Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(12):1696-1708. doi:10.7150/ijbs.27774 This issue Cite
Research Paper
GLP-1 treatment protects endothelial cells from oxidative stress-induced autophagy and endothelial dysfunction
Xiangsheng Cai1,3, Miaoqin She2, Mingyu Xu3, Huiying Chen3, Jingjing Li3, Xinglu Chen1, Dianpeng Zheng3, Jun Liu4, Shangliang Chen5, Jianbin Zhu6, Xiaosong Xu1, Ruiying Li1, Jinlong Li3, Shaolian Chen1 ![]() , Xiaorong Yang1
, Xiaorong Yang1 ![]() , Hongwei Li3
, Hongwei Li3 ![]()
1. Clinical Laboratory, The First Affiliated Hospital of Guangdong Pharmaceutical University, Guangzhou, 510080, People's Republic of China.
2. Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences. Guangzhou, 510660, People's Republic of China.
3. Institute of Biotherapy, Southern Medical University, Guangzhou, 510515, People's Republic of China.
4. Dermatology Hospital of Southern Medical University, Guangzhou, 510091, People's Republic of China.
5. ShenZhen Hospital, Southern Medical University, ShenZhen 518101, People's Republic of China.
6. Technology Center, Guangdong Vitalife Bio-tech Co.,LTD, FoShan, 528200, People's Republic of China.
Received 2018-6-10; Accepted 2018-8-22; Published 2018-9-7
Abstract

Endothelial dysfunction and excessively stimulated autophagy, often caused by oxidant injury or inflammation, will lead to atherosclerosis development and progression in diabetes. The aim of this study is to investigate the protective effect of glucagon-like peptide-1 (GLP-1) treatment on preventing oxidative stress-induced endothelial dysfunction and excessively stimulated autophagy. Treatment of endothelial cells with GLP-1 significantly attenuated oxidative stress-induced endothelial dysfunction and autophagy, which was associated with the reduction of intracellular reactive oxygen species (ROS) levels. These protective effects of GLP-1 were likely mediated by reducing phosphorylation of ERK1/2. We further demonstrated that GLP-1 treatment could reverse downregulation of epigenetic factor histone deacetylase 6 (HDAC6), a downstream molecular of the EKR1/2, induced by oxidant injury. In conclusion, our results suggest that GLP-1 produces a protective effect on endothelial cells from oxidant injury by preventing endothelial dysfunction and autophagy, which may be dependent on restoring HDAC6 through a GLP-1R-ERK1/2-dependent manner.
Keywords: oxidative stress, endothelial dysfunction, GLP-1, autophagy, HDAC6
Introduction
The prevalence of type 2 diabetes mellitus (T2DM) in China is 11.6% of the general adult population [1]. Cardiovascular complications are recognized as the leading cause for morbidity and mortality in the diabetic population [2]. Diabetes mellitus is proposed to accelerate arteriosclerosis which has high risk at coronary heart disease and stroke [3].
It is well known that diabetes could lead to upregulation of intracellular reactive oxygen species (ROS), subsequently inducing inflammation, injury in endothelial cells and apoptotic cell death [4, 5]. Previous study indicated that elevated generation of reactive oxygen species (ROS) and increased oxidative stress lead to vascular dysfunction [6]. In mammalian cells, mitochondrial ROS accumulation and lipids oxidation seem to play a key role in autophagy, since both O2• - and H2O2 could induce autophagy [7].
Through degradation of damaged intracellular materials caused by ROS, autophagy plays a key role in the maintenance of normal cellular homeostasis [8, 9]. It is clear that basal autophagy can protect cells against oxidative stress by eliminating damaged intracellular materials [10]. However, it may cause autophagic death in cells due to excessively stimulated autophagy [11]. Endothelial cell death may be unfavorable for the structure of plaque because endothelial injury or death will promote lesional thrombosis [12, 13]. Numerous data suggest that autophagy is tightly associated with atherosclerosis [14, 15]. In addition, dysregulated or impaired autophagy has been associated with cardiovascular diseases such as atherosclerosis [16], cardiomyopathy [17], heart failure [18], and hypoperfusion injury [19].
Glucagon-like peptide-1 (GLP-1), released from L-cells of the intestine, is a peptide with several protective biological functions. Besides the insulin tropic effects of GLP-1 on insulin-secreting cells, numerous evidences demonstrated that GLP-1 has cardiovascular effects and antioxidant effect [20-22]. Recent study indicated that GLP-1 could inhibit advanced glycation end products (AGEs) -induced increase of reactive oxygen species in HUVECs [23]. In addition, GLP-1 could improve H2O2-induced cellular injury [24]. GLP-1 also exerts anti-apoptotic effects in various cell types, including cardiomyocytes [25], neuronal cells [26-28], cholangiocytes [29] and pancreatic β cells [30-32]. However, there is limited report about the effect of GLP-1 on dysfunction and autophagy of endothelial cells subjected to oxidative stress.
Epigenetic factor HDAC6 has been reported to play a key role in redox regulation and oxidative stress response [33]. HDAC6 regulates various cellular processes, including cell motility, cell migration, endocytosis, aggresome formation and autophagy etc., by deacetylating cytoplasmic proteins, such as cortactin, heat shock protein 90 (HSP90) and α-tubulin [34].
In this study, we first investigated the effects of GLP-1 on preventing dysfuction and autophagy in endothelial cells induced through oxidative stress. We further revealed the possible mechanism behind anti-autophagic effects of GLP-1 by assessing the roles of ERK1/2 and HDAC6.
Materials and Methods
Reagents
GLP-1 and Ex-9 (Exendin-[9-39], a GLP-1 receptor antagonist) were purchased from Booteck Company (Shanghai, China). Rohdamine123, DHE (Dihydroethidium), compound LY294002 (PI3K inhibitor) and compound U0126 (ERK1/2 inhibitor) were purchased from Biyuntian Company (Hangzhou, China). A fluorescent probe DCFH-DA (St. Louis, MO, USA) was initially made in DMSO at 10 mM and then sub stocked in PBS at 0.5 μM. The antibodies (rabbit anti-human) against LC3, ATG7, Akt, p-Akt, ERK1/2, p-ERK1/2, HDAC6 and acetyl-α-tubulin were purchased from Cell Signaling Technology (Beverly, MA, USA). GAPDH antibody (rabbit anti-human) and horseradish peroxidase (HRP)-conjugated second antibody (goat anti-rabbit) were purchased from Fude Company (Wuhan, China).
Cell culture and treatments
HUVECs (Human umbilical vein endothelial cells) were purchased from American Tissue Culture Consortium (ATCC) and cultured in RIPM 1640 medium supplemented with 10% FBS (Gibco, Invitrogen, NY, USA) and 1% penicillin-streptomycin. HUVECs were used from passages 2 and 6 in this study.
The cells were grown to 70% confluence and cultured in serum-free medium for 4 h before treatments. To investigate the ROS generation, the cells were cultured in serum-free medium containing high glucose (HG, 45 mM D‑glucose) , normal glucose (NG, 5 mM D‑glucose) and 1 mM H2O2 for 12 h. Then, the protective effects of GLP-1 were evaluated. The cells were pretreated with various concentrations (1, 10, 100 and 1000 nM) of GLP-1 for 1 h prior to exposure to H2O2.
To reveal the possible mechanism of protective effects of GLP-1, the cells were randomly assigned to one of the following 7 groups: i) the control group: cells were cultured under normal incubation conditions; ii) the H2O2 group: cells were subjected to H2O2 without pretreatment with GLP-1; iii) the H2O2 + GLP-1 group: cells were pretreated with GLP-1 for 1 h prior to exposure to H2O2; iv) the GLP-1 + Ex-9 group: cells were pretreated with GLP-1 in addition with Ex-9 for 1 h prior to exposure to H2O2; v) the H2O2 + LY294002 group: cells were pretreated with LY294002 for 1 h prior to exposure to H2O2; vi) the H2O2 + U0126 group: cells were pretreated with U0126 for 1 h prior to exposure to H2O2. vii) the H2O2 + GLP-1 + U0126 group: cells were pretreated with GLP-1 in addition with U0126 for 1 h prior to exposure to H2O2.
Afterwards, the cells were cultured in serum-free medium containing 1 mM H2O2 for 12 h.
Measurement of Reactive Oxygen Species
The intracellular ROS level was detected by labeling with DCFH-DA or DHE. The cells were plated in 6-well plates. After reaching 60-70 % confluent, they were treated with different conditions respectively. After incubation for 12h, the cells were washed with PBS (pH 7.4) and stained with DCFH-DA or DHE at 37 °C for 20 min. Then the cells were examined under a fluorescence microscope (Olympus FV1000, Japan). The averaging fluorescence intensity of numerous outlined cells was calculated using Image-Pro Plus software version 6.0.
Measurement of mitochondrial membrane potential (MMP)
The mitochondrial membrane potential (ΔΨm) was measured by loading cells with 5 μM Rhodamine123, a cationic lipophilic fluorochrome taken up by mitochondria in proportion to the ΔΨm. After 20 min incubation in the dark, the cells were washed twice with PBS. Rhodamine123 staining was visualized under a fluorescence microscope. Then, mean fluorescence intensity of cells was analyzed with Image-Pro Plus software version 6.0.
Immunofluorescence
The cells were treated with different conditions respectively. Following 30 min fixation in 4 % paraformaldehyde, they were washed with PBS, and then permeabilized with 0.5 % Triton X-100 in PBS. After washing with PBS and blocking with 10 % NGS for 2 h at room temperature, the cells were incubated with LC3B or HDAC6 primary antibody (1:200). After washing with PBS, they were incubated with the FITC-conjugated goat anti-rabbit IgG secondary antibody (1:1000). Thereafter, the cell nuclei were stained by DAPI. Samples were examined under a fluorescence microscope (Olympus FV1000, Japan) or a confocal microscope (Nikon Eclipse Ti, Japan).
Western blot analysis
Western blotting was performed using routine protocols. Briefly, treated cells were harvested and lysed in RIPA lysis buffer (Fude, Wuhan, China). Protein samples were electrophoresed, transferred to PVDF membrane (Millipore, Bedford, MA, USA) and then blocked. Membranes were then incubated overnight at 4℃ with primary antibody (1:1000 dilution). After washing, the membranes were incubated with peroxidase-conjugated secondary antibodies (1:5000). The membranes were then washed and detected using a chemiluminescence detection kit (ECL; Pierce, Rockford, USA) and exposed to X-ray film. Bands of interest were quantified by densitometry using Gel-Pro analysis software.
Statistical analysis
All analyses were carried out with the GraphPad Prism 4.0 software (GraphPad Software, San Diego, CA, USA). Data were expressed as the mean ±SD and then analyzed using one-way ANOVA followed by Tukey's Multiple Comparison Test. p value < 0.05 was considered statistically significant. The results are representative an average of at least 3 experiments.
Results
GLP-1 treatment attenuates intracellular ROS accumulation
Mitochondrial production of ROS induced by hyperglycemia plays a key role in the pathogenesis of diabetes. Herein, to investigate the production of ROS, HUVECs were subjected to high glucose medium (HG, 45 mM D‑glucose). As shown in Fig. 1A, compared with cells cultured in serum-free medium (Ctrl group) or the normal glucose medium (NG group), the cells cultured in the medium with high glucose (HG group) or H2O2 (H2O2 group) increased the ROS production significantly (p<0.01).
GLP-1 treatment attenuates intracellular ROS accumulation. (A) Intracellular ROS generation in HUVECs in each experimental group. (B) Cells were pretreated with or without 1, 10, and 100 nM GLP-1 for 1 h, incubated with H2O2 for 12 h, and then labeled with DCFH-DA (30 μM) prior to fluorescence microscopic analysis. Production of ROS was quantified by the amount of cellular DCF synthesis. n = 6. (C) Cells were pretreated with or without GLP-1 (10 nM) for 1 h in addition of Ex-9 (10 nM), incubated with H2O2 (1 mM) for 12 h, and then labeled with DCFH-DA. (D, E, F) Cells were pretreated with 1, 10 and 100 nM GLP-1 for 1 h, and incubated with H2O2 (1 mM) for 12 h, and then labeled with the fluorescent dyes Rhodamine 123, superoxide indicator DHE, and nucleic acid stain Hoechst 33342. All data are expressed as mean ±SD, **p<0.01 as compared with the control values; #p<0.05, ##p<0.05 as compared with the values of cells treated with H2O2; &&p<0.01 as compared with the values of cells treated with H2O2 plus GLP-1. Scale bar=100μm.
