Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(12):1732-1744. doi:10.7150/ijbs.24232 This issue Cite
Research Paper
The small molecule NSM00191 specifically represses the TNF-α/NF-кB axis in foot and ankle rheumatoid arthritis
Xiaodong Wen, Xun Chen, Xiaojun Liang, Hongmou Zhao, Yi Li, Xiangxiang Sun ![]() , Jun Lu
, Jun Lu ![]()
Department of Orthopaedics, HongHui Hospital, Xi'an Jiaotong University, Xi'an 710054, Shaanxi, China.
Received 2017-12-6; Accepted 2018-8-23; Published 2018-10-3
Abstract

The activation of TNF-α/NF-кB signaling is involved in the regulation of a wide range of biological processes, such as cell proliferation, differentiation and apoptosis, eventually causing a number of diseases, such as cancer and inflammation. Here, we found that TNF-α/NF-кB signaling was activated in a large number of blood samples taken from foot and ankle rheumatoid arthritis (RA) patients. By applying a microarray assay to the human synovial sarcoma cell line SW982 and the human fibroblast-like synoviocyte cell line HFLS-RA, as well as in their corresponding p65 knockdown and -overexpressing cells, we identified and verified the activation of many p65 targets, including cytokines (e.g., TNF-α and IL-6), chemokines (e.g., MCP-1 and PANTES), protein receptors (e.g., CD-40 and MHC-1), and inducible enzymes (e.g., COX2). In addition, we subjected microRNAs from foot and ankle RA patients to a microRNA-specific microarray and found that miR-7-5p targeted the 3'-UTR of p65, negatively regulating its expression. By applying an in vitro screen to identify small molecules that specifically inhibited the interaction between TRADD and TNFR2, we found that NSM00191 strongly inhibited the activation of TNF-α/NF-кB signaling in vitro and in vivo, causing the downregulation of NF-кB targets and the decrease of arthritis scores. Collectively, our findings shed new light on the regulation of the TNF-α/NF-кB axis and might provide a new avenue for RA treatment.
Keywords: rheumatoid arthritis, miR-7-5p, TNF-α, NF-кB, small molecule
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that occurs when the immune system attacks multiple joints throughout the body [1-3]. Over 90% of RA patients have symptoms in the foot and ankle [1-3]. The chronic inflammatory phase of RA is tightly controlled by a number of inflammatory cytokines (e.g., tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1)) and anti-inflammatory cytokines (e.g., IL-4 and IL-10) [4-6]. Of these cytokines, TNF-α plays a dominant role and functions as an autocrine stimulator or a paracrine inducer of other inflammatory cytokines, such as IL-1, IL-6 and IL-8 [7, 8]. An excessive amount of TNF-α has been identified in the blood and joint of RA patients, especially in the synovial fluid and synovium, where it can affect various cells, including synoviocytes, macrophages, chondrocytes and osteoclasts, resulting in local inflammation and eventually leading to cartilage erosion and bone destruction [9-11]. Moreover, the TNF-α-mediated signaling pathways involved in inflammation have been well characterized. Briefly, TNF-α initiates downstream signaling through two distinct cell-surface receptors, TNFR1 and TNFR2 [12, 13]. The binding of TNF-α causes conformational changes of these receptors, which enables the adaptor protein TRADD (Tumor necrosis factor receptor type 1-associated DEATH domain protein) to bind to the death domain. TRADD further serves as a platform for the binding of other proteins, including receptor-interacting protein (RIP) and TNF receptor-associated factor 2 (TRAF2) [12, 13]. TRAF2 recruits IκB kinase (IKK), which can be activated by RIP [11, 12]. IKK subsequently phosphorylates and causes the degradation of IκBα, which releases its inhibition of nuclear factor of kappa light-chain enhancer of activated B-cells (NF-κB) [12, 13]. The released NF-κB translocates from cytoplasm to the nucleus and regulates the transcription of a number of genes involved in the inflammatory response, such as those encoding cytokines (e.g., TNF-α, IL-2 and IL-6), chemokines (e.g., IL8, MCP-1 (monocyte chemoattractant protein 1) and RANTES (regulated on activation, normal T cell expressed and secreted)), adhesion molecules (e.g., ICAM-1 (intercellular adhesion molecule 1) and VCAM-1 (vascular cell adhesion protein 1)), and some inducible enzymes (e.g., COX-2 (cyclooxygenase-2) and iNOS (inducible nitric oxide synthase)) [12-17]. Although elevated TNF-α has been identified in RA patients [18, 19], whether TNF-α-mediated downstream signaling is also activated in this process and, more importantly, how the signaling is regulated are still unclear.
