Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(12):1686-1695. doi:10.7150/ijbs.27599 This issue Cite
Research Paper
Paired box 5 is a novel marker of breast cancers that is frequently downregulated by methylation
Xia Li1,3, Jianbo Huang1,2, Xinrong Luo1,2, Dejuan Yang1, Xuedong Yin1,2, Weiyan Peng1, Can Bi1, Guosheng Ren1,2 ![]() , Tingxiu Xiang1
, Tingxiu Xiang1 ![]()
1. Chongqing Key Laboratory of Molecular Oncology and Epigenetics, the First Affiliated Hospital of Chongqing Medical University, Chongqing, China;
2. Department of Endocrine and Breast Surgery, the First Affiliated Hospital of Chongqing Medical University, Chongqing, China
3. Department of Oncology, the First Affiliated Hospital of Chongqing Medical University, Chongqing, China
Received 2018-6-1; Accepted 2018-7-15; Published 2018-9-7
Abstract

Identifying markers for breast cancer is important for both diagnosis and the design of treatment strategies. Recent studies have implicated Paired box 5 (PAX5) as a suppressor in various cancer types, where it is silenced by hypermethylation. However, determining the role of PAX5 in breast cancer requires further study, and the relationship between PAX5 methylation and breast cancer remains unclear. In this study, we found that PAX5 expression was frequently silenced or reduced by methylation in breast cancer cell lines as well as in breast cancer tissues. Restoring expression of PAX5 in breast cancer cells led to tumor suppression through inhibited proliferation and invasion, which resulted from modulation of the cell cycle and altered vascular endothelial growth factor (VEGF) expression. Most importantly, we found that PAX5 methylation status in breast cancer tissues was significantly correlated with patients' age, estrogen receptor (ER) status, progesterone receptor (PR) status, indicating that PAX5 could serve as a marker for breast cancer diagnosis and treatment strategy design.
Keywords: PAX5, Breast cancer, biomarker
Introduction
Although the breast cancer cases vary worldwide, it remains the most common cancer as well as the leading cause of cancer-related death in women [1]. Despite progress in the diagnosis and treatment of breast cancer in recent decades, the mechanism of tumorigenesis remains unclear. Tumor suppressor silencing by promoter hypermethylation is thought to play a crucial role in breast cancer tumorigenesis and progression. Tumor suppressors that are silenced by aberrant promoter methylation in early stages of breast cancer progression, such as APC, CDH1, and CTNNB1, can be used as cancer biomarkers to aid in early detection [2]. The identification of novel genes silenced by aberrant promoter hypermethylation can therefore contribute to our understanding of the molecular mechanisms of breast cancer tumorigenesis, providing potential candidates for use in diagnosis and treatment. Our team has previously identified important tumor suppressors that are silenced by methylation in breast cancer [3-5].
The Paired box 5 (PAX5) gene, located on chromosome 9p13, belongs to a family of nuclear transcription factors termed the paired box genes, which consists of nine members [6]. PAX5 plays an important role as an activator during B lymphoid cell commitment. It is also known to be a commonly altered gene and target of genetic lesions in B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL) [6-8]. Previous studies have reported that the deletion of PAX5 in hepatocellular carcinoma [9], gastric carcinoma [10] and lung cancer [11] is associated with DNA promoter hypermethylation. Frequent aberrant methylation of PAX5 has also been reported in both female and male breast cancers [12-14]. PAX5 has also been identified as an inhibitor of epithelial-to-mesenchymal transition (EMT) progression in breast cancer cells [15, 16]. Recently, Branham et al. reported that the methylation state of PAX5 is a strong predictor of BRCAness tumors [17]. However, the relationship between PAX5 methylation and breast cancer, particularly the correlation between PAX5 methylation and the clinical parameters of breast cancer have yet to be determined. In this study, we find that promoter methylation can silence or reduce PAX5 expression in multiple breast cancer cell lines and primary tumors. Further functional studies demonstrated that the ectopic expression of PAX5 leads to tumor suppression via the inhibition of proliferation and invasion, a result of alterations in the cell cycle pathway and the expression of vascular endothelial growth factor (VEGF). These results suggest that PAX5 is a potential tumor suppressor in breast cancer. Most importantly, we find that PAX5 methylation status in breast cancer tissue is significantly correlated with age, estrogen receptor (ER) status, progesterone receptor (PR) status and breast cancer molecular subtype, indicating that PAX5 could be used as a biomarker in breast cancer diagnosis and treatment strategy design.
Materials and Methods
Cell lines, tumor samples and control tissues
Nine breast cancer cell lines (BT549, MDA-MB-231, MDA-MB-468, MCF-7, SK-BR-3, T47D, YYC-B1, YCC-B3 and ZR-75-1) were examined. All cell lines were maintained at 37°C in RPMI 1640 supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) and 100 U/ml of penicillin and streptomycin. Human breast tissue RNA samples were purchased commercially (Stratagene, La Jolla, CA, USA; Millipore Chemicon, Billerica, MA, USA and BioChain Institute, Hay-ward, CA, USA). Primary breast tumor samples, breast cancer adjacent tissues and breast normal tissues were obtained from the First Affiliated Hospital of Chongqing Medical University. Breast normal tissues were obtained from breast benign lesion tissues and were identified as breast normal tissues by pathologists. All samples were evaluated and histologically classified by pathologists. Clinical data were also collected for the majority of tumor cases. All patients provided informed consent to their participation in this study in order to ensure that their tissues were for research purposes only. Our research was approved by the Institutional Review Board of Chongqing Medical University.
Semi-quantitative RT-PCR analysis
Information about the relationship between PAX5 expression and survival rate of breast cancer patients was extracted from The Cancer Genome Atlas (TCGA) breast cancer database using GEPIA online software.
Total RNA was isolated from cell lines using TRI Reagent (Invitrogen, Carlsbad, CA, USA). Semi-quantitative RT-PCR was performed as described previously [3]. GAPDH was used as a control. Primer sequences used were as follows: PAX5-F: GTCCATTCCATCAAGTCCTG, PAX5-R: TTGCTGACACAACCATGGCT, GAPDH-F: GGAGTCAACGGATTTGGT, GAPDH-R: GTGATGGGATTTCCATTGAT. RT-PCR was performed with 32 cycles for PAX5, and 23 cycles for GAPDH using Go-Taq (Promega, Madison, WI, USA).
DNA bisulfite treatment and methylation-specific PCR (MSP)
Bisulfite treatment and methylation-specific PCR (MSP) were performed as described previously [11]. Bisulfite-treated DNA was amplified in order to evaluate the methylation status of PAX5 by MSP with primers PAX5-m1 and PAX5-m2 to detect methylated DNA, and PAX5-u1 and PAX5-u2 to detect unmethylated DNA. The methylation- specific primers used were as follows: m1: AAATAAAAATTCGGTTT GCGTTC, m2: AAACATACGCTTAAAAATCGCG. The unmethylation -specific primers used were as follows u1: TAAAAATAAAAATTTG GTTTGTGTTT, u2: TTAAAACATACACTTAAAAATCACA.
5-aza-2-deoxycytidine (Aza) and trichostatin A (TSA) treatment
Cells were treated with 10 µM 5-aza-2'-deoxycytidine (Aza; Sigma-Aldrich, St. Louis, MO, USA) for 3 days and then treated with 100 nM trichostatin A (TSA; Cayman Chemical Co, Ann Arbor, MI, USA) for an additional 1 day.
Quantitative RT-PCR analysis
Quantitative RT-PCR was performed using a SYBR® Green PCR Master Mix kit (Invitrogen) and the Applied Biosystem 7500 Real-time PCR System (Applied Biosystems, Foster City, CA, USA). Relative expression levels of PAX5 in breast tissues were standardized to β-actin levels. The primers used were as follows: PAX5-F: TGGCAGGTATTATGAGAC AGG, PAX5-R: CAGGCAAACATGGTGGGATT. β-actin F: GTCTTCC CCTCCATCGTG, β-actin R: AGGGTGAGGATGCCTCTCTT.
Cell culture and transfection
MDA-MB-231 and BT549 cell lines were cultured as previously described. Tumor cells were seeded in 6-well plates at a density of 1x105 cells per well. Lipofectamine-2000 (Invitrogen) was used to transfect NEG and PAX5 plasmids. G418 was used to select for PAX5 stably expressing cells 48 hrs post-transfection.
Cell viability assay
Tumor cells were seeded into 96-well plates at a density of 1×103 cells per well. Cell viability was determined at 24 hr, 48 hr and 72 hr time points with 450 nm absolution using a Cell Counting Kit-8 (CCK-8) assay (Beyotime Institute of Biotechnology, Jiangsu, China).
Flow Cytometry assay
MDA-MB-231 and BT549 cells were transfected as previously described. Transfection was stopped after 48 hrs and cells were digested using 0.1% trypsin. Cells were centrifuged at 4°C at 1000 rpm for 5 mins. The pellets were fixed in ice-cold 70% ethanol and stained with propidium iodide. Data were analyzed using the CELL Quest kit (BD Biosciences, San Jose, CA, USA).
Colony formation assay
Stably transfected cells were seeded in 6-well plates at a density of 200, 400 or 800 cells per well. The resulting colonies were fixed in 4% Paraformaldehyde, stained with crystal violet solution and counted under the microscope after 14 days.
Soft agar colony formation assay
Each 6-well plate was lined with a mix of 1 ml of media containing 0.7% low-melting-temperature agarose, 10% serum and 100 µg/ml penicillin and streptomycin. Cells were re-suspended in media containing 0.35% low-melting-temperature agarose, 10% serum and 100 µg/ml penicillin and streptomycin at a density of 1×104 cells/ml, and 1 ml of this solution was poured into each well once the bottom agar layer was polymerized. After 10 days, cell colonies ≥50 μm in size were detected and counted on an Axiovert 40 CFL microscope.
Wound healing assay
BT549 stably transfected cells were seeded in a 6-well plate. Once confluent, the cell layer was wounded, washed with PBS, then re-cultured in fresh complete media. Images were acquired following an additional 36 hrs of incubation.
Transwell assay
Cell culture well inserts were set in 24-well plates and coated with basement matrigel (BD Biosciences). Stably transfected cells were starved for 24 hrs and plated on inserts with 600 μl serum-free media. The bottom of the well was filled with 800 μl fresh complete media. After 24 hrs, the cells that passed through the membrane were fixed in 4% Paraformaldehyde, stained with crystal violet solution and detected on an Axiovert 40 CFL microscope.
In vivo tumor model
Stably transfected MDA-MB-231 cells and control cells (5×106 cells in 0.2 ml PBS) were hypodermically injected into the thigh flanks of female nude mice (aged 6 weeks old, six mice per group). Mice were weighed every 4 days from the 10th day post-injection onwards for 16 days. Mice were sacrificed on day 26 post-injection, and the weight of tumors was measured. The protocols for in vivo animal experimentation were approved by the Committee on the Ethical Use of Animals of the First Affiliated Hospital of Chongqing Medical University.
Immunohistochemistry (IHC)
Tumor tissue obtained from mice was embedded in paraffin. Antigen retrieval was performed by heating sections in Tris-EDTA buffer (pH=9) for 20 min in a microwave oven. Endogenous peroxidase activity was inhibited by incubating sections in 3% H2O2 for 15 min. Non-specific binding sites were blocked for 30 min. Tissue sections were incubated with primary antibodies (PAX5, Ki67, VEGF) overnight at 4°C. Sections were then incubated with secondary antibodies in PBST for 30 min at room temperature. Slides were developed for 5 min with 3-3'-diaminobenzidine (DAB) chromogen and counterstained with hematoxylin. Images were obtained on microscope.
Western blotting
Protein extraction from transfected cells was performed 48 hrs following transfection with a PAX5 plasmid or empty vector control. Cell lysates were then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer to polyvinylidene fluoride (PVDF) membranes. After blocking with 5% milk in TBST for 1 hr, membranes were incubated with PAX5 primary antibody (Abcam, Cambridge, UK) as well as primary antibodies against p53, p21, p27, cyclin D1, CDK4 (Cell Signaling Technology, Danvers, MA, USA) and VEGF (Abcam). Membranes were then incubated in secondary antibody solution, and immuno-blots were revealed using an enhanced chemiluminescence detection system. GAPDH (Epitomics, Burlingame, CA, USA) was used as a control.
Statistical Analysis
Statistical analyses were performed with SPSS version 17.0 (SPSS Inc., Chicago, IL). A student's t-test was used to analyze the differences in PAX5 expression between breast cancer tissues and surgical margin tissues, and the differences in biological behaviors of PAX5-expressing cells vs. controls. A chi-squared (χ2) or Fisher's exact test was used to determine the correlation between PAX5 methylation status and a given clinical parameter. For all statistical tests, p <0.05 was the cut-off for statistical significance.
Results
PAX5 expression is reduced in breast cancer cell lines by epigenetic regulation
By using the GEPIA online software, we analyzed breast cancer database in TGCA and we found that patients with low expression of PAX5 have less DFS than the patients who have high PAX5 expression (p< 0.05) (Supplementary Fig 1), meaning that PAX5 is a potential biomarker for breast cancer outcome.
We first tested the expression of PAX5 in 20 paired breast tumor tissues and adjacent tissues from patients by quantitative RT-PCR (Fig. 1A). The expression of PAX5 was significantly lower in breast tumors relative to adjacent tissues (p<0.05)(Fig. 1B). We then determined the expression level of PAX5 in breast cancer cell lines vs. normal breast tissue using semi-quantitative RT-PCR (Fig. 1C). Compared to breast normal tissues, PAX5 expression was more frequently silenced or reduced in breast cancer cell lines, implicating PAX5 as a potential tumor suppressor gene that is altered in breast cancer.
The expression and methylation status of PAX5 in breast cancer. (A) 20 pairs of primary breast cancer and adjacent tissues are tested by q-PCR. (B) The same data with (A) shows that the expression of PAX5 in breast cancer tissues is lower than the adjacent tissues (*, p<0.05). (C) The expression of PAX5 in breast cancer cell lines is detected by RT-PCR and the methylation status of PAX5 in breast cancer cell lines is detected by MSP. Methylation of PAX5 induced PAX5 silent or expression reduction in breast cancer cell lines. (D) Demethylation of PAX5 with Aza combined with TSA (A+T) restores PAX5 expression in breast cancer cell lines.
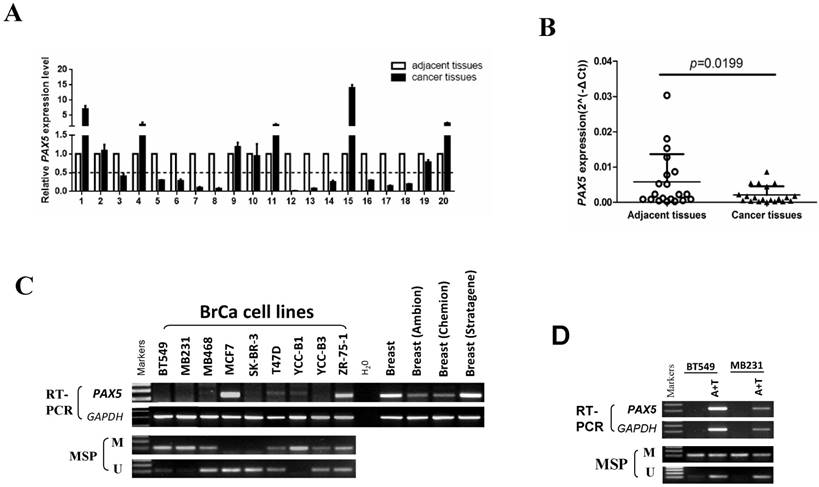
An MSP analysis revealed that PAX5 was hypermethylated in seven of nine breast cancer cell lines that display little or no PAX5 expression. To further investigate whether the observed downregulation of PAX5 is caused by methylation, we chose two hypermethylated cell lines to perform a de-methylation treatment with the agent Aza, in combination with TSA. PAX5 expression was restored in both two breast cancer cell lines following de-methylation treatment (Fig. 1D), suggesting that the downregulation of PAX5 in breast cancer is a result of hypermethylation.
PAX5 methylation status in breast cancer tissues is correlated with clinical parameters
In order to study the PAX5 methylation status in breast cancer, an MSP analysis was performed to examine 258 primary breast tumor tissues, 32 breast cancer adjacent tissues and 45 breast normal tissues (Fig 2). The methylation status of PAX5 in these conditions is shown in Table 1 and 2. PAX5 was methylated in 68.6% (177/258) of breast tumors, 50% (16/32) of breast cancer adjacent tissues and 18.4% (7/45) of normal breast tissues, showing a decreasing methylation grade from breast cancer tissues to breast normal tissues. We also examined the correlation between PAX5 methylation status and clinical parameters including age, clinical stage, tumor size, presence of lymph node metastasis, estrogen receptor status, progesterone receptor status, HER2 status, Ki67 status, p53 status. We found that PAX5 methylation status significantly correlated with age, estrogen receptor status (ER), progesterone receptor status (PR).
Clinicopatholog features of PAX5 methylation in breast cancer
| Clinicopathological features | Number (n=258) | PAX5 promoter methylated status | p Value | |||
|---|---|---|---|---|---|---|
| Methylated | Unmethylated | |||||
| Age | ||||||
| ≥55 | 102 | 79 | 23 | 0.0132 | ||
| <55 | 153 | 96 | 57 | |||
| Unknown | 3 | 2 | 1 | |||
| Stage | ||||||
| I | 55 | 34 | 21 | 0.2712 | ||
| II | 107 | 76 | 31 | |||
| III | 16 | 8 | 8 | |||
| IV | 4 | 2 | 2 | |||
| Unknown | 76 | 57 | 19 | |||
| Size | ||||||
| ≤2cm | 123 | 86 | 37 | 0.8781 | ||
| >2cm,≤5 | 93 | 63 | 30 | |||
| >5 | 29 | 21 | 8 | |||
| unknown | 13 | 7 | 6 | |||
| Lymph node metastasis | ||||||
| Positive | 99 | 69 | 30 | 0.3902 | ||
| Negative | 97 | 62 | 35 | |||
| unknown | 62 | 46 | 16 | |||
| Estrogen receptor status | ||||||
| Positive | 135 | 99 | 36 | 0.0334 | ||
| Negative | 97 | 58 | 39 | |||
| unknown | 26 | 20 | 6 | |||
| Progesterone receptor status | ||||||
| Positive | 111 | 83 | 28 | 0.0253 | ||
| Negative | 120 | 73 | 47 | |||
| unknown | 27 | 21 | 6 | |||
| HER2 status | ||||||
| Positive | 186 | 22 | 4 | 0.1091 | ||
| ++ | 43 | 72 | 34 | |||
| Negative | 29 | 61 | 36 | |||
| unknown | 29 | 22 | 7 | |||
| Ki67 status | ||||||
| <14% | 86 | 60 | 26 | 0.5522 | ||
| >14% | 120 | 79 | 41 | |||
| unknown | 52 | 38 | 14 | |||
| p53 status | ||||||
| Positive | 119 | 77 | 42 | 0.2353 | ||
| Negative | 94 | 68 | 26 | |||
| Unknown | 45 | 32 | 13 | |||
The methylation status of PAX5 in primary breast tumor samples, breast cancer adjacent tissues and breast normal tissues. As the representative results shown, the methylation status of PAX5 is tested in primary breast tumor samples, breast cancer adjacent tissues and breast normal tissues.
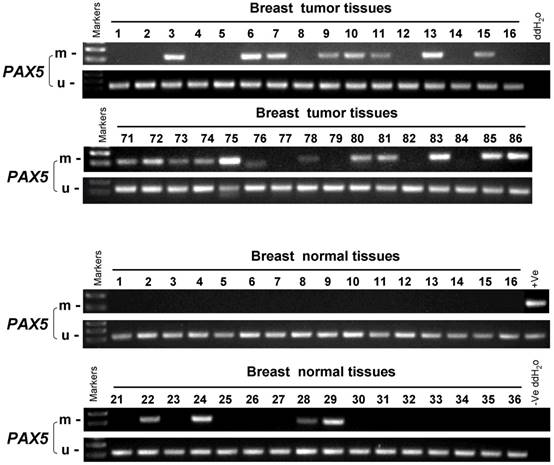
Methylation status of the PAX5 promoter in primary breast tumors
| Samples | PAX5 promoter | Frequency of methylation | |
|---|---|---|---|
| methylation unmethylation | |||
| BrCa (n= 258) | 177 | 81 | 68.6% |
| BA (n= 32) | 16 | 16 | 50% |
| BN (n= 45) | 7 | 38 | 18.4% |
Note: BrCa, breast cancer; BA, breast cancer adjacent (surgical margin tissues); BN, breast normal tissues
PAX5 inhibits proliferation by inducing cell cycle arrest in breast cancer cells
Our observation of PAX5 silencing by hypermethylation in breast cancer cells indicated that it may act as a tumor suppressor. We, therefore, examined the influence of PAX5 on tumor cell proliferation by restoring PAX5 expression in two PAX5 silenced cell lines (MDA-MB-231 and BT549). We validated the re-expression of PAX5 in transiently transfected cell lines by RT-PCR and western-blot (Fig. 3A). A CCK-8 assay revealed that ectopic PAX5 expression led to decreased cell viability at 48 hr and 72 hr time points compared to control vectors in both cell lines (p<0.01) (Fig. 3B). Additionally, a colony formation assay revealed that colony numbers were markedly reduced to 49% in MDA-MB-231 cells (p<0.01) and 15% in BT549 (p<0.01), relative to control cells (Fig. 3C). The inhibitory effect of PAX5 on tumor cell proliferation was further demonstrated by a soft agar colony formation assay in MDA-MB-231 cells. The number of resultant colonies (≥50 μm in size) from transient transfected cells was significantly decreased to 15% (p<0.01) relative to control transfected cells (Fig. 3D).
PAX5 inhibits cell proliferation by inducing cell cycle arrest in breast cancer cells. (A) After transfection of PAX5 plasmid and the control, the expression of PAX5 in BT549 and MB231 is detected by RT-PCR and Western-blot. (B) Inhibitory effect of PAX5 in breast cancer cell (BT549 and MB231) viability is detected by CCK-8 assays. (C) (D) The suppressive function of PAX5 on cell clone formation is tested respectively by clone formation assays and soft agar colony formation assays. (E) As the left representative illustration, PAX5 induced cell cycle G0/G1 phase arrest in breast cancer cells (BT549 and MB231). All the values are exhibited as the mean ± SD from three independent experiments (**, p < 0.01).
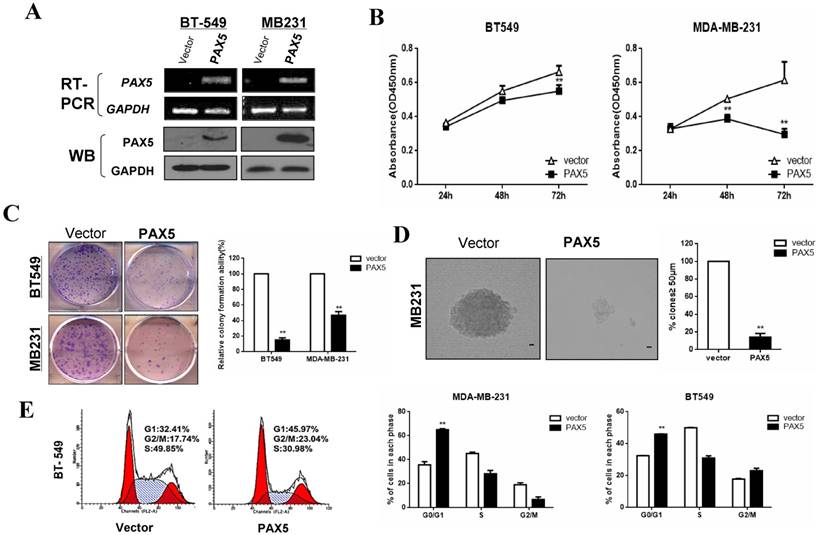
Additionally, we studied the effect of PAX5 on cell cycle regulation using flow cytometry. An increased number of cells were found in the G1 phase of PAX5-transfected cells compared to controls, in both MDA-MB-231 (35.73% vs. 65.09%, p<0.001) and BT549 cell lines (32.41% vs. 45.79%, p<0.01) (Fig. 3E). PAX5-transfected BT549 cells also displayed an increase in the number of cells in the G2 phase, compared to controls (17.74% vs. 23.04%, p<0.01). These results indicate that PAX5 is able to inhibit tumor cell proliferation by inducing cell cycle arrest in breast cancer cells.
PAX5 suppresses breast cancer cell migration and invasion
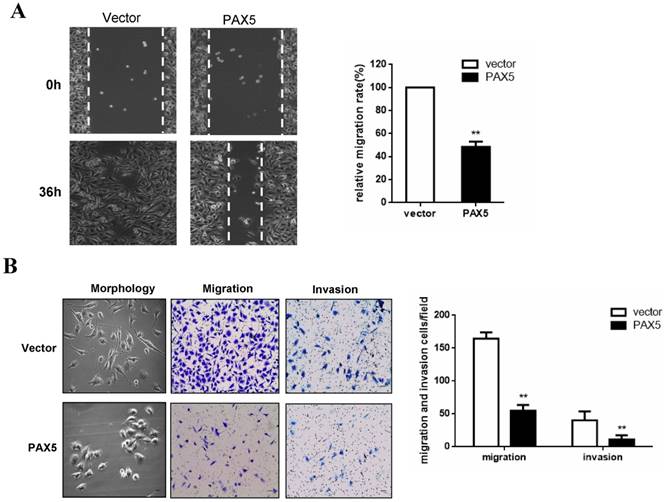
To investigate the effect of PAX5 on breast cancer cell migration, we performed a wound-healing assay. A significant delay in the confluence of wound gaps in PAX5-transfected BT549 cells was detected at 36 hrs, relative to controls (Fig. 4A). Next, we used a Transwell assay to quantitatively test cell migration and invasion (Fig. 4B). The number of invaded cells found in PAX5- transfected BT549 cells was dramatically lower relative to controls (p<0.01) (Fig. 4B). We also found that PAX5 transfected BT549 cells showed a less invasive morphology than control transfected cells (Fig. 4B).
PAX5 suppress breast cancer cells migration and invasion. (A) The wound gap confluence is delayed by PAX5 after 36h, compared with controls. (B) PAX5 modified the morphology of transfected cells and suppressed cell migration and invasion. The representative images are shown in both panels and the values are exhibited as the mean ± SD from three independent experiments. All the values are exhibited as the mean ± SD from three independent experiments (**, p < 0.01).
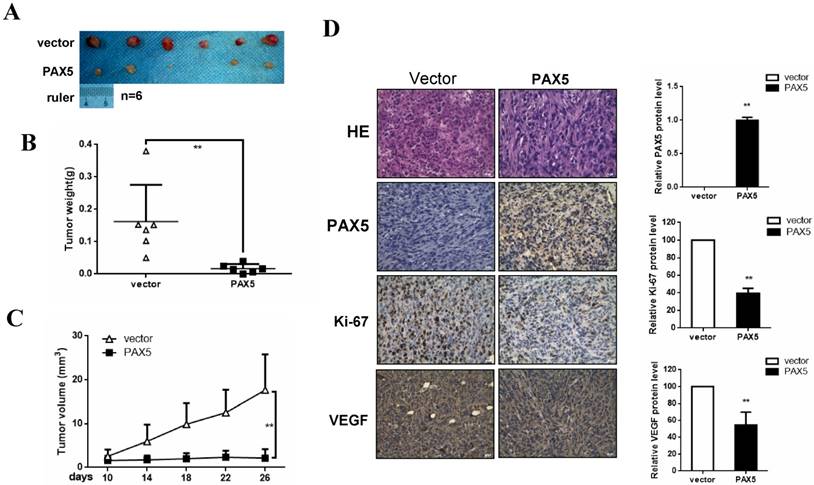
PAX5 inhibits breast cancer growth in vivo
To investigate the effect of PAX5 on breast cancer growth in vivo, we injected mice with PAX5-transfected MDA-MB-231 cells and vector-transfected cells. After 26 days, mice were sacrificed and their tumor weights were obtained. We found that both tumor size and weight were dramatically decreased in PAX5-transfected cell injections, compared to controls (p<0.01) (Fig. 5A&5B&5C). To further qualify these results, we also performed immunohistochemistry on histological sections taken from these tumors (Fig. 5D). We confirmed that the expression of PAX5 in PAX5-transfected MDA-MB-231 tumors was increased relative to control tumors (p<0.01). Interestingly, along with the expression of PAX5, expression of both the proliferation marker Ki67 and VEGF was increased in PAX5-transfected MDA-MB-231 tumors, relative to controls (p<0.01).
PAX5 inhibit breast cancer growth in mouse model. (A) Nude mice are injected with PAX5 transfected MB231cells and controls. (B) (C) Mice are sacrificed after 26 days and the weight of the tumors are measured. PAX5 induces a decrease in the growth of the tumors and the weight of tumors, compared with controls (**, p < 0.01). (D) The expression of PAX5, Ki67 and VEGF in tumors are confirmed by immunochemistry (**, p < 0.01).
PAX5 is involved in cell cycle regulation and VEGF expression
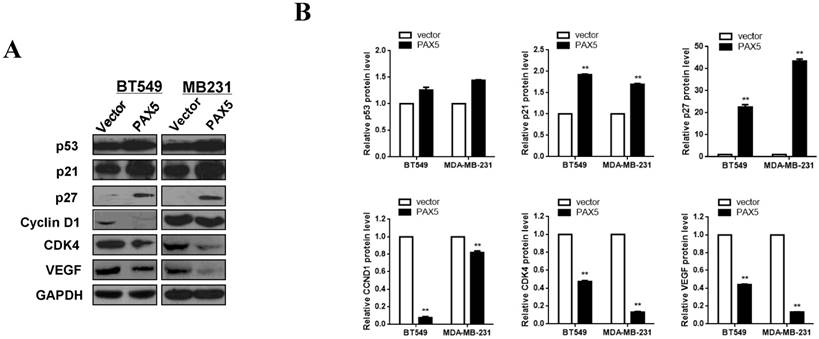
In order to further investigate the mechanism of cell cycle arrest and inhibition of cell invasion induced by PAX5, we performed western blots against known cell-cycle regulators (Fig. 6A). As p53 is a key regulator of cell proliferation and cell cycle progression, we first examined its expression and found that p53 was more strongly expressed in PAX5-transfected cells relative to controls. We then tested the expression of G0/G1 phase inhibitory factors, and found that the expression of both p21 and p27 was increased in PAX5-transfected cells. We also found that the expression levels of CyclinD1 and CDK4 were decreased in PAX5-transfected cells, performed in parallel to the previous assay (Fig. 6B). As VEGF is an important regulator of tumor angiogenesis and metastasis, as well as being down-regulated by p53 [18], we investigated the effect of PAX5 on VEGF expression level (Fig. 6A). We found that VEGF expression was decreased in PAX5-transfected cells relative to control transfected cells (Fig. 6B). Thus, our results demonstrate that PAX5 induces cell cycle arrest and inhibits cellular proliferation and invasion, likely by increasing the activity of cell cycle inhibitors and decreasing the expression of VEGF.
PAX5 involved in the regulation of cell cycle pathway and VEGF expression. (A) Western-blot is performed to test the effect of PAX5 on cell cycle pathway and the expression of VEGF. (B) It is shown that PAX5 increases the expression of p53, p21, p27 and decreases the expression of cyclin D1, CDK4, VEGF in breast cancer cells (BT549 and MB231). The values are exhibited as the mean ± SD from three independent experiments (**, p < 0.01).
Discussion
We report here that PAX5 is diffusely expressed in normal breast and paracarcinoma tissues, while frequently deleted or reduced in breast cancer cell lines and primary breast cancer tissues. This indicates that PAX5 may play an important role in suppressing breast cancer tumorigenesis. We further observed that the deletion or reduction of PAX5 might be due to aberrant promoter hypermethylation, which was confirmed by methylation specific PCR, and validated with a demethylation treatment assay. However, one breast cancer cell line, SK-BR-3, displayed unmethylated PAX5 along with an absence of detectable PAX5 expression, suggesting that other mechanisms may also contribute to the downregulation of PAX5 in breast cancer. The loss of PAX5 transcriptional activation has been reported in B-Cell Acute Lymphoblastic Leukemia (B-ALL), and was found to be due to either the loss of the PAX5 paired domain or frameshift mutations [19].
We demonstrate here an inhibitory role for PAX5 in breast cancer using both in vitro and in vivo assays. Ectopic expression of PAX5 in two silent cell lines (MDA-MB-231 and BT-549) suppressed cellular proliferation by inhibiting cell viability and colony formation. Other reports have shown contradictory roles for PAX5 in different cancers; PAX5 increases cellular proliferation in neuroblastoma cells [20], yet decreases proliferation in hepatocellular carcinoma [9], gastric carcinoma [10] and lung cancer [11]. Thus, PAX5 seems to play differing roles in various tissues, and its effect on malignant multiplication in solid tumors requires further study.
The tumor suppressor p53 is a key regulator of tumor growth, where it exerts its effects by modulating the cell cycle. Swaminathan et al. reported that BACH2 mediates PAX5-dependent tumor suppression in pre-B ALL through the activation of p53 [21]. In the present study, we found that PAX5 in two breast cancer cell lines (MDA-MB-231 and BT-549) is able to promote G1/G0 arrest. Correspondingly, PAX5 is able to increase the expression of p53, p21 and p27. The cell cycle inhibitor p21 has been reported to play a key role in the p53-dependent anti-cancer pathway [22], and is also a mediator of cell cycle arrest by inhibiting the activity of cyclin-dependent kinase (CDK)-cyclin complexes [23]. Additionally, p27, which belongs to the kinase inhibitor protein (KIP) family (whose members include p21CIP1, p27Kip1 and p57Kip2), induces G1 phase arrest by attenuating cyclin E/CDK2 and cyclin A/CDK2 activity [24]. p53 is also independently capable of inhibiting cyclin D1 promoter activity [25]. As key regulators of the transition from the G0/G1 phase to the synthesis (S) phase, cyclinD1 and CDK4 have already been used as targets in breast cancer treatment [26]. In our study, we found that PAX5 is able to decrease the expression of both cyclinD1 and CDK4.
Furthermore, we found that ectopic expression of PAX5 represses cell migration and invasion in breast cancer cell lines. Recently, VEGF was identified as a migration activator in breast cancer cell lines [27], and it is known that VEGF is downregulated by p53 via the inhibition of hypoxia-induced Src kinase activity [18]. Previous reports have also shown that PAX5 can attenuate breast cancer cell migration and invasion by inhibiting EMT [15, 16]. In our study, PAX5 is able to inhibit breast cancer cells migration and invasion, likely due to a down-regulation of VEGF. As VEGF is known to be an important activator of cancer angiogenesis [28], the role of PAX5 in angiogenesis should be the subject of further investigation.
In this study, we not only demonstrate a role for PAX5 in breast cancer progression, but also show that the methylation status of PAX5 is associated with several clinical parameters of breast cancer. First of all, A decreasing methylation grade of PAX5 was found from breast cancer tissues to breast normal tissues, suggesting that PAX5 methylation has a role in breast tumorigenesis. Most interestingly, PAX5 methylation status statistically correlates with the ER and PR which two are already applied in determining of endocrine therapy of breast cancer. There is a rising fact that ER and PR also important for the breast cancer molecular subtype classification and breast cancer molecular subtypes were already reported to be associated with breast cancer prognosis in several studies [29, 30] and breast cancer chemoresistance [31]. Thus, further investigation into the role of PAX5 methylation in different breast cancer subtypes is required to determine any direct link to cancer prognosis or cancer chemoresistance.
In conclusion, our study demonstrates that PAX5 plays a role as a tumor suppressor in breast cancer, and that the methylation status of PAX5 significantly correlates with several clinical features including age, estrogen receptor status, progesterone receptor status. Together, this indicates that PAX5 may serve as a potential breast cancer biomarker.
Supplementary Material
Supplementary figure.
Acknowledgements
The authors thank Prof. Qian Tao (Cancer Epigenetics laboratory, department of Clinical Oncology, the Chinese University of Hong Kong, Hong Kong, China) for generously providing cell lines, plasmids, primers and technical assistance.
Funding
This study was supported by the National Natural Science Foundation of China (No 81572769), the Natural Science Foundation of Chongqing Health and Family Planning Commission (2016ZDXM006) and Yuzhong Qu of Chongqing (2017-18).
Author Contributions
TX: conception and design of the study. XL, JH: performed experiments and analyzed data; WP, CB: contributed to RNA and DNA extraction. DY, XL, XY: collected samples. XL, TX: prepared figures and drafted the manuscript; XL, TX, GR: reviewed the manuscript; XL, TX: finalized the manuscript. All authors reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Servick K. Breast cancer. Breast cancer: a world of differences. Science. 2014;343:1452-3
2. Hoque MO, Prencipe M, Poeta ML, Barbano R, Valori VM, Copetti M. et al. Changes in CpG islands promoter methylation patterns during ductal breast carcinoma progression. Cancer Epidemiol Biomarkers Prev. 2009;18:2694-700
3. Xiang T, Li L, Yin X, Zhong L, Peng W, Qiu Z. et al. Epigenetic silencing of the WNT antagonist Dickkopf 3 disrupts normal Wnt/beta-catenin signalling and apoptosis regulation in breast cancer cells. J Cell Mol Med. 2013;17:1236-46