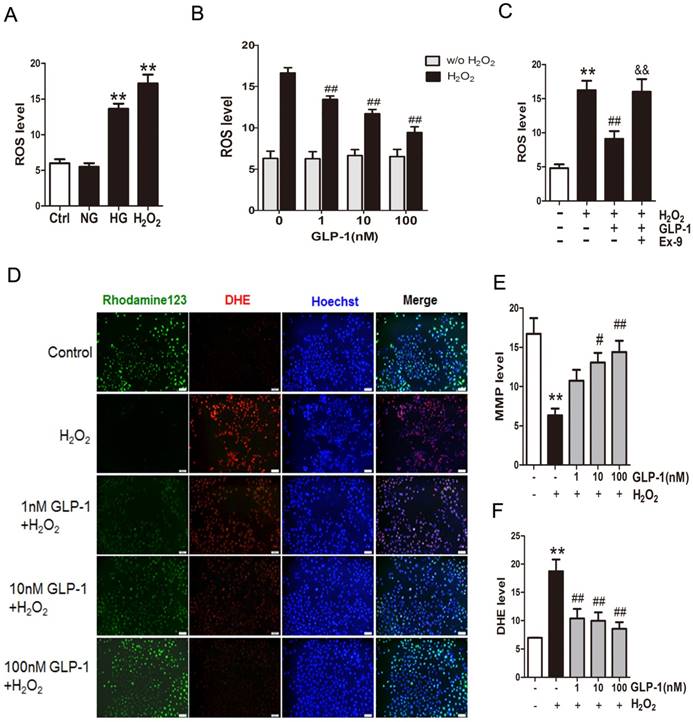
Then, in order to observe whether GLP-1 can reduce oxidative-induced ROS generation, HUVECs were incubated with increasing concentrations of GLP-1 in the presence or absence of H2O2. As shown in Fig. 1B, ROS significantly increased in HUVECs upon subjected to 1 mM of H2O2 for 12 h (p<0.01), while GLP-1 treatment reduced the ROS generation in a dose dependent manner, and this decrease was statistically significant at concentrations of GLP-1(1,10 and 100 nM) compared with the control group (p<0.01). Additionally, as shown in Fig. 1C, the ROS level was significantly increased through H2O2 stimulation (p<0.01), which was attenuated by GLP-1 (p<0.01). This effect was abolished by Ex-9 (p<0.01).
Mitochondrial membrane potential (MMP) dissipation is a common early event during apoptosis. In this study, MMP dissipation was induced by exposure to 1mM H2O2 for 12 h. As shown in Fig. 1C and 1D, exposure to 1mM H2O2 for 12 h led to MMP dissipation (p<0.01), while pretreated with 1, 10, and 100 nM GLP-1 for 1h prior to H2O2 exposure attenuated the changes in MMP.
In addition, the redox-sensitive probe DHE were employed to test if the protective effect of GLP-1 on HUVECs is associated with a decreased vascular pro-oxidant response. Thus, we investigated significantly decreased levels of DHE fluorescence in GLP-1-pretreated cells upon oxidative stress compared with cells not treated by GLP-1 (p<0.01) (Fig. 1C and 1E).
GLP-1 treatment attenuates oxidative stress-mediated injury in endothelial cells
To investigate the possible protective effects of GLP-1 on oxidative stress-mediated injury, the cells were pretreated with GLP-1 for 1h prior to exposure to H2O2. We found that pretreatment with GLP-1 increased cell viability in a dose dependent manner, and this increase was statistically significant (p<0.05) at the concentrations of GLP-1 (10 and 100nM) (Fig. 2A). Our results suggest that GLP-1 exerts protective effects against oxidative stress-caused damage in HUVECs cells. Moreover, as shown in Fig. 2B, the cell viability was significantly decreased through H2O2 stimulation (p<0.01), which was attenuated by GLP-1 (p<0.01). This effect was abolished by Ex-9 (p<0.01).
GLP-1 alleviates oxidative stress-mediated injury in endothelial cells. (A) After pretreatment with GLP-1 of different concentration and induction by H2O2, cell viability was assessed with MTT assay, n = 6. (B) Cells were pretreated with or without GLP-1 (10 nM) for 1 h in addition of Ex-9 (10 nM), incubated with H2O2 (1 mM) for 12 h, and then cell viability was assessed with MTT assay, n = 6. (C) Cells were treated as (B), then stained with primary antibodies of BCL2 and Bax followed by staining with secondary antibodies. Cell nuclei were labeled with DAPI and immunofluorescence analysis was performed under the fluorescence microscope. (D, E, F) Cell was treated as (B), BCL2 and Bax were detected by Western blot. All data are expressed as mean ± SD, **p < 0.01 as compared with the control values; #p<0.05, ##p<0.01 as compared with the values of cells treated with H2O2 group; &&p<0.01 as compared with the values of cells treated with H2O2 plus GLP-1. Scale bar =100μm.
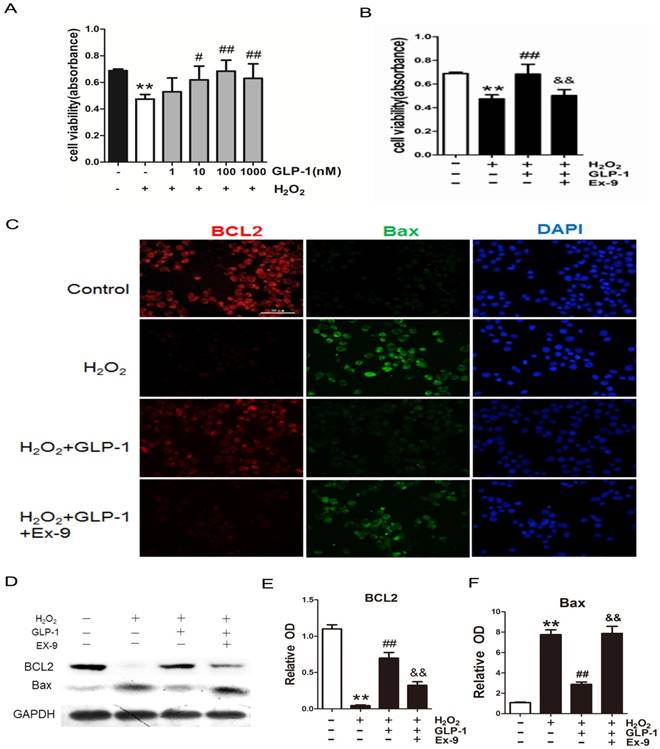
The expression of two cell death regulators, BCL2 and Bax was evaluated by immunofluorescence staining and Western blot. As shown in Fig. 2C -F, we observed a significantly decrease signal of BCL2 when the cells were subjected to H2O2 (p<0.01). However, BCL2 expression was significantly restored by 1 h pretreatment with GLP-1 (p<0.01). This effect was inhibited by Ex-9 (p<0.01). Conversely, the expression of Bax was increased in HUVECs exposure to H2O2 (p<0.01), and then reversed by GLP-1 pretreatment at 10 nM (p<0.01). This effect was also abolished by Ex-9 (p<0.01).
GLP-1 treatment attenuates oxidative stress-mediated autophagic response
We first examined whether H2O2 could induce autophagy in HUVECs. Autophagy was assessed by LC3-II fluorescent staining of cells to measure the number of autophagosomes. As shown in Fig. 3A and 3B, H2O2-induced autophagic flux was observed, and was then sustained at high levels during 12 h treatment (p<0.01). Next, we observed the effect of GLP-1 on autophagy caused by H2O2 (p<0.01). The result showed that GLP-1 pretreatment notably decreased the rate of autophagy, which was inhibited by Ex-9 (p<0.01).
GLP-1 alleviates oxidative stress-mediated autophagy in endothelial cells. (A) Cells were pretreated with or without GLP-1 (10 nM) for 1 h in the presence or absence of Ex-9 (10 nM), incubated with H2O2 (1 mM) for 12 h, and then stained with primary antibodies of LC3II followed by staining with secondary antibodies. Cell nuclei were labeled with DAPI and immunofluorescence analysis was performed under the confocal. (B) Quantification of the number of autophagosomes. (C, D, E) Cell was treated as (A), relative changes in ATG7 and LC3II protein levels were analyzed by Western blot analysis, GAPDH was used as a loading control. The blots shown are the representative average of three experiments. All data are expressed as mean ±SD, *p<0.05, **p<0.01 as compared with the control values; #p<0.05, ##p<0.01 as compared with the values of cells treated with H2O2; &p<0.05, &&p<0.01 as compared with the values of cells treated with H2O2 plus GLP-1. Scale bar = 50μm.
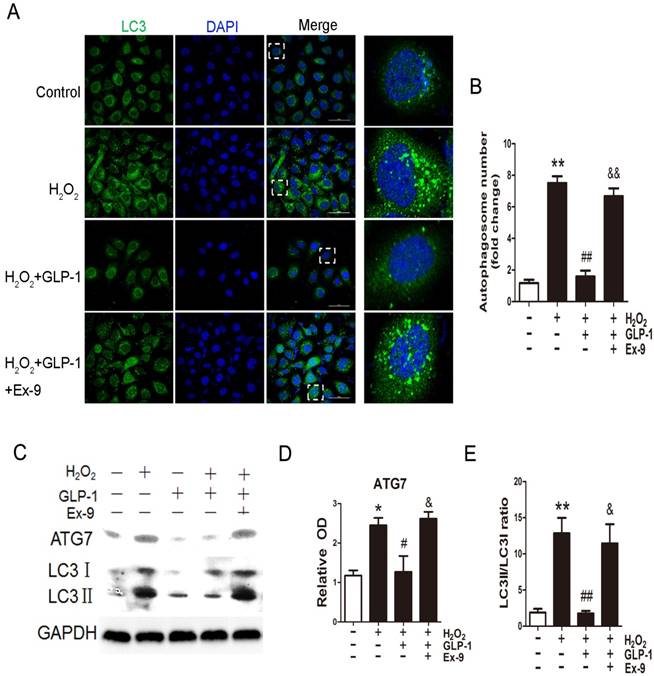
Then, LC3II/I and ATG7 levels were investigated through Western blot analysis, which are both markers of autophagosome formation. As shown in Fig. 3C-E, ATG7 and LC3II/I levels were significantly increased in HUVECs exposure to H2O2, which were attenuated by GLP-1 treatment. This effect was blocked by Ex-9.
GLP-1 treatment reduces phosphorylation of AKT and ERK1/2 induced by oxidative stress
To elucidate the possible mechanism of GLP-1 against oxidative stress-mediated autophagy, we further assessed the effects of GLP-1 on phosphorylated Akt and phosphorylated ERK1/2 levels in HUVECs cells. As shown in Fig. 4A-C, H2O2 loading were associated with an increase in the phosphorylation of Akt and ERK1/2 (p<0.01), which were attenuated by GLP-1 pretreatment (p<0.05). These effects were abolished in addition of Ex-9 (p<0.05).
GLP-1 treatment reduces phosphorylation of AKT and ERK1/2 induced by oxidative stress. (A, B, C) Cells were pretreated with or without GLP-1 (10 nM) for 1 h in the presence or absence of Ex-9 (10 nM), then incubated with H2O2 (1 mM) for 12 h, expressions of phosphorylated Akt and ERK1/2 were detected by Western blot. The blots shown are the representative average of three experiments. (D) Cells were pretreated with or without GLP-1 (10 nM), LY294002 (20 μM) or U0126 (20 μM) for 1h, incubated with H2O2 (1 mM) for 12 h, then stained with primary antibodies of LC3 followed by staining with secondary antibodies. (E) Quantification of the number of autophagosomes. All data are expressed as mean ±SD, **p<0.01 as compared with the control values; #p<0.05, ##p<0.01 as compared with the values of cells treated with H2O2; &p<0.05 as compared with the values of cells treated with H2O2 plus GLP-1. Scale bar = 50 μm.
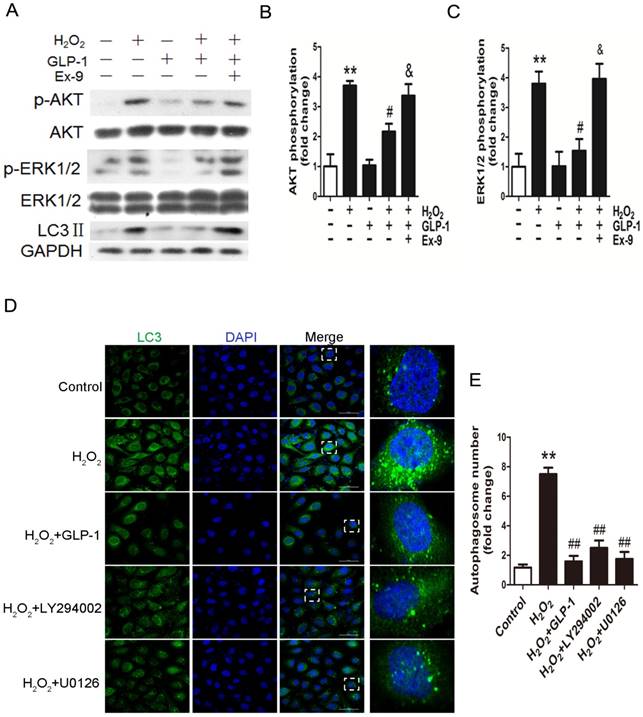
In addition, as shown in Fig. 4D-E, LY294002 and U0126, the inhibitors of PI3K and ERK1/2, could decrease the LC3 levels and autophagosomes in HUVECs subjected to H2O2. Our results suggest that GLP-1 treatment attenuates H2O2-mediated autophagy at least partially through Akt/ERK pathway.
GLP-1 treatment improves migratory and adhesive ability of HUVECs through increasing HDAC6 expression
HDAC6 has been reported as the downstream molecular of the EKR1/2. Herein, HDAC6 levels were investigated through Western blot analysis. As shown in Fig. 5A and B, HDAC6 levels were significantly increased in HUVECs exposure to GLP-1 (p<0.01). Correspondingly, as shown in Fig. 5A and C, the levels of acetylated α-tubulin, which was the substrate of HDAC6, were markedly decreased in HUVECs subjected to GLP-1 (p<0.01).
GLP-1 treatment improves migratory and adhesive ability of HUVECs through increasing HDAC6 expression. (A, B, C) Cells were pretreated with 1, 10, 100 and 1000 nM GLP-1 for 12 h, HDAC6 and ac-α-tubulin were detected by Western blot. (D) Cell was treated as (A), then stained with primary antibodies of ac-α-tubulin followed by staining with secondary antibodies. Cell nuclei were labeled with DAPI and immunofluorescence analysis was performed under the fluorescence microscope. (E, G) A Boyden chamber assay was used to evaluate the effect of GLP-1 on HUVECs migration. The numbers of migrated cells were quantified by performing cell counts of 5 random fields. (F, H) A fibronectin adhesion assay was used to evaluate the effect of GLP-1 on HUVECs adhesive function. Cells were stained with crystal violet. Quantification of attached cells was determined by cell counting of 5 random fields. All data are expressed as mean ±SD, **p<0.01 as compared with the control values. Scale bar in D = 50 μm, in E, F=100 μm.
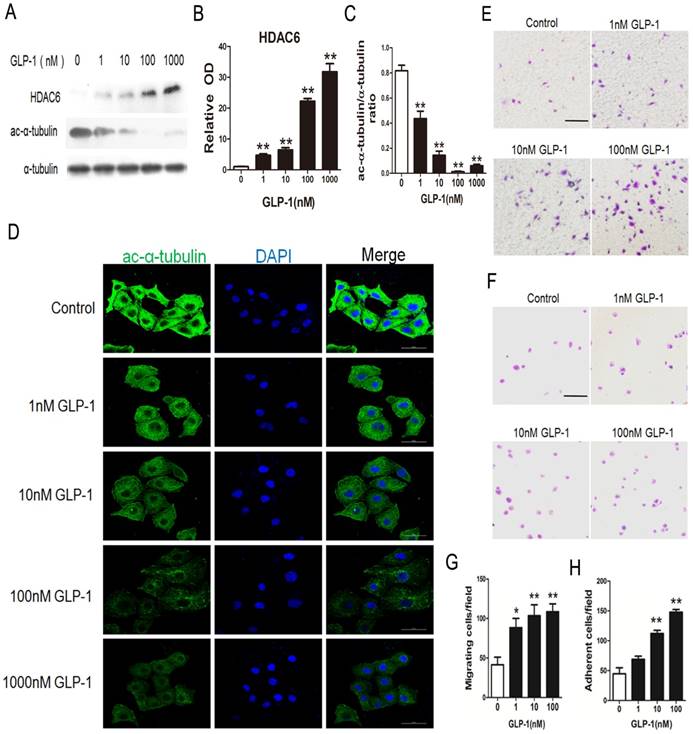
In addition, acetylated α-tubulin levels were detected by immunofluorescence staining. As shown in Fig. 5D, we observed a notably decreased signal of acetylated α-tubulin in HUVECs exposure to GLP-1.
HDAC6 and acetylated α-tubulin were tightly associated with the mobility. Thus, a Boyden chamber assay was performed to assess the effect of GLP-1 on HUVECs migration. As shown in Fig.5E and 5G, GLP-1 treatment increased the number of migrated cells in a dose dependent manner, and this increase was statistically significant (p<0.01) at higher concentrations of GLP-1(10 and 100 nM) compared with the control group.
Then, a fibronectin adhesion assay was employed to assess the effect of GLP-1 on HUVECs adhesive function. As shown in Fig.5F and 5H, GLP-1 treatment increased the number of adherent cells in a dose dependent manner, and this increase was statistically significant (p<0.01) at higher concentrations of GLP-1(10 and 100 nM) compared with the control group.
GLP-1 provides cellular protection against oxidative stress through restoring HDAC6 expression
Expression of HDAC6 was evaluated by immunofluorescence staining (Fig. 6A). We observed a dramatically decreased signal of HDAC6 when the cells were exposed to H2O2. However, the reduction of HDAC6 expression was significantly attenuated by 1 h pretreatment with GLP-1. This effect was inhibited by Ex-9.
GLP-1 provides cellular protection against oxidative stress in endothelial cells through restoring HDAC6 expression. (A) Cells were pretreated with or without GLP-1 (10 nM) for 1 h in the presence or absence of Ex-9 (10 nM) and incubated with H2O2 (1 mM) for 12 h, HDAC6 were detected by immunofluorescence analysis. (B, C, D) Cells were treated as (A), HDAC6, ac-α-tubulin and LC3 were detected by Western blot. (E, F) Cells were treated as (A), cell migratory and adhesive ability was determined by the methods as described above. Data are expressed as mean ± SD, **p<0.01 as compared with the control values; #p<0.05, ##p<0.01 as compared with the values of cells treated with H2O2; &p<0.05, &&p<0.01 as compared with the values of cells treated with H2O2 plus GLP-1. Scale bar = 50 μm.
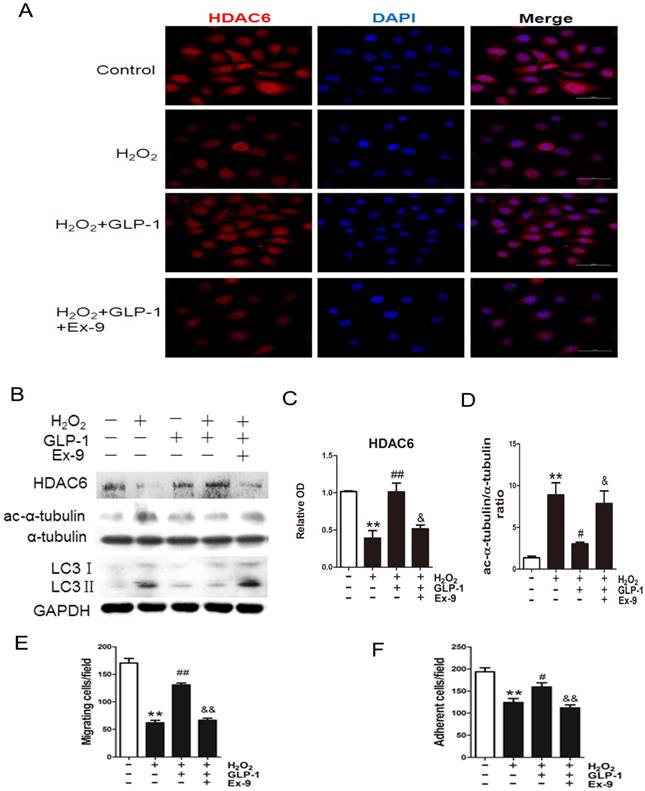
Western blot analysis indicated that GLP-1 substantially reversed the H2O2-induced reduction of HDAC6 (Fig. 6B and 6C). HDAC6 level was significantly reduced after 12 h in HUVECs in response to H2O2 stimulation (p<0.01), while GLP-1 pretreatment notably restored expression of HDAC6 (p<0.05) and decreased the LC3II/LC3I ratio. This effect was abolished by Ex-9 (p<0.01), implying that GLP-1 restores HDAC6 levels reduced by oxidative stress, which was tightly associated with autophagy.
We also examined changes in the levels of acetylated α-tubulin, which was reported as a substrate of HDAC6 (Fig. 6B and 6D). Western blot results showed that acetylated α-tubulin level was significantly increased after 12 h in HUVECs in response to H2O2 stimulation (p<0.01), while GLP-1 pretreatment notably decreased the expression of acetylated α-tubulin (p<0.05). This effect was abolished by Ex-9 (p<0.05).
Acetylation of α-tubulin is tightly associated with the mobility of the cells. As shown in Fig. 6E, H2O2 stimulation significantly reduced the number of migrated cells (p<0.01), which was attenuated through GLP-1 treatment (p<0.01). This effect was inhibited by Ex-9 (p<0.01). Additionally, as shown in Fig. 6F, the number of adherent cells were significantly reduced through H2O2 stimulation (p<0.01), which were attenuated by GLP-1 (p<0.05). This effect was abolished by Ex-9 (p<0.01).
Blockade of ERK1/2 phosphorylation by GLP-1 restores HDAC6 expression
We have confirmed that GLP-1 attenuated the increase of ERK1/2 phosphorylation stimulated by H2O2, while ERK1/2 has been reported to interact with HDAC6. Herein, we hypothesize that GLP-1 restores HDAC6 expression through attenuating increase of ERK1/2 phosphorylation.
As shown in Fig. 7A-E, H2O2 loading reduced HDAC6 protein expression, while pretreatment with GLP-1 and U0126 both partially restore HDAC6 levels, and attenuated increase of ATG7 and LC3II levels. H2O2-induced autophagic responses were attenuated when ERK1/2 activity were suppressed by their inhibitor U0126, suggesting that ERK activation are required for autophagy induced by H2O2 in HUVECs, implying that an ERK1/2-HDAC6 pathway is at least partially involved in H2O2-mediated autophagy.
Blockade of ERK1/2 phosphorylation induced by GLP-1 restores HDAC6 expression. (A) Cells were pretreated with or without GLP-1 (10 nM) and U0126 (20 μM) for 1 h, incubated with H2O2 (1 mM) for 12 h, HDAC6 were detected by immunofluorescence analysis. (B, C, D, E) Cells were treated as (A), HDAC6, ac-α-tubulin and LC3Ⅱ were detected by Western blot. The blots shown are the representative average of three experiments. (F, G, H, I) Cells were treated as (A), cellular ROS level, cellular viability, cell migratory and adhesive ability were determined by the methods as described above. Data are expressed as mean ± SD, *p<0.05, **p<0.01 as compared with the control values; #p<0.05, ##p<0.01 as compared with the values of cells treated with H2O2. Scale bar = 50 μm.
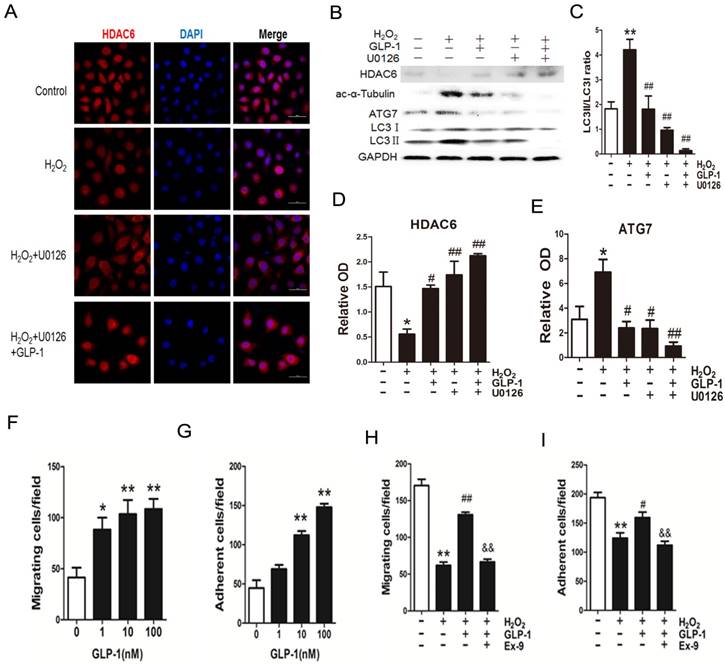
Moreover, ERK1/2 inhibition by U0126 and GLP-1, which restored the HDAC6 expression, resulted in declining ROS production, increasing cell survival, increasing cell migratory and adhesive ability upon H2O2-induced oxidative stress (Fig. 7F-I), illustrating that the loss of ERK1/2 activity accompanied with restore of HDAC6 is protective during H2O2 stimulation.
Discussion
Type 2 diabetes mellitus generates chronic and acute toxicity to vascular endothelium in patients, including dyslipidemia, hyperglycemia, and increased oxidative stress. This culminates in the development of microvascular (i.e., neuropathy, nephropathy, retinopathy) and macrovascular (i.e., myocardial infarction, peripheral vascular disease, stroke) complications [35].
Besides potent insulin-releasing and glucose-lowering effects, GLP-1 has been proved of its cardiovascular effects and antioxidant effect. However, GLP-1 is unstable for it would be degraded by dipeptidyl peptidase (DPP)-4 which make its bioactivity lose rapidly [36]. To avoid the degradation, we applied the serum-free medium which didn't contain dipeptidyl peptidase (DPP)-4. In addition, cells were pretreated with GLP-1 for 1 h before added H2O2 to avoid the degradation.
Excessive ROS generation response to hyperglycemia plays a pivotal role in the onset and development of diabetes complications. Our study has proved that high glucose increased the ROS generation of HUVECs in vitro. Previous studies showed that H2O2 could induce oxidative damage by enhancing ROS level [37, 38]. Our results showed that GLP-1 treatment could decrease the levels of ROS in HUVECs enhanced by H2O2. It suggests that GLP-1 is of benefit to the H2O2-treated HUVECs cells through decreasing ROS level.
It is well known that excessive ROS accumulation may lead to cell apoptosis and the mitochondrial pathway of apoptosis is regulated by Bcl-2 family members. Thus, we studied the expression of anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax after incubation with H2O2. Our study showed that H2O2 treatment induced cells apoptosis and the GLP-1 treatment reduced the apoptotic levels significantly.
Blood vessels injury occurs early in the disease process, thus, patients often present with cardiovascular disease and its associated complications before being diagnosed with type 2 diabetes mellitus [39-41]. Autophagy is a potent adaptive mechanism, which protects cells against the stress-caused injury by the lysosome-mediated degradation of damaged or dysfunctional organelles and protein aggregates. However, excessively stimulated autophagy in endothelial cells may cause autophagic death. Moreover, the death of endothelial cells may be detrimental for the structure of plaque, because endothelial injury or death will promote lesion thrombosis, which may lead to the occurrence of acute vascular events [12, 13].
LC3 is a crucial autophagy marker. Cytosolic LC3-I is conjugated with phosphatidylethanolamine to form LC3-II during the formation of autophagosomes. This lipidated form remains associated with the autophagosome membranes. Hence, an increased conversion of LC3-I to LC3-II is an indicator of autophagy [42].Our results showed that H2O2 treatment could increase LC3-II/ LC3-I ratio, which indicated that H2O2 activated autophagy pathway, would possibly become excessively stimulated autophagy. This oxidative stress-mediated autophagy effect was significantly attenuated by GLP-1 treatment. To our best knowledge, it is the first report on the effect of GLP-1 on reducing autophagy of endothelial cells subjected to oxidative stress. We further explored the mechanism underlying the effect of GLP-1 treatment on preventing autophagy of endothelial cells and found that H2O2 promoted autophagy in HUVECs cells through activation of the ROS dependent Akt and ERK1/2 signaling pathway. In our study, GLP-1 attenuated H2O2-induced autophagy, as identified by reducing autophagosome formation, accompanied with reducing Akt and ERK1/2 phosphorylation, and this effect was blocked by Ex-9.
As the final effectors of ERK-MAPK pathway, ERK1/2 was reported to interact with HDAC6 [43]. One study showed that reduction of HDAC6 by LBH589 showed elevated ERK1/2 phosphorylation in a prostate cancer cell [44]. In our study, through oxidative stress injury, ERK1/2 phosphorylation was increased with reduction of HDAC6 (Figure 8). The upregulation of ERK1/2 phosphorylation was attenuated by GLP-1 treatment while levels of HDAC6 expression exhibited a contrary tendency.
Schematic diagram of possible mechanism of oxidative stress-induced autophagy and endothelial dysfunction in HUVECs. GLP-1 treatment may protect endothelial cells from oxidative stress-induced autophagy and endothelial dysfunction via GLP-1R-Erk1/2-HDAC6 signaling pathway.
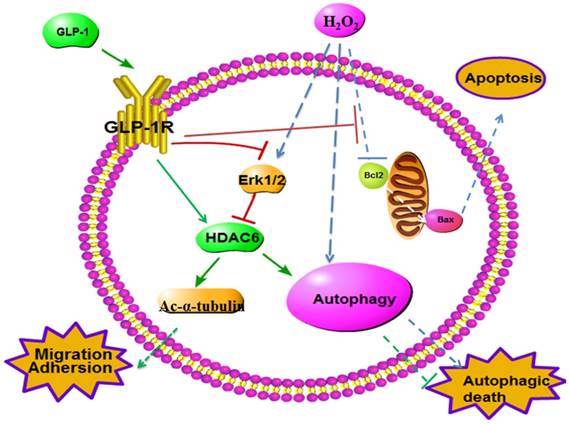
Oxidant injury or inflammation induced by diabetes often causes endothelial dysfunction, subsequently lead to cardiovascular disease [6, 7]. In present study, our findings confirmed that H2O2 impaired the migration and adhesion of HUVECs. Whereas GLP-1 could improve migration and adhesion in H2O2-treated HUVECs, restoring cellular function to similar levels as the non-H2O2-treated cells. To our best knowledge, it is the first report on the effects of GLP-1 on improving endothelial dysfunction induced by oxidative stress.
Epigenetic factor HDAC6 has been reported to be associated tightly with both cell migration and autophagy among all histone deacetylases. Previous study revealed that HDAC6 promoted cell migration by deacetylated cytoplasmic proteins such as α-tubulin [45]. In our study, the reduction of HDAC6 levels induced by oxidative stress led to migration and adhesion impairment, which was reversed by GLP-1 treatment. In addition, HDAC6 mediated retrograde transport of inclusion bodies containing aggregate to be degraded by autophagy through deacetylating α-tubulin [46]. In line with our study, reduction of HDAC6 expression induced by oxidative stress resulted in aggravating autophagy, which was reversed through GLP-1 treatment.
In conclusion, our findings suggest that GLP-1 prevent oxidative stress-induced dysfunction and autophagy in endothelial cells. This anti-autophagic effect of GLP-1 on endothelial cells at an early stage may be crucial for preventing arteriosclerosis which will further lead to serious cardiovascular complications. The protective effects of GLP-1 may be dependent on downstream restore of HDAC6 through a GLP-1R-ERK1/2-dependent manner. These findings suggest the potential therapeutic application of GLP-1 in the prevention and treatment of endothelial damage induced by oxidative stress in diabetic patients.
Acknowledgements
This work was supported by grants from Guangdong Province Science and Technology Fund (No: 2017ZC0208).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xu Y, Wang LM, He J, Bi YF, Li M, Wang TG. et al. Prevalence and Control of Diabetes in Chinese Adults. Jama-J Am Med Assoc. 2013;310:948-58
2. Mazzone T. Intensive Glucose Lowering and Cardiovascular Disease Prevention in Diabetes Reconciling the Recent Clinical Trial Data. Circulation. 2010;122:2201-11
3. Hamamdzic D, Wilensky RL. Porcine Models of Accelerated Coronary Atherosclerosis: Role of Diabetes Mellitus and Hypercholesterolemia. Journal of diabetes research. 2013
4. Chang YC, Chuang LM. The role of oxidative stress in the pathogenesis of type 2 diabetes: from molecular mechanism to clinical implication. Am J Transl Res. 2010;2:316-31
5. Naudi A, Jove M, Ayala V, Cassanye A, Serrano J, Gonzalo H. et al. Cellular Dysfunction in Diabetes as Maladaptive Response to Mitochondrial Oxidative Stress. Exp Diabetes Res. 2012
6. Madamanchi NR, Vendrov A, Runge MS. Oxidative Stress and Vascular Disease. Arteriosclerosis Thrombosis & Vascular Biology. 2000;34:953
7. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo Journal. 2007;26:1749-60
8. Lavandero S, Troncoso R, Rothermel BA, Martinet W, Sadoshima J, Hill JA. Cardiovascular autophagy Concepts, controversies, and perspectives. Autophagy. 2013;9:1455-66
9. Chen YQ, Klionsky DJ. The regulation of autophagy - unanswered questions. Journal of cell science. 2011;124:161-70
10. Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Sign. 2006;8:152-62
11. Xie CM, Chan WY, Yu S, Zhao J, Cheng CHK. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radical Biology & Medicine. 2011;51:1365
12. Liao XH, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS. et al. Macrophage Autophagy Plays a Protective Role in Advanced Atherosclerosis. Cell metabolism. 2012;15:545-53
13. Schrijvers DM, De Meyer GRY, Martinet W. Autophagy in Atherosclerosis A Potential Drug Target for Plaque Stabilization. Arterioscl Throm Vas. 2011;31:2787-91
14. Perrotta I. The use of electron microscopy for the detection of autophagy in human atherosclerosis. Micron. 2013;50:7-13
15. Maiuri MC, Grassia G, Platt AM, Carnuccio R, Ialenti A, Maffia P. Macrophage Autophagy in Atherosclerosis. Mediat Inflamm. 2013