MicroRNAs (miRNAs) are a class of small endogenous noncoding RNAs (18-25 nucleotides) that bind to the 3′-untranslated region (3′-UTR) of a target mRNA to influence the stability and translation of the mRNA [20]. A variety of miRNAs regulate many physiological actions in RA [21, 22]. For instance, miR-30a, which directly targets the B lymphocyte-activating factor of the tumor necrosis factor family (BAFF), is downregulated in RA synovial tissue [21], which is a critical factor for the autoimmune responses occurring in RA. In RA synoviocytes, miR-124a is downregulated, and this miRNA normally functions to repress the expression of cyclin-dependent kinase 2 (CDK2) and MCP-1 [24]. In addition, some miRNAs, such as miR-16, miR-146b, miR-132, and miR-155, are upregulated in the circulation of RA patients but have unknown targets [25]. Although TNF-α and its mediated signaling pathways play important roles in the pathogenesis of RA, no miRNA has been reported to play a role in the pathogenesis of RA through targeting TNF-α or other members of TNF-α-mediated signaling pathway.
In the past decades, a great number of medications have been developed and widely used for the treatment of RA. These medications mainly include steroids, non-steroidal anti-inflammatory drugs (NSAIDs), disease-modifying anti-rheumatic drugs (DMARDs), TNF-α inhibitors, Janus kinase (JAK) inhibitors, and IL-6 inhibitors [26-28]. However, it may develop resistance after taking these drugs for a long time. On the other hand, these medications have serious side effects, such as tinnitus and heart, liver and kidney damage [26-28]. To overcome these side effects, new medications must be developed.
In the current study, we made three important advancements in RA: (1) verified the activation of TNF-α/NF-κB axis signaling in foot and ankle RA patients; (2) identified a miRNA known as miR-7-5p, which can bind to the 3'-UTR of p65 and negatively regulate its expression; and (3) obtained a small molecule named NSM00191 through high-throughput screening (HTS) that can repress the activation of TNF-α/NF-κB signaling, causing the downregulation of NF-κB targets and the decrease of arthritis scores.
Material and methods
Cells and Cell culture
The Human Fibroblast-Like Synoviocytes: Rheumatoid Arthritis (HFLS-RA) cell line was purchased from Cell Applications (USA). The human synovial cell line SW982 was obtained from the American Type Culture Collection (ATCC, USA). Cells were first grown in synoviocyte growth medium (Cell Applications) for 24 h but were then switched to RPMI 1640 medium (Gibco, USA), supplemented with 10% heat-inactivated fetal bovine serum (FBS), 4 mM L-glutamine (Thermo Fisher Scientific, USA) and 100 U ml-1 penicillin-streptomycin (Thermo Fisher Scientific, USA) at 37°C with 5% CO2.
Blood samples and Joint tissue specimens
Blood samples and joint tissue specimens were obtained from RA patients who underwent surgery and therapy from September 2015 to July 2016 at the Department of Orthopedics, Hong-hui Hospital, School of Medicine, Xi'an Jiao Tong University in Shaanxi, China. The clinical characteristics of the patients are summarized in Supplementary Table 1. Blood samples were immediately placed in glass tubes containing EDTA, followed by centrifugation to isolate the plasma. The concentrations of TNF-α, IL-1, IL-6 and IL-8 in plasma were measured with enzyme-linked immunosorbent assay (ELISA) kits (Sigma, USA). The clinical samples were obtained in accordance with written informed consent from all the participants following protocols approved by the ethical board of Xi'an Jiao Tong University. All experimental procedures used in this study were carried out in accordance with the approved guidelines of the Ethics Committee of Xi'an Jiao Tong University.
Immunofluorescence (IMF) and Immunohistochemistry (IHC)
For IMF staining, joint tissue samples were fixed in 10% formalin at 4°C overnight, embedded in paraffin, and sectioned to 5-μm thickness from the midline of the samples. The tissue sections were mounted on slides and cleared with xylene twice for 20 min. The slides were then rinsed with 100% alcohol (18:1:1 ethanol:methanol:isopropanol) twice for 2 min, followed by rinsing with 95% and 80% alcohol twice for 2 min. The slides were blocked with 1% BSA for 30 min at room temperature. After incubation with the primary antibody against p65 (rabbit, Abcam, USA) