedback and eventually amplify the fibrotic response [28,29].
The pro-fibrotic growth factors and cytokines
TGF-β
Up to mow, TGF-β is the best-known fibrogenic growth factor during cardiac fibrosis. TGF-β exists in 3 isoforms (TGF-β1, TGF-β2 and TGF-β3) in mammals, which are encoded by 3 different genes [30,31], among which TGF-β1 is the predominant form. Previous studies in humans and experimental models have shown increased TGF-β expression during cardiac fibrosis [32,33]. Increased TGF-β binds to TGF-β receptor I (TβRI, also called ALK5) and the type II receptor TβRII to activate Smad2/3 pathway, thus promoting the process of cardiac fibrosis [34,35]. TGF-β is a pleiotropic mediator with potent and diverse effects on many cell types involved in cardiac fibrosis (Figure 3). TGF-β could induce the transformation from fibroblasts to myofibroblasts [36], and increase ECM gene expression, thus promoting ECM deposition. TGF-β also inhibited degradation of ECM by regulating the level of plasminogen activator inhibitor (PAI)-1 and TIMPs [37]. In addition, TGF-β was also involved in the synthesis and secretion of other profibrotic cytokines [38]. TGF-β receptors were expressed in almost all the inflammatory cells subsets, therefore TGF-β also had profound effects on inflammatory cells. TGF-β bidirectionally regulated function of macrophage depending on the cytokine microenvironment, differentiation status and the tissue origin of the cells. Intradermal or intraarticular injection of TGF-β induced monocyte infiltration and matrix deposition [39].TGF-β mediated monocytes adhesion via enhancing the expression of specific integrin [40]. In addition, TGF-β stimulated monocytes to secret a variety of profibrotic cytokines [41,42]. Inconsistent with these studies, TGF-β could suppress pro-inflammatory cytokines and chemokines synthesis [43]. TGF-β suppressed T cell proliferation by inhibiting interleukin (IL)-2 production [44,45]. TGF-β1 inhibited the T helper type 1 differentiation, which has been found to be profibrotic in our recent study using T-bet deficiency rat [46,47]. In addition, TGF-β also induced migration of mast cell [48]. Beyond its effects on inflammatory cells, TGF-β also controlled cardiomyocyte phenotype. TGF-β stimulation resulted in the hypertrophy of cardiomyocytes [49,50]. The precise role of TGF-β in endothelial cells and angiogenesis has proved to be dual, dependent on contextual and environmental cues [51,52]. We previously demonstrated that TGF-β1 induced endothelial-to-mesenchymal transition of human umbilical vein endothelial cells [53]. Heterozygous TGF- β1 deficient mice showed decreased age-associated myocardial fibrosis and improved cardiac compliance, indicating endogenous TGF-β may be involved in the pathogenesis of cardiac fibrosis [54]. Conversely, mice with TGF-β1 overexpression had significant ventricular fibrosis, with an increase in the fractional area of cardiac fibroblasts [55]. An orally available TGF-β receptor antagonist (NP-40208) attenuated myocardial fibrosis in mice with cardiac-restricted overexpression of tumor necrosis factor (TNF) [56]. Inconsistent with these studies, transgenic mice that carrying a mutated human TGF-β1 under the control of α-myosin heavy chain (MHC) promoter had a marked increase in active TGF-β1 levels in adult hearts and overt fibrosis only in the atria, implying that increased TGF-β1 activity by itself is insufficient to promote ventricular fibrosis in the adult mouse [57]. Of note, inhibition of TGF-β signaling via dominant negative mutation of the TGF-β type II receptor attenuated pressure overload-induced interstitial fibrosis, but promoted cardiac dysfunction [58], implying double-faced role of TGF-β in cardiac remodeling. These findings suggested that sustained and excessive TGF-β activation may be deleterious, whereas, a baseline level of TGF-β signaling or an early-responsive increase of TGF-β may protect the hearts from the acute injury.
Fibrotic signaling mechanisms between different cardiac populations. Fibrosis can be augmented by proinflammatory cytokines and profibrotic and molecules. Angiotensin II, Ang II; AT1, angiotensin II type 1; extracellular matrix, ECM; interleukin, IL; transforming growth factor-β, TGF-β; TGF-β receptor, TβR.
Angiotensin II (Ang II)
Ang II is a critical mediator of cardiac fibrosis (Figure 3). Ang II acts through 2 specific receptors: angiotensin type (AT) 1 and AT2. Elevated Ang II level was found in hypertrophic hearts with fibrosis. Both AT1 and AT2 have been shown to be increased during the process of cardiac fibrosis [59,60]. AT1 stimulation promoted the activation of profibrotic downstream effects, whereas activation of AT2 counteracted the profibrotic effects of AT1 [61,62]. Ang II enhanced human CFs growth, increased the secretion of TGF-β1, PAI-1 and ECM, and promoted CFs adhesion [63]. Ang II also induced the hypertrophy of cardiomyocytes, and enhanced the secretion of profibrotic growth factor by myocytes [64,65]. Ang II induced activation of inflammatory cells via direct chemotaxis and production of proinflammatory cytokine [66]. Study in vitro demonstrated that Ang II increased leukocyte adhesion to the endothelium [67]. The profibrotic effects of Ang II were confirmed by a number of studies. De Mello et al. found that chronic blockade of AT1 reduced fibrosis in the failing heart via increasing cell-to-cell communication [68]. Ang II blockade reversed cardiac fibrosis in a human hypertrophic cardiomyopathy model using transgenic mouse model without affecting myocyte disarray [69]. Losartan, an Ang II receptor antagonist decreased myocardial fibrosis in patients with end-stage renal disease [70]. Ang II-induced fibrotic response was mediated by TGF-β. Providing a support for this notion, a study reported Ang II was not able to induce cardiac fibrosis in vivo in TGF-β1-deficient mice, indicating that Ang II may not directly induce cardiac fibrosis in vivo, but indirectly promoting secretion of growth factors, which subsequently function on CFs in autocrine or paracrine dependent manners[71].
Connective tissue growth factor (CTGF)
CTGF, a cysteine-rich 36-38 kDa secreted protein, belongs to the CCN family and is also referred to as CCN2. Though CTGF is abundantly expressed in the fetal myocardium, its expression is restricted to the atria and large blood vessels in the adult heart [72,73]. Increased expression of CTGF were detected in the infarcted hearts [74]. Increased CTGF levels were observed in cardiac samples obtained from patients with heart failure and the CTGF-stained area correlated with myocardial fibrosis area [75]. In addition, as an immediate early gene, CTGF expression could be induced by TGF-β and Ang II in CFs and cardiomyocytes [76]. Though accumulating evidence demonstrated that CTGF induced the proliferation of fibroblasts, promoted the transformation from CFs to myofibroblasts and increased ECM production [77-80], CTGF is considered to have limited capacity to induce cardiac fibrosis. CTGF was to create a fibrotic environment and modulated the activity of growth factors in the ECM [81]. Providing evidence for this notion, cardiac-specific overexpression CTGF using myosin light chain-2 promoter did not induce cardiac fibrosis in murine or rat hearts [82]. Moreover, transgenic mice overexpressing CTGF had similar amount of fibrosis after cardiac injury compared to control mice [82-84]. Consistent with these findings, Fontes et al. found that CTGF global knockout does not affect cardiac hypertrophy and fibrosis induced by chronic pressure overload [85]. However, there sounds quite a different voice that CTGF cardiac transgenic mice showed significantly increased fibrosis in response to pressure overload [86]. For the sake of deciphering the role of CTGF, It would be certainly interesting to manipulate the expression of CTGF specifically in CFs.
Endothelin-1 (ET-1)
ET-1, a protein secreted from endothelial cells, has been shown to play a key role in cardiac remodeling. ET-1 binds to ET receptor A (ETA) and ETB to act a powerful vasoconstrictor. ET-1 could induce cardiac fibroblast proliferation, enhances types I and III collagen synthesis, resulting in myofibroblast differentiation [87,88]. ET-1 was also involved in the fibrotic responses of TGF-β [89]. The role of ET-1 in cardiac fibrosis was evident by a study that an ETA receptor antagonist, BQ123, blocked the collagen synthesis response in cardiac fibroblasts induced by TGF-β [90].
Inflammatory cytokine
Upon cardiac injury, the inflammatory signaling molecules immediately increased. The inflammatory factors could be released by inflammatory cells, CFs or even cardiomyocytes [91]. The levels of TNF-α, IL-6, and IL-1β correlated with disease severity in patients with heart failure [92]. The role of TNF-α in cardiac fibrosis were confirmed by a study reported that TNF-α global knockout mice showed attenuated inflammatory response and cardiac fibrosis induced by pressure overload [93]. Conversely, cardiac-restricted overexpression of TNF-α induced cardiac fibrosis, eventually resulting in dilated cardiomyopathy [94]. Unexpectedly, TNF-α inhibition in clinical studies had no benefit in patients with HF [95]. Previous studies also demonstrated a controversial of role IL-1β in cardiac fibrosis. IL-1β could promote rat CFs migration [96]. Inconsistent with this study, IL-1β inhibited myofibroblast differentiation induced by TGF-β [97]. This observation was also supported by a study in vivo using mice with IL-1β deficiency. This study found that mice with IL-1β deficiency had reduced cardiac dysfunction, but exacerbated cardiac fibrosis [98]. However, the dispute is going to be settled by a clinical trial, in which the improvement in left ventricular function was observed in patients with rheumatoid arthritis after inhibition of IL-1 [99]. Current studies also demonstrated a pleiotropic role of IL-6 in cardiac fibrosis. IL-6 alone resulted in cardiac fibrosis [100]. In agreement with this view, Ma et al. reported that IL-6 produced by CFs that activated by macrophage is essential for Ang II-induced cardiac fibrosis [101]. Another study also supported the notion that IL-6 had a detrimental role during cardiac fibrosis, using IL-6 knockout mice the author found that genetic deletion of IL-6 attenuated myocardial fibrosis [102]. Contrary to the notion that IL-6 induced cardiac fibrosis, Lai and colleagues reported that IL-6 deficiency was not sufficient to attenuate cardiac fibrosis [103], and loss of IL-6 led to cardiac dysfunction via altering the ECM and blocking critical cell-cell interactions [104]. IL-6 may exert different biological functions during the different phase of cardiac fibrosis, therefore further exploration is still needed. Extensive evidence also implicates chemokines in the pathogenesis of cardiac fibrosis. Anti-monocyte chemotactic protein-1 (MCP-1) gene therapy attenuated interstitial fibrosis after myocardial infarction [105]. Absence of MCP-1 resulted in attenuated myofibroblasts accumulation and improved cardiac function [106]. Interestingly, cardiac overexpression of MCP-1 resulted in α-SMA-positive myofibroblasts at 14 days after myocardial infarction, and prevented cardiac dysfunction [107]. These inconsistent results indicated a bidirectional role of MCP-1 in cardiac fibrosis.
Signaling pathway involved in cardiac fibrosis
Smad-dependent and Smad-independent pathway
TGF-β binds to the type II receptor, which can phosphorylate the type I receptor. Phosphorylation of type I receptor leads to the recruitment and activation of receptor-regulated Smads (mainly Smad2 and Smad3). Smad2 and Smad3 are released from the receptors and bind to Smad4 to form a heterotrimeric complex. This complex translocates into nucleus and recruits the related coactivators CREB-binding protein or p300 to regulate the transcription of target genes (Figure 4). Smad6 and Smad7 are inhibitory Smads, which can interact with type I receptor and competitively inhibit transcription of Smad2 and Smad3. Smad6 and Smad7 also promote degradation of active type I receptors.
Transforming growth factor-β (TGF-β) signaling pathway through Smad-dependent and Smad-independent pathways. TGF-β induced the translocation of Smad2 and Smad3 complex into nucleus to amplify the fibrotic response. Apart from this, MAPK, AKT and AMPK were also activated during fibrotic response.
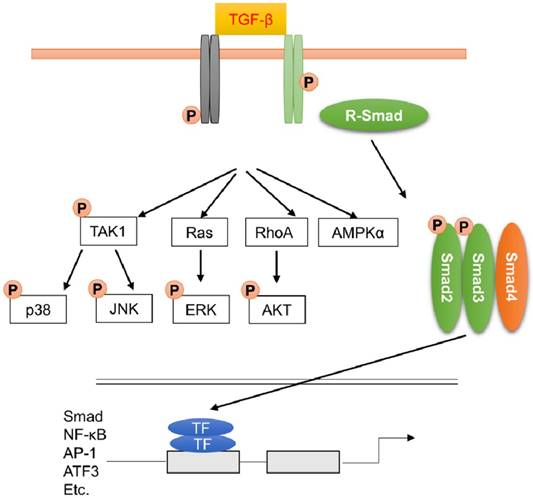
The essential role of Smad3 in fibrotic remodeling has been confirmed by a study using Smad3-null mice. Bujak and the colleagues found that mice with Smad3 deficiency had reduced fibrotic remodeling after myocardial infarction. Unexpectedly, increased myofibroblast density was observed in Smad3 knockout mice due to the abrogation of Smad3-dependent antiproliferative effects on CFs [28,108]. In some cases, TGF-β-mediated signaling pathway could be rapidly activated, implying that this activation was not the result of Smad-dependent transcription response [109]. TGF-β also activated other signaling cascades, including MAPKs (Figure 4). Activation of MAPKs after TGF-β treatment even in Smad4-deficient cells, or cell with dominant-negative Smads, implying that the activation of MAPKs was not Smads-dependent [110]. The activated type II receptor resulted in the activation of TGF-β-activated kinase 1 (TAK1), which then acted on MKK3/6 or MKK4 and led to the phosphorylation of JNK or p38, respectively [111]. Zhang et al. found that cardiomyocytes-specific overexpression of TAK1 resulted in myocyte disorganization and interstitial fibrosis [112]. The profibrotic effect of p38 was supported by a study found that transgenic mice with cardiac-specific expression of dominant negative forms of p38α and p38β were resistant to cardiac fibrosis in response to pressure overload [113]. Using inducible periostin knock-in Cre mice, Molkentin et al found that fibroblast-specific genetic depletion of p38 blocked CFs differentiation into myofibroblasts and reduced fibrosis in response to ischemic injury through the transcription factor serum-response factor (SRF) and the signaling effector calcineurin [114]. TGF-β also induced the activation of extracellular regulated protein kinases (ERK) via the small GTPase Ras or direct phosphorylation of ShcA [115]. Recent study also found that nonmyocyte ERK signaling promoted load-induced cardiac fibrosis in Marfan mice [116]. MAPKs also took in charge of the activation of TGF-β-Smads axis. ERK and JNK pathways could result in Smads phosphorylation [117,118]. TGF-β-induced activation of ERK resulted in the elevated expression of TGF-β, thereby amplifying the TGF-β response. Activation of p38 and JNK resulted in the activation of activating transcriptional factor 2 (ATF2), which is a Smad-interacting transcription factor [109].
AKT was also the downstream of TGF-β signaling pathway (Figure 4). TGF-β activated AKT pathway via a RhoA-dependent manner, and inhibition of AKT reduced Smad2 phosphorylation and transcription, and epithelial-to-mesenchymal transdifferentiation. Consistent with this, we also found that inhibition of AKT by piperine or leflunomide could suppress pressure overload or isoprenaline (ISO)-induced cardiac fibrosis [15,119]. Using two fibroblast-specific knockout mouse models, Lal et al found that CFs-specific deletion of glycogen synthase kinase 3 (GSK-3) β, a downstream of AKT, in CFs resulted in fibrogenesis, cardiac fibrosis and excessive scarring in the ischemic heart [120]. This study also showed that GSK-3β inhibited pro-fibrotic TGF-β-Smad3 signaling via interaction with Smad3.
AMP-activated protein kinase α (AMPKα) signaling pathway
AMPKα signaling pathway is also closely involved in the process of cardiac fibrosis. AMPKα could be activated by many profibrotic conditions, such as pressure overload, ischemia, TGF-β, Ang II and ISO. AMPKα also regulated signaling pathways that has been demonstrated to regulate fibrotic response. AMPKα attenuated cardiac fibrosis via a cross-talk with ERK in CFs [121]. AMPK inhibited TGF-β-induced Smad3-dependent transcription [122]. The results in our lab demonstrated that activation of AMPKα led to a decrease in the activation of ERK and p70 during fibrotic response [123]. AMPKα also phosphorylated β-catenin in mesenchymal stem cells [124], loss of which in resident cardiac fibroblasts attenuated cardiac fibrosis induced by pressure overload in mice [125]. The importance of AMPKα in the regulation of cardiac fibrosis has been confirmed using AMPKα knockout mice showing that deletion of AMPKα2 promoted fibrosis and decreased cardiac function [126]. Consistent with this, we also found that activation of AMPKα2 could protect pressure overload-induced cardiac fibrosis [123,127]. However, there sounds a quite opposite voice that AMPKα1, not AMPKα2, was involved in the process of cardiac fibrosis due to the reason that CFs mainly express the AMPKα1 catalytic isoform. This view was supported by the studies that AMPKα1 deficiency did not affect capillary density or inflammation in the infarcted myocardium but changed the fibrotic properties of CFs [128]. However, one fact cannot be ignored that the evidence that supporting the profibrotic effect of AMPKα is only circumstantial. Almost all the studies using pharmacological drugs, which could activate AMPKα combined with off-target effects. Fibroblast-specific manipulation of AMPKα catalytic isoform could end this question.
Wnt signaling
Wnt signaling controls the proliferation, differentiation and and migration of cells. The most understood Wnt-mediated signaling cascade is the canonical Wnt/β-catenin pathway. In the absence of Wnt ligands, cytosolic β-catenin is degraded by the destruction complex, which is composed of GSK-3β, the scaffolding proteins adenomatous polyposis coli (APC) and casein kinase (CK)-1α. Upon binding of Wnt proteins to Frizzled (Fz) receptor and lipoprotein receptor-related protein 5/6 (LRP5/6) coreceptor, cytoplasmic β-catenin is stabilized. β-Catenin then enters the nucleus and interacts with T-cell factor (TCF) to regulate Wnt-related genes [129-131]. The non-canonical Wnt signaling pathways act independently of β-catenin and LRP-5/6 and activate protein kinase C (PKC), calcium/calmodulin-dependent kinase II (CamKII) or calcineurin (CaN). The binding of a Wnt ligand resulted in the activation of Frizzled and Dishevelled, leading to activation of Rho/ROCK and Rac/JNK [132]. Several publications have indicated a crucial role for Wnt signaling in the development of cardiac fibrosis. Previous study revealed an association between canonical Wnt/β-catenin signaling and epicardial fibrosis of failed pediatric heart allografts with diastolic dysfunction [133]. Upon cardiac injury, the epicardium was activated in a Wnt-dependent manner, and underwent epithelial-mesenchymal transition to generate CFs [134]. Disruption of downstream Wnt signaling in epicardial cells decreased epicardial expansion, epithelial-mesenchymal transition and resulted in cardiac dysfunction and ventricular dilatation [135]. Wnt signaling pathway was also closely associated with the proliferation and transformation of CFs. Wnt 1 could induce CFs to proliferate and express pro-fibrotic genes [25]. Chronic activation of Wnt signaling induced transformation of fibroblasts into myofibroblasts and contributed to cardiac fibrosis in the aged heart [129]. Secreted Frizzled-related proteins (sFRPs) have emerged as key regulators of fibrotic response. CFs lacking endogenous sFRP-1 showed increased α-SMA, higher proliferation ability, and increased collagen accumulation [136]. sFRP2 is one of the best-known inhibitor of Wnt signaling. sFRP2 could inhibit cardiac fibrosis and improve cardiac function in infarcted hearts [137]. Lal et al. suggested a key role of GSK-3β in the process of fibrotic response. Lal and his colleagues achieved CF-specific deletion of GSK-3β by employing periostin-Cre mice and tamoxifen-inducible Col1α2-cre mice. Lal and his colleagues found that deletion of GSK-3β resulted in hyperactivation of pro-fibrotic TGF-β-Smad-3 signaling which causes excessive fibrosis [120]. Mechanistically, they found GSK-3β could interact with Smad-3. Recently, the role of β-catenin in resident and activated CFs after cardiac pressure overload were also examined. Pressure overload resulted in increased β-catenin signaling in CFs, and fibroblast-specific loss of β-catenin led to improved cardiac function, reduced interstitial fibrosis and decreased expression of fibrotic ECM protein genes via directly regulating the gene expression of Col1α1, Col3α1, and periostin [125].
Transcription factors activated in the fibrosis
Smad
After TGF-β binds to its ligand, activated TGF-β receptor phosphorylates Smad2 and Smad3, which interact with Smad4, resulting in the translocation into the nucleus to control the level of targeted genes. To precisely control the transcription, Smad associated with coactivators and corepressors. The coactiavtors included CBP/p300, SMIF, MSG1 and ARC105. The corepressors currently found were c-Ski, cMyc, ATF3, SNIP1, SIP1, Tob. Smad complexes bind to specific DNA sequences in the promoters or enhancers of target genes. The Smad binding element (SBE), which binds Smad complexes via MH1 domains, was first identified in the PAI-1 promoter [138,139]. The DNA binding site of Smad is defined as the sequence 5′-AGAc-3′ or its reverse complement 5′-GTcT-3′[140]. This short SBE only allow low-affinity binding, and interaction with coactivators and corepressors is required for the specific recruitment of Smads to response elements [141]. The targeted genes of Smad complex included PAI-1, junB [142], collagen 3 [143].
Peroxisome proliferators-activated receptor gamma (PPAR-γ) signaling pathway
PPAR-γ was also closely involved in many fibrotic responses, including hypertension, atherosclerosis, HF, diabetic cardiomyopathy. A PPAR-γ agonist significantly reduced the expression of TGF-β [144]. PPAR-γ agonists inhibited TGF-β-driven myofibroblasts differentiation and collagen I protein production [145]. Activation of PPAR-γ negatively regulated the activities of other fibrosis-related transcription factors, such as activated protein-1 (AP-1), NF-κB, nuclear factor of activated T-cells (NFAT) or signal transducer and activator of transcription (STAT) via a ligand-dependent manner [146]. Treatment with the PPAR-γ activators resulted in the reduction of ECM deposition and cardiac fibrosis [147,148]. Consistent with these studies, we also found that pioglitazone reduced pressure overload-induced cardiac fibrosis [149]. However, there was a different voice that pioglitazone inhibited pressure overload-induced cardiac remodeling even in heterozygous PPAR-γ-deficient mice, suggesting that pioglitazone inhibited fibrotic response was not dependent on PPAR-γ [150]. In addition, Duan et al found that rosiglitazone causes cardiac hypertrophy at least partially independent of PPAR-γ in cardiomyocytes [151]. Later studies demonstrated that the side effects of thiazolidinedione are largely attributed to its high affinity for PPAR-γ and overactivation of the PPAR-γ pathway [152,153]. Therefore, drugs that can activated moderately PPAR-γ would be of great significance. In our previous study, we found that piperine could reduce pressure overload or ISO-caused cardiac fibrosis without any side effect via modestly activating PPAR-γ [154].
Activating protein-1 (AP-1)
AP-1 proteins are homodimers and heterodimers composed of basic region-leucine zipper (bZIP) proteins, which were composed of Jun, Fos, and Jun dimerization partners and activating transcription factors [155]. Pressure overload, Ang II, and hypoxia could activate AP-1 in CFs [156,157]. Potential AP-1 binding sites have been identified in many signaling molecules that were closely associated with fibrosis, including transcription factors, ECM proteins, MMPs. In cardiac fibroblasts, the potential target genes include collagen [158], fibronectin [159], intercellular cell adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) [160].
NF-κB
NF-κB is one of the most well-known transcription factors that control a number of inflammation-related genes. The mammalian NF-κB super family consists of 5 genes encoding RelA, RelB, c-Rel, p50, and p52. NF-κB is composed as a heterodimer of these members, and regulates the transcription of genes that contain κB binding sites. In the absence of activating signal, NF-κB exists in the cytoplasm of cells and is bound by an inhibitory IκB family protein. The DNA-binding activity of NF-κB could be rapidly induced by proinflammatory factors in all the cells [161]. NF-κB can be activated by reactive oxygen species (ROS), hypoxia, hyperoxia, cytokines, growth factors which produced by profibrotic stimuli. These stimuli led to free NF-κB translocating into the nucleus and regulating the transcription of genes. NF-κB dimers bind a consensus DNA element called the “κB site” (5'-GGGRNWYYCC-3'; R, A or G; N, any nucleotide; W, A or T; Y, C or T). NF-κB directly regulated the expression fibrosis-related genes, including PAI-1, fibronectin and MMPs. Ang II-stimulated pro-fibrotic process was regulated by NF-κB activation [162]. NF-κB also suppressed the expression of miR-26, thus increasing collagen I and CTGF gene expression in the presence of Ang II [163]. NF-κB could regulate the mRNA levels of several profibrotic cytokines (TNF-α, IL-1β, IL-6, IL-10 and interferon-γ), cyclooxygenase-2 and inducible nitric oxide synthase, leukocyte adhesion molecules (ICAM-1, VCAM-1 and E-selectin) [164]. These factors further activated NF-κB, forming a positive feedback loop to amplify the fibrotic response.
ATF3
ATF3, a member of the activating transcription factor/cAMP responsive element-binding protein (ATF/CREB) family of transcription factors, binds to a consensus DNA sequence (TGACGTCA)[165,166]. ATF3 could be induced by profibrotic conditions, such as injury, ischemia, chemical toxin, Ang II and H2O2 [167,168]. Using transgenic mice with cardiac-specific expression of ATF3, Okamoto et al found that this mice exhibited fibrosis [169]. However, the finding in our study demonstrated that ATF3 deficiency promotes pathological hypertrophy and fibrosis [170].The first possibility for the discrepancy due to consistent high expression level of ATF3 in the transgenic mice. This hypothesis was supported by another study that adenovirus-mediated expression of ATF3 could inhibit doxorubicin-induced cardiomyocytes apoptosis [171]. A recent study reported using a miRNA-aided/lentivirus to overexpress ATF3 in the cardiac fibroblast and using periostin-Cre mice to construct a fibroblast-specific ATF3 deficiency mice found that ATF3 protected against cardiac fibrosis via suppression of Map2K3 expression and p38 MAPK signaling [172].
Myocardin-related transcription factor/SRF
SRF, is a globally expressed transcription factor, which regulates cardiac differentiation and maturation. SRF regulates gene expression via binding to CArG box [CC(A/T)6GG] [173]. Specificity of gene activation by SRF is regulated by its interactions with tissue-specific or signal-responsive transcriptional coregulators [174]. One of the best-known cofactors responsible for this activity is myocardin-related transcription factor (MRTF) family. The activity of MRTF is regulated by variations in the cellular concentration of G-actin. TGF-β1 could induce the nuclear localization and activity of MRTF via modulating Rho-mediated actin dynamics [175]. In addition, MRTFs and Smad3 synergistically activated CArG element-containing promoters during myofibroblasts activation [176]. Exogenous expression of MRTF-A in fibroblasts or epithelial cells could induce phenotypic transformation into myofibroblasts [177,178]. Forced expression of MRTF-A in cardiac fibroblasts promoted the production of robust SMA-positive fibers [174]. Conversely, genetic deletion of MRTF-A in mice led to reduced scar formation following ischemic injury or Ang II treatment via reducing SMA-positive myofibroblasts and suppressed fibrosis-associated genes [175]. SRF overexpression promoted cardiac fibrosis and increases CTGF expression by inducing miR-133a expression [179]. Currently, mice with CF-specific depletion of SRF has not yet been reported, therefore, future study focusing this area would be of great importance. Esnault et al. showed that MMP-2, MMP-9, and MMP-14 were all MRTF/SRF targets in fibroblasts [180]. MRTF-A/SRF directly regulated Col1α2 gene expression to promote cardiac fibrosis [175].
Nuclear factor of activated T-cells
Sustained, low-level calcium influx resulted in the activation of calcineurin, which dephosphorylated NFATc family members and promoted nuclear translocation to regulate gene expression [181]. Mechanical stress and alteration in intracellular calcium levels could activate NFAT in fibroblasts. Activated NFAT regulated the gene expression of collagen III and MRTF-A in CFs [182,183]. Activation of NFAT via calcineurin overexpression was sufficient to induce myofibroblasts differentiation both in vivo and in vitro [184,185]. Further study using mice with CFs-specific depletion of NFAT to investigate the precise role of NFAT would be of great interest.
Potential therapeutic targets for cardiac fibrosis
Pirfenidone is a small molecule that can inhibit TGF-β activity. Pirfenidone has been approved for idiopathic pulmonary fibrosis in Japan [186]. However, in a mouse model of myocardial infarction, TGF-β antibody treatment resulted in increased mortality and worsened ventricular remodeling[187], suggesting that broad targeting of TGF-β activity might not be a viable antifibrotic strategy. Drugs that inhibit the angiotensin pathway (angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists) have been widely used in clinical for the treatment of heart failure. In a human experiment, patients with hypertension and hypertrophy had significant regression of fibrosis and increased cardiac function after a 6-month course of lisinopril treatment[188], implying that angiotensin pathway antagonists may be useful approaches to control fibrotic disease. There are strong in vivo evidence directly that support the key roles of inflammatory factors in development of cardiac fibrosis. Tanercept, an anti-TNF-α agent, prevented TNF-α-mediated cardiac fibrosis, but unexpectedly augmented TNF-α cytotoxicity [189]. Smad3 signaling plays an essential role in fibrotic response. Bujak and the colleagues found that mice with Smad3 deficiency had reduced fibrotic remodeling. Unexpectedly, increased myofibroblast density was also observed in Smad3 knockout mice [28,108], suggesting that targeting Smad3 might not be a viable antifibrotic strategy. AMPKα activation might be considered as an appropriate therapy for the fibrosis. The finding from our lab found that activation of AMPKα prevented cardiac fibrosis induced by pressure overload in mice [190]. Moreover, metformin, a best-known AMPKα activator, has been safely used in clinical for a long time. In addition, the development of specific inhibitors of MRTF/SRF signaling, such as CCG-1423 104 and CCG-203971, is of particular interest [191,192]. CCG-1423 inhibited TGF-β induced myofibroblasts differentiation in a fibrosis model [193]. Future study should validate safety and efficacy of the specific inhibitors in the clinical cardiac fibrosis setting.
Future challenges
Although fibrosis has been documented in several cardiac conditions, the mechanisms responsible for this disease remain unknown. Moreover, our knowledge about the mechanisms of cardiac fibrosis predominantly came from experiments performed in cell-culture systems or global knockout mice. Understanding how the process of CF proliferation and transformation into myofibroblasts are regulated upon injury are hampered by the lack of suitable fibroblast markers and appropriate lineage mapping tools. Another decade may have to elapse before a satisfactory level of understanding about the mechanism of cardiac fibrosis is achieved.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No: 81470516, 81500184 and 81700254), the Key Project of the National Natural Science Foundation (No. 81530012), the Fundamental Research Funds for the Central Universities (No. 2042017kf0085), National Major Scientific Instrument and Equipment Development Projects (2013YQ03092306) and Development Center for Medical Science and Technology National Health and Family Planning Commission of the People's Republic of China (The prevention and control project of cardiovascular disease, 2016ZX-008-01).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83:1849-65
2. Banerjee I, Yekkala K, Borg TK, Baudino TA. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann N Y Acad Sci. 2006;1080:76-84
3. Chilton L, Giles WR, Smith GL. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol. 2007;583:225-36
4. Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103-13
5. Sabbah HN, Sharov VG, Lesch M, Goldstein S. Progression of heart failure: a role for interstitial fibrosis. Mol Cell Biochem. 1995;147:29-34
6. Mork C, van Deurs B, Petersen OW. Regulation of vimentin expression in cultured human mammary epithelial cells. Differentiation. 1990;43:146-56
7. Weber KT. Monitoring tissue repair and fibrosis from a distance. Circulation. 1997;96:2488-92
8. Azuma K, Ichimura K, Mita T. et al. Presence of alpha-smooth muscle actin-positive endothelial cells in the luminal surface of adult aorta. Biochem Biophys Res Commun. 2009;380:620-6
9. Strutz F, Okada H, Lo CW. et al. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393-405
10. Moore-Morris T, Guimaraes-Camboa N, Banerjee I. et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124:2921-34
11. Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883-91
12. Vogel WF, Abdulhussein R, Ford CE. Sensing extracellular matrix: an update on discoidin domain receptor function. Cell Signal. 2006;18:1108-16
13. Snider P, Hinton RB, Moreno-Rodriguez RA. et al. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res. 2008;102:752-60
14. Kanisicak O, Khalil H, Ivey MJ. et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260
15. Ma ZG, Yuan YP, Zhang X, Xu SC, Wang SS, Tang QZ. Piperine Attenuates Pathological Cardiac Fibrosis Via PPAR-gamma/AKT Pathways. EBioMedicine. 2017;18:179-87
16. Ljungqvist A, Unge G. The proliferative activity of the myocardial tissue in various forms of experimental cardiac hypertrophy. Acta Pathol Microbiol Scand A. 1973;81:233-40
17. Mandache E, Unge G, Appelgren LE, Ljungqvist A. The proliferative activity of the heart tissues in various forms of experimental cardiac hypertrophy studied by electron microscope autoradiography. Virchows Arch B Cell Pathol. 1973;12:112-22
18. Zeisberg EM, Tarnavski O, Zeisberg M. et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952-61
19. Liu Y, Hu ZF, Liao HH. et al. Toll-like receptor 5 deficiency attenuates interstitial cardiac fibrosis and dysfunction induced by pressure overload by inhibiting inflammation and the endothelial-mesenchymal transition. Biochim Biophys Acta. 2015;1852:2456-66
20. Alva JA, Zovein AC, Monvoisin A. et al. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235:759-67
21. Lugus JJ, Park C, Ma YD, Choi K. Both primitive and definitive blood cells are derived from Flk-1+ mesoderm. Blood. 2009;113:563-6
22. Fang M, Xiang FL, Braitsch CM, Yutzey KE. Epicardium-derived fibroblasts in heart development and disease. J Mol Cell Cardiol. 2016;91:23-7
23. Zhou B, von Gise A, Ma Q, Hu YW, Pu WT. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev Biol. 2010;338:251-61
24. Braitsch CM, Kanisicak O, van Berlo JH, Molkentin JD, Yutzey KE. Differential expression of embryonic epicardial progenitor markers and localization of cardiac fibrosis in adult ischemic injury and hypertensive heart disease. J Mol Cell Cardiol. 2013;65:108-19
25. Duan J, Gherghe C, Liu D. et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. Embo J. 2012;31:429-42
26. van Amerongen MJ, Bou-Gharios G, Popa E. et al. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J Pathol. 2008;214:377-86
27. Moore-Morris T, Guimaraes-Camboa N, Banerjee I. et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124:2921-34
28. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549-74
29. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation. 2015;131:1019-30
30. Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184-95
31. Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600-6
32. Kuwahara F, Kai H, Tokuda K. et al. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130-5
33. Villarreal FJ, Dillmann WH. Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am J Physiol. 1992;262:H1861-6
34. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685-700
35. Heldin CH, Miyazono K, Ten DP. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465-71
36. Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103-11
37. Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83-92
38. Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675-80
39. Luger TA, Charon JA, Colot M, Micksche M, Oppenheim JJ. Chemotactic properties of partially purified human epidermal cell-derived thymocyte-activating factor (ETAF) for polymorphonuclear and mononuclear cells. J Immunol. 1983;131:816-20
40. Wahl SM, Allen JB, Weeks BS, Wong HL, Klotman PE. Transforming growth factor beta enhances integrin expression and type IV collagenase secretion in human monocytes. Proc Natl Acad Sci U S A. 1993;90:4577-81
41. Wahl SM, Hunt DA, Wakefield LM. et al. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci U S A. 1987;84:5788-92
42. Turner M, Chantry D, Feldmann M. Transforming growth factor beta induces the production of interleukin 6 by human peripheral blood mononuclear cells. Cytokine. 1990;2:211-6
43. Ashcroft GS. Bidirectional regulation of macrophage function by TGF-beta. Microbes Infect. 1999;1:1275-82
44. Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392-404
45. Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99-146
46. Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med. 2002;195:1499-505
47. Ma ZG, Dai J, Yuan YP. et al. T-bet deficiency attenuates cardiac remodelling in rats. Basic Res Cardiol. 2018;113:19
48. Olsson N, Piek E, Sundstrom M, Ten DP, Nilsson G. Transforming growth factor-beta-mediated mast cell migration depends on mitogen-activated protein kinase activity. Cell Signal. 2001;13:483-90
49. Takahashi N, Calderone A, Izzo NJ, Maki TM, Marsh JD, Colucci WS. Hypertrophic stimuli induce transforming growth factor-beta 1 expression in rat ventricular myocytes. J Clin Invest. 1994;94:1470-6
50. Lim JY, Park SJ, Hwang HY. et al. TGF-beta1 induces cardiac hypertrophic responses via PKC-dependent ATF-2 activation. J Mol Cell Cardiol. 2005;39:627-36
51. Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600-6
52. Goumans MJ, Lebrin F, Valdimarsdottir G. Controlling the angiogenic switch: a balance between two distinct TGF-b receptor signaling pathways. Trends Cardiovasc Med. 2003;13:301-7
53. Liu Y, Hu ZF, Liao HH. et al. Toll-like receptor 5 deficiency attenuates interstitial cardiac fibrosis and dysfunction induced by pressure overload by inhibiting inflammation and the endothelial-mesenchymal transition. Biochim Biophys Acta. 2015;1852:2456-66
54. Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. J Mol Cell Cardiol. 2000;32:187-95
55. Rosenkranz S, Flesch M, Amann K. et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol. 2002;283:H1253-62
56. Zhang W, Chancey AL, Tzeng HP. et al. The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell-fibroblast interactions. Circulation. 2011;124:2106-16
57. Nakajima H, Nakajima HO, Salcher O. et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000;86:571-9
58. Lucas JA, Zhang Y, Li P. et al. Inhibition of transforming growth factor-beta signaling induces left ventricular dilation and dysfunction in the pressure-overloaded heart. Am J Physiol Heart Circ Physiol. 2010;298:H424-32
59. Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press. 2003;12:70-88
60. Busche S, Gallinat S, Bohle RM. et al. Expression of angiotensin AT(1) and AT(2) receptors in adult rat cardiomyocytes after myocardial infarction. A single-cell reverse transcriptase-polymerase chain reaction study. Am J Pathol. 2000;157:605-11
61. Rompe F, Artuc M, Hallberg A. et al. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappaB. Hypertension. 2010;55:924-31
62. Meffert S, Stoll M, Steckelings UM, Bottari SP, Unger T. The angiotensin II AT2 receptor inhibits proliferation and promotes differentiation in PC12W cells. Mol Cell Endocrinol. 1996;122:59-67
63. Kawano H, Do YS, Kawano Y. et al. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation. 2000;101:1130-7
64. Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977-84
65. Kamo T, Akazawa H, Komuro I. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ Res. 2015;117:89-98
66. Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertension. 2001;38:635-8
67. Kim JA, Berliner JA, Nadler JL. Angiotensin II increases monocyte binding to endothelial cells. Biochem Biophys Res Commun. 1996;226:862-8
68. De Mello WC, Specht P. Chronic blockade of angiotensin II AT1-receptors increased cell-to-cell communication, reduced fibrosis and improved impulse propagation in the failing heart. J Renin Angiotensin Aldosterone Syst. 2006;7:201-5
69. Lim DS, Lutucuta S, Bachireddy P. et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789-91
70. Shibasaki Y, Nishiue T, Masaki H. et al. Impact of the angiotensin II receptor antagonist, losartan, on myocardial fibrosis in patients with end-stage renal disease: assessment by ultrasonic integrated backscatter and biochemical markers. Hypertens Res. 2005;28:787-95
71. Schultz JJ, Witt SA, Glascock BJ. et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787-96
72. Chuva DSLS, Feijen A, Korving J. et al. Connective tissue growth factor expression and Smad signaling during mouse heart development and myocardial infarction. Dev Dyn. 2004;231:542-50
73. Friedrichsen S, Heuer H, Christ S, Cuthill D, Bauer K, Raivich G. Gene expression of connective tissue growth factor in adult mouse. Growth Factors. 2005;23:43-53
74. Ohnishi H, Oka T, Kusachi S. et al. Increased expression of connective tissue growth factor in the infarct zone of experimentally induced myocardial infarction in rats. J Mol Cell Cardiol. 1998;30:2411-22
75. Koitabashi N, Arai M, Kogure S. et al. Increased connective tissue growth factor relative to brain natriuretic peptide as a determinant of myocardial fibrosis. Hypertension. 2007;49:1120-7
76. Zhang J, Chang L, Chen C. et al. Rad GTPase inhibits cardiac fibrosis through connective tissue growth factor. Cardiovasc Res. 2011;91:90-8
77. Lam S, van der Geest RN, Verhagen NA. et al. Connective tissue growth factor and igf-I are produced by human renal fibroblasts and cooperate in the induction of collagen production by high glucose. Diabetes. 2003;52:2975-83
78. Duncan MR, Frazier KS, Abramson S. et al. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. Faseb J. 1999;13:1774-86
79. Daniels JT, Schultz GS, Blalock TD. et al. Mediation of transforming growth factor-beta(1)-stimulated matrix contraction by fibroblasts: a role for connective tissue growth factor in contractile scarring. Am J Pathol. 2003;163:2043-52
80. Grotendorst GR, Rahmanie H, Duncan MR. Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation. Faseb J. 2004;18:469-79
81. Liu S, Shi-wen X, Abraham DJ, Leask A. CCN2 is required for bleomycin-induced skin fibrosis in mice. Arthritis Rheum. 2011;63:239-46
82. Panek AN, Posch MG, Alenina N. et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. Plos One. 2009;4:e6743
83. Gravning J, Ahmed MS, von Lueder TG, Edvardsen T, Attramadal H. CCN2/CTGF attenuates myocardial hypertrophy and cardiac dysfunction upon chronic pressure-overload. Int J Cardiol. 2013;168:2049-56
84. Gravning J, Orn S, Kaasboll OJ. et al. Myocardial connective tissue growth factor (CCN2/CTGF) attenuates left ventricular remodeling after myocardial infarction. Plos One. 2012;7:e52120
85. Fontes MS, Kessler EL, van Stuijvenberg L. et al. CTGF knockout does not affect cardiac hypertrophy and fibrosis formation upon chronic pressure overload. J Mol Cell Cardiol. 2015;88:82-90
86. Yoon PO, Lee MA, Cha H. et al. The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J Mol Cell Cardiol. 2010;49:294-303
87. Piacentini L, Gray M, Honbo NY, Chentoufi J, Bergman M, Karliner JS. Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase C. J Mol Cell Cardiol. 2000;32:565-76
88. Shi-Wen X, Denton CP, Dashwood MR. et al. Fibroblast matrix gene expression and connective tissue remodeling: role of endothelin-1. J Invest Dermatol. 2001;116:417-25
89. Shi-wen X, Kennedy L, Renzoni EA. et al. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum. 2007;56:4189-94
90. Hafizi S, Wharton J, Chester AH, Yacoub MH. Profibrotic effects of endothelin-1 via the ETA receptor in cultured human cardiac fibroblasts. Cell Physiol Biochem. 2004;14:285-92
91. Kamo T, Akazawa H, Komuro I. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ Res. 2015;117:89-98
92. Testa M, Yeh M, Lee P. et al. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol. 1996;28:964-71
93. Sun M, Chen M, Dawood F. et al. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115:1398-407
94. Kubota T, McTiernan CF, Frye CS. et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res. 1997;81:627-35
95. Bozkurt B, Torre-Amione G, Warren MS. et al. Results of targeted anti-tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation. 2001;103:1044-7
96. Mitchell MD, Laird RE, Brown RD, Long CS. IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am J Physiol Heart Circ Physiol. 2007;292:H1139-47
97. Bronnum H, Eskildsen T, Andersen DC, Schneider M, Sheikh SP. IL-1beta suppresses TGF-beta-mediated myofibroblast differentiation in cardiac fibroblasts. Growth Factors. 2013;31:81-9
98. Honsho S, Nishikawa S, Amano K. et al. Pressure-mediated hypertrophy and mechanical stretch induces IL-1 release and subsequent IGF-1 generation to maintain compensative hypertrophy by affecting Akt and JNK pathways. Circ Res. 2009;105:1149-58
99. Ikonomidis I, Lekakis JP, Nikolaou M. et al. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117:2662-9
100. Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225-31
101. Ma F, Li Y, Jia L. et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. Plos One. 2012;7:e35144
102. Zhao L, Cheng G, Jin R. et al. Deletion of Interleukin-6 Attenuates Pressure Overload-Induced Left Ventricular Hypertrophy and Dysfunction. Circ Res. 2016;118:1918-29
103. Lai NC, Gao MH, Tang E. et al. Pressure overload-induced cardiac remodeling and dysfunction in the absence of interleukin 6 in mice. Lab Invest. 2012;92:1518-26
104. Banerjee I, Fuseler JW, Intwala AR, Baudino TA. IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am J Physiol Heart Circ Physiol. 2009;296:H1694-704
105. Hayashidani S, Tsutsui H, Shiomi T. et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2003;108:2134-40
106. Dewald O, Zymek P, Winkelmann K. et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881-9
107. Morimoto H, Takahashi M, Izawa A. et al. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ Res. 2006;99:891-9
108. Bujak M, Ren G, Kweon HJ. et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127-38
109. Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169-78
110. Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274:37413-20
111. Wang W, Zhou G, Hu MC, Yao Z, Tan TH. Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK) pathway by transforming growth factor beta (TGF-beta)-activated kinase (TAK1), a kinase mediator of TGF beta signal transduction. J Biol Chem. 1997;272:22771-5
112. Zhang D, Gaussin V, Taffet GE. et al. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556-63
113. Zhang S, Weinheimer C, Courtois M. et al. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Invest. 2003;111:833-41
114. Molkentin JD, Bugg D, Ghearing N. et al. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation. 2017;136:549-61
115. Lee MK, Pardoux C, Hall MC. et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. Embo J. 2007;26:3957-67
116. Rouf R, MacFarlane EG, Takimoto E. et al. Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice. JCI Insight. 2017:2
117. Funaba M, Zimmerman CM, Mathews LS. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J Biol Chem. 2002;277:41361-8
118. Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274:37413-20
119. Ma ZG, Zhang X, Yuan YP. et al. A77 1726 (leflunomide) blocks and reverses cardiac hypertrophy and fibrosis in mice. Clin Sci (Lond). 2018;132:685-99
120. Lal H, Ahmad F, Zhou J. et al. Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation. 2014;130:419-30
121. Du J, Guan T, Zhang H, Xia Y, Liu F, Zhang Y. Inhibitory crosstalk between ERK and AMPK in the growth and proliferation of cardiac fibroblasts. Biochem Biophys Res Commun. 2008;368:402-7
122. Mishra R, Cool BL, Laderoute KR, Foretz M, Viollet B, Simonson MS. AMP-activated protein kinase inhibits transforming growth factor-beta-induced Smad3-dependent transcription and myofibroblast transdifferentiation. J Biol Chem. 2008;283:10461-9
123. Ma ZG, Dai J, Wei WY. et al. Asiatic Acid Protects against Cardiac Hypertrophy through Activating AMPKalpha Signalling Pathway. Int J Biol Sci. 2016;12:861-71
124. Zhao J, Yue W, Zhu MJ, Sreejayan N, Du M. AMP-activated protein kinase (AMPK) cross-talks with canonical Wnt signaling via phosphorylation of beta-catenin at Ser 552. Biochem Biophys Res Commun. 2010;395:146-51
125. Xiang FL, Fang M, Yutzey KE. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun. 2017;8:712
126. Zhang P, Hu X, Xu X. et al. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918-24
127. Ma ZG, Dai J, Zhang WB. et al. Protection against cardiac hypertrophy by geniposide involves the GLP-1 receptor / AMPKalpha signalling pathway. Br J Pharmacol. 2016;173:1502-16
128. Noppe G, Dufeys C, Buchlin P. et al. Reduced scar maturation and contractility lead to exaggerated left ventricular dilation after myocardial infarction in mice lacking AMPKalpha1. J Mol Cell Cardiol. 2014;74:32-43
129. Naito AT, Shiojima I, Komuro I. Wnt signaling and aging-related heart disorders. Circ Res. 2010;107:1295-303
130. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781-810
131. Rao TP, Kuhl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res. 2010;106:1798-806
132. Veeman MT, Axelrod JD, Moon RT. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev Cell. 2003;5:367-77
133. Ye B, Ge Y, Perens G. et al. Canonical Wnt/beta-catenin signaling in epicardial fibrosis of failed pediatric heart allografts with diastolic dysfunction. Cardiovasc Pathol. 2013;22:54-7
134. Acharya A, Baek ST, Huang G. et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139-49
135. von Gise A, Pu WT. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ Res. 2012;110:1628-45
136. Sklepkiewicz P, Shiomi T, Kaur R. et al. Loss of secreted frizzled-related protein-1 leads to deterioration of cardiac function in mice and plays a role in human cardiomyopathy. Circ Heart Fail. 2015;8:362-72
137. He W, Zhang L, Ni A. et al. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci U S A. 2010;107:21110-5
138. Dennler S, Itoh S, Vivien D, Ten DP, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. Embo J. 1998;17:3091-100
139. Zawel L, Dai JL, Buckhaults P. et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611-7
140. Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970-82
141. Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970-82
142. Jonk LJ, Itoh S, Heldin CH, Ten DP, Kruijer W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem. 1998;273:21145-52
143. Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature. 1998;394:909-13
144. Kawai T, Masaki T, Doi S. et al. PPAR-gamma agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-beta. Lab Invest. 2009;89:47-58
145. Burgess HA, Daugherty LE, Thatcher TH. et al. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1146-53
146. Ahmadian M, Suh JM, Hah N. et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557-66
147. Tanaka T, Yamamoto J, Iwasaki S. et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U S A. 2003;100:15924-9
148. Gong K, Chen YF, Li P. et al. Transforming growth factor-beta inhibits myocardial PPARgamma expression in pressure overload-induced cardiac fibrosis and remodeling in mice. J Hypertens. 2011;29:1810-9
149. Wei WY, Ma ZG, Xu SC, Zhang N, Tang QZ. Pioglitazone Protected against Cardiac Hypertrophy via Inhibiting AKT/GSK3beta and MAPK Signaling Pathways. Ppar Res. 2016;2016:9174190
150. Asakawa M, Takano H, Nagai T. et al. Peroxisome proliferator-activated receptor gamma plays a critical role in inhibition of cardiac hypertrophy in vitro and in vivo. Circulation. 2002;105:1240-6
151. Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res. 2005;97:372-9
152. Elasy TA, Griffin M. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association: response to Nesto. Diabetes Care. 2004;27:2096 2096
153. Larsen PJ, Lykkegaard K, Larsen LK. et al. Dissociation of antihyperglycaemic and adverse effects of partial perioxisome proliferator-activated receptor (PPAR-gamma) agonist balaglitazone. Eur J Pharmacol. 2008;596:173-9
154. Ma ZG, Yuan YP, Zhang X, Xu SC, Wang SS, Tang QZ. Piperine Attenuates Pathological Cardiac Fibrosis Via PPAR-gamma/AKT Pathways. EBioMedicine. 2017;18:179-87
155. Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390-400
156. van Wamel JE, Ruwhof C, van der Valk-Kokshoorn EJ, Schrier PI, van der Laarse A. Rapid gene transcription induced by stretch in cardiac myocytes and fibroblasts and their paracrine influence on stationary myocytes and fibroblasts. Pflugers Arch. 2000;439:781-8
157. Sadoshima J, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. 1993;73:413-23
158. Weber KT, Swamynathan SK, Guntaka RV, Sun Y. Angiotensin II and extracellular matrix homeostasis. Int J Biochem Cell Biol. 1999;31:395-403
159. Moriguchi Y, Matsubara H, Mori Y. et al. Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-beta synthesis via transcriptional and posttranscriptional mechanisms. Circ Res. 1999;84:1073-84
160. Kacimi R, Karliner JS, Koudssi F, Long CS. Expression and regulation of adhesion molecules in cardiac cells by cytokines: response to acute hypoxia. Circ Res. 1998;82:576-86
161. Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221-7
162. Thakur S, Li L, Gupta S. NF-kappaB-mediated integrin-linked kinase regulation in angiotensin II-induced pro-fibrotic process in cardiac fibroblasts. Life Sci. 2014;107:68-75
163. Wei C, Kim IK, Kumar S. et al. NF-kappaB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol. 2013;228:1433-42
164. Valen G, Yan ZQ, Hansson GK. Nuclear factor kappa-B and the heart. J Am Coll Cardiol. 2001;38:307-14
165. Thompson MR, Xu D, Williams BR. ATF3 transcription factor and its emerging roles in immunity and cancer. J Mol Med (Berl). 2009;87:1053-60
166. Liang G, Wolfgang CD, Chen BP, Chen TH, Hai T. ATF3 gene. Genomic organization, promoter, and regulation. J Biol Chem. 1996;271:1695-701
167. Lv D, Meng D, Zou FF. et al. Activating transcription factor 3 regulates survivability and migration of vascular smooth muscle cells. Iubmb Life. 2011;63:62-9
168. Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1-11
169. Okamoto Y, Chaves A, Chen J. et al. Transgenic mice with cardiac-specific expression of activating transcription factor 3, a stress-inducible gene, have conduction abnormalities and contractile dysfunction. Am J Pathol. 2001;159:639-50
170. Zhou H, Shen DF, Bian ZY. et al. Activating transcription factor 3 deficiency promotes cardiac hypertrophy, dysfunction, and fibrosis induced by pressure overload. Plos One. 2011;6:e26744
171. Nobori K, Ito H, Tamamori-Adachi M. et al. ATF3 inhibits doxorubicin-induced apoptosis in cardiac myocytes: a novel cardioprotective role of ATF3. J Mol Cell Cardiol. 2002;34:1387-97
172. Li Y, Li Z, Zhang C. et al. Cardiac Fibroblast-Specific Activating Transcription Factor 3 Protects Against Heart Failure by Suppressing MAP2K3-p38 Signaling. Circulation. 2017;135:2041-57
173. Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35:577-93
174. Small EM. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res. 2012;5:794-804
175. Small EM, Thatcher JE, Sutherland LB. et al. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294-304
176. Qiu P, Feng XH, Li L. Interaction of Smad3 and SRF-associated complex mediates TGF-beta1 signals to regulate SM22 transcription during myofibroblast differentiation. J Mol Cell Cardiol. 2003;35:1407-20
177. O'Connor JW, Gomez EW. Cell adhesion and shape regulate TGF-beta1-induced epithelial-myofibroblast transition via MRTF-A signaling. Plos One. 2013;8:e83188
178. Elberg G, Chen L, Elberg D, Chan MD, Logan CJ, Turman MA. MKL1 mediates TGF-beta1-induced alpha-smooth muscle actin expression in human renal epithelial cells. Am J Physiol Renal Physiol. 2008;294:F1116-28
179. Angelini A, Li Z, Mericskay M, Decaux JF. Regulation of Connective Tissue Growth Factor and Cardiac Fibrosis by an SRF/MicroRNA-133a Axis. Plos One. 2015;10:e139858
180. Esnault C, Stewart A, Gualdrini F. et al. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014;28:943-58
181. Beals CR, Clipstone NA, Ho SN, Crabtree GR. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 1997;11:824-34
182. Herum KM, Lunde IG, Skrbic B. et al. Syndecan-4 signaling via NFAT regulates extracellular matrix production and cardiac myofibroblast differentiation in response to mechanical stress. J Mol Cell Cardiol. 2013;54:73-81
183. Gonzalez BL, Layne JJ, Nelson MT, Hill-Eubanks DC. Nuclear factor of activated T cells and serum response factor cooperatively regulate the activity of an alpha-actin intronic enhancer. J Biol Chem. 2005;280:26113-20
184. Molkentin JD, Lu JR, Antos CL. et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215-28
185. Lighthouse JK, Small EM. Transcriptional control of cardiac fibroblast plasticity. J Mol Cell Cardiol. 2016;91:52-60
186. Schelbert EB, Fonarow GC, Bonow RO, Butler J, Gheorghiade M. Therapeutic targets in heart failure: refocusing on the myocardial interstitium. J Am Coll Cardiol. 2014;63:2188-98
187. Frantz S, Hu K, Adamek A. et al. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. 2008;103:485-92
188. Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102:1388-93
189. Mann DL, Bozkurt B, Torre-Amione G, Soran OZ, Sivasubramanian N. Effect of the soluble TNF-antagonist etanercept on tumor necrosis factor bioactivity and stability. Clin Transl Sci. 2008;1:142-5
190. Zhang X, Ma ZG, Yuan YP. et al. Rosmarinic acid attenuates cardiac fibrosis following long-term pressure overload via AMPKalpha/Smad3 signaling. Cell Death Dis. 2018;9:102
191. Evelyn CR, Wade SM, Wang Q. et al. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6:2249-60
192. Bell JL, Haak AJ, Wade SM, Kirchhoff PD, Neubig RR, Larsen SD. Optimization of novel nipecotic bis(amide) inhibitors of the Rho/MKL1/SRF transcriptional pathway as potential anti-metastasis agents. Bioorg Med Chem Lett. 2013;23:3826-32
193. Sandbo N, Ngam C, Torr E, Kregel S, Kach J, Dulin N. Control of myofibroblast differentiation by microtubule dynamics through a regulated localization of mDia2. J Biol Chem. 2013;288:15466-73
Author contact
![]() Corresponding author: Qi-Zhu Tang, Department of Cardiology, Renmin Hospital of Wuhan University, Cardiovascular Research Institute, Hubei Key Laboratory of Cardiology, Wuhan University at Jiefang Road 238, Wuhan 430060, RP China. Tel.: +86 27 88073385; Fax: +86 27 88042292; E-mail: qztangedu.cn
Corresponding author: Qi-Zhu Tang, Department of Cardiology, Renmin Hospital of Wuhan University, Cardiovascular Research Institute, Hubei Key Laboratory of Cardiology, Wuhan University at Jiefang Road 238, Wuhan 430060, RP China. Tel.: +86 27 88073385; Fax: +86 27 88042292; E-mail: qztangedu.cn