4. Shao B, Feng Y, Zhang H, Yu F, Li Q, Tan C. et al. The 3p14.2 tumour suppressor ADAMTS9 is inactivated by promoter CpG methylation and inhibits tumour cell growth in breast cancer. J Cell Mol Med. 2018;22:1257-71
5. Luo X, Xiong X, Shao Q, Xiang T, Li L, Yin X. et al. The tumor suppressor interferon regulatory factor 8 inhibits beta-catenin signaling in breast cancers, but is frequently silenced by promoter methylation. Oncotarget. 2017;8:48875-88
6. Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. 2007;8:463-70
7. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD. et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758-64
8. Familiades J, Bousquet M, Lafage-Pochitaloff M, Bene MC, Beldjord K, De Vos J. et al. PAX5 mutations occur frequently in adult B-cell progenitor acute lymphoblastic leukemia and PAX5 haploinsufficiency is associated with BCR-ABL1 and TCF3-PBX1 fusion genes: a GRAALL study. Leukemia. 2009;23:1989-98
9. Liu W, Li X, Chu ES, Go MY, Xu L, Zhao G. et al. Paired box gene 5 is a novel tumor suppressor in hepatocellular carcinoma through interaction with p53 signaling pathway. Hepatology (Baltimore, Md). 2011;53:843-53
10. Li X, Cheung KF, Ma X, Tian L, Zhao J, Go MY. et al. Epigenetic inactivation of paired box gene 5, a novel tumor suppressor gene, through direct upregulation of p53 is associated with prognosis in gastric cancer patients. Oncogene. 2012;31:3419-30
11. Zhao L, Li S, Gan L, Li C, Qiu Z, Feng Y. et al. Paired box 5 is a frequently methylated lung cancer tumour suppressor gene interfering β-catenin signalling and GADD45G expression. Journal of Cellular and Molecular Medicine. 2016;20:842-54
12. Palmisano WA, Crume KP, Grimes MJ, Winters SA, Toyota M, Esteller M. et al. Aberrant promoter methylation of the transcription factor genes PAX5 alpha and beta in human cancers. Cancer research. 2003;63:4620-5
13. Moelans CB, Verschuur-Maes AH, van Diest PJ. Frequent promoter hypermethylation of BRCA2, CDH13, MSH6, PAX5, PAX6 and WT1 in ductal carcinoma in situ and invasive breast cancer. The Journal of pathology. 2011;225:222-31
14. Kornegoor R, Moelans CB, Verschuur-Maes AH, Hogenes M, de Bruin PC, Oudejans JJ. et al. Promoter hypermethylation in male breast cancer: analysis by multiplex ligation-dependent probe amplification. Breast Cancer Research: BCR. 2012;14:R101
15. Vidal LJ, Perry JK, Vouyovitch CM, Pandey V, Brunet-Dunand SE, Mertani HC. et al. PAX5alpha enhances the epithelial behavior of human mammary carcinoma cells. Molecular cancer research: MCR. 2010;8:444-56
16. Benzina S, Beauregard AP, Guerrette R, Jean S, Faye MD, Laflamme M. et al. Pax-5 is a potent regulator of E-cadherin and breast cancer malignant processes. Oncotarget. 2017;8:12052-66
17. Branham MT, Campoy E, Laurito S, Branham R, Urrutia G, Orozco J. et al. Epigenetic regulation of ID4 in the determination of the BRCAness phenotype in breast cancer. Breast cancer research and treatment. 2016;155:13-23
18. Pal S, Datta K, Mukhopadhyay D. Central role of p53 on regulation of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in mammary carcinoma. Cancer research. 2001;61:6952-7
19. Dang J, Wei L, de Ridder J, Su X, Rust AG, Roberts KG. et al. PAX5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood. 2015;125:3609-17
20. Baumann Kubetzko FB, Di Paolo C, Maag C, Meier R, Schafer BW, Betts DR. et al. The PAX5 oncogene is expressed in N-type neuroblastoma cells and increases tumorigenicity of a S-type cell line. Carcinogenesis. 2004;25:1839-46
21. Swaminathan S, Huang C, Geng H, Chen Z, Harvey R, Kang H. et al. BACH2 mediates negative selection and p53-dependent tumor suppression at the pre-B cell receptor checkpoint. Nature medicine. 2013;19:1014-22
22. Efeyan A, Collado M, Velasco-Miguel S, Serrano M. Genetic dissection of the role of p21Cip1/Waf1 in p53-mediated tumour suppression. Oncogene. 2007;26:1645-9
23. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nature reviews Cancer. 2009;9:400-14
24. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & development. 1999;13:1501-12
25. Rocha S, Martin AM, Meek DW, Perkins ND. p53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-kappaB subunit with histone deacetylase 1. Molecular and cellular biology. 2003;23:4713-27
26. Edessa D, Sisay M. Recent advances of cyclin-dependent kinases as potential therapeutic targets in HR+/HER2- metastatic breast cancer: a focus on ribociclib. Breast Cancer (Dove Med Press). 2017;9:567-79
27. Perrot-Applanat M, Di Benedetto M. Autocrine functions of VEGF in breast tumor cells: adhesion, survival, migration and invasion. Cell adhesion & migration. 2012;6:547-53
28. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nature medicine. 1995;1:27-31
29. Bertucci F, Finetti P, Birnbaum D. Basal breast cancer: a complex and deadly molecular subtype. Curr Mol Med. 2012;12:96-110
30. Bertucci F, Finetti P, Cervera N, Charafe-Jauffret E, Buttarelli M, Jacquemier J. et al. How different are luminal A and basal breast cancers? International journal of cancer. 2009;124:1338-48
31. Rouzier R, Perou CM, Symmans WF, Ibrahim N, Cristofanilli M, Anderson K. et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res. 2005;11:5678-85
Author contact
![]() Corresponding authors: Xiang T and Ren G, the First Affiliated Hospital of Chongqing Medical University, Chongqing, China. Tel: (86) 23-89011904, Fax: (86) 23- 89012558, Email: larissaxiangcom; and rengs726com
Corresponding authors: Xiang T and Ren G, the First Affiliated Hospital of Chongqing Medical University, Chongqing, China. Tel: (86) 23-89011904, Fax: (86) 23- 89012558, Email: larissaxiangcom; and rengs726com