16. Ouimet M. Autophagy in obesity and atherosclerosis: Interrelationships between cholesterol homeostasis, lipoprotein metabolism and autophagy in macrophages and other systems. Bba-Mol Cell Biol L. 2013;1831:1124-33
17. Takemura G, Miyata S, Kawase Y, Okada H, Maruyama R, Fujiwara H. Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy. 2006;2:212-4
18. Saito T, Asai K, Sato S, Hayashi M, Adachi A, Sasaki Y. et al. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy. 2016;12:579-87
19. Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA. et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell death and differentiation. 2007;14:146-57
20. Aaboe K, Krarup T, Madsbad S, Holst JJ. GLP-1: physiological effects and potential therapeutic applications. Diabetes Obesity & Metabolism. 2008;10:994-1003
21. Nystrom T. The potential beneficial role of glucagon-like peptide-1 in endothelial dysfunction and heart failure associated with insulin resistance. Horm Metab Res. 2008;40:593-606
22. Nystrom T, Gutniak MK, Zhang QM, Zhang F, Holst JJ, Ahren B. et al. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am J Physiol-Endoc M. 2004;287:E1209-E15
23. Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Glucagon-like peptide-1 (GLP-1) inhibits advanced glycation end product (AGE)-induced up-regulation of VCAM-1 mRNA levels in endothelial cells by suppressing AGE receptor (RAGE) expression. Biochemical and biophysical research communications. 2010;391:1405-8
24. Oeseburg H, de Boer RA, Buikema H, van der Harst P, van Gilst WH, Sillje HHW. Glucagon-Like Peptide 1 Prevents Reactive Oxygen Species-Induced Endothelial Cell Senescence Through the Activation of Protein Kinase A. Arterioscl Throm Vas. 2010;30:1407-U356
25. Ravassa S, Zudaire A, Carr RD, Diez J. Antiapoptotic effects of GLP-1 in murine HL-1 cardiomyocytes. Am J Physiol-Heart C. 2011;300:H1361-H72
26. Liu JH, Yin F, Guo LX, Deng XH, Hu YH. Neuroprotection of geniposide against hydrogen peroxide induced PC12 cells injury: involvement of PI3 kinase signal pathway. Acta pharmacologica Sinica. 2009;30:159-65
27. Kimura R, Okouchi M, Fujioka H, Ichiyanagi A, Ryuge F, Mizuno T. et al. Glucagon-like peptide-1 (GLP-1) protects against methylglyoxal-induced PC12 cell apoptosis through the PI3K/Akt/mTOR/GCLc/redox signaling pathway. Neuroscience. 2009;162:1212-9
28. Poornima I, Brown SB, Bhashyam S, Parikh P, Bolukoglu H, Shannon RP. Chronic Glucagon-Like Peptide-1 Infusion Sustains Left Ventricular Systolic Function and Prolongs Survival in the Spontaneously Hypertensive, Heart Failure-Prone Rat. Circ-Heart Fail. 2008;1:153-60
29. Marzioni M, Alpini G, Saccomanno S, Candelaresi C, Venter J, Rychlicki C. et al. Exendin-4, a glucagon-like peptide 1 receptor agonist, protects cholangiocytes from apoptosis. Gut. 2009;58:990-7
30. Cunha DA, Ladriere L, Ortis F, Igoillo-Esteve M, Gurzov EN, Lupi R. et al. Glucagon-Like Peptide-1 Agonists Protect Pancreatic beta-Cells From Upotoxic Endoplasmic Reticulum Stress Through Upregulation of BiP and JunB. Diabetes. 2009;58:2851-62
31. Blandino-Rosano M, Perez-Arana G, Mellado-Gil JM, Segundo C, Aguilar-Diosdado M. Anti-proliferative effect of pro-inflammatory cytokines in cultured beta cells is associated with extracellular signal-regulated kinase 1/2 pathway inhibition: protective role of glucagon-like peptide-1. J Mol Endocrinol. 2008;41:35-44
32. Tews D, Lehr S, Hartwig S, Osmers A, Passlack W, Eckel J. Anti-apoptotic Action of Exendin-4 in INS-1 Beta Cells: Comparative Protein Pattern Analysis of Isolated Mitochondria. Horm Metab Res. 2009;41:294-301
33. Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P. et al. HDAC6 Is a Specific Deacetylase of Peroxiredoxins and Is Involved in Redox Regulation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9633-8
34. Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18:291-7
35. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813-20
36. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131-57
37. Wang Y, Liu F, Wei YJ, Liu DC. The effect of exogenous melamine on rat hippocampal neurons. Toxicol Ind Health. 2011;27:571-6
38. Guo CZ, Yuan H, He ZP. Melamine causes apoptosis of rat kidney epithelial cell line (NRK-52e cells) via excessive intracellular ROS (reactive oxygen species) and the activation of p38 MAPK pathway. Cell biology international. 2012;36:383-9
39. Brunner EJ, Shipley MJ, Witte DR, Fuller JH, Marmot MG. Relation between blood glucose and coronary mortality over 33 years in the Whitehall study. Diabetes care. 2006;29:26-31
40. Borch-Johnsen K, Neil A, Balkau B, Larsen S, Nissinen A, Pekkanen J. et al. Glucose tolerance and mortality: comparison of WHO and American Diabetic Association diagnostic criteria. Lancet. 1999;354:617-21
41. Satar N, Gaw A, Scherbakova O. Metabolic syndrome with and without C-reactive protein as a predictor of coronary heart disease and diabetes in the west of Scotland coronary prevention study ☆. Circulation. 2003;12:414-9
42. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542-5
43. Williams KA, Zhang M, Xiang SY, Hu C, Wu JY, Zhang SP. et al. Extracellular Signal-regulated Kinase (ERK) Phosphorylates Histone Deacetylase 6 (HDAC6) at Serine 1035 to Stimulate Cell Migration. Journal of Biological Chemistry. 2013;288:33156-70
44. Chuang MJ, Wu ST, Tang SH, Lai XM, Lai HC, Hsu KH. et al. The HDAC Inhibitor LBH589 Induces ERK-Dependent Prometaphase Arrest in Prostate Cancer via HDAC6 Inactivation and Down-Regulation. PloS one. 2013:8
45. Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S. et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. Embo J. 2003;22:1168-79
46. Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated Huntingtin. Journal of Biological Chemistry. 2005;280:40282-92
Author contact
![]() Corresponding authors: Hongwei Li, Email: lihwedu.cn, Xiaorong Yang, Email: 122424818com, Shaolian Chen, Email: 491274985com
Corresponding authors: Hongwei Li, Email: lihwedu.cn, Xiaorong Yang, Email: 122424818com, Shaolian Chen, Email: 491274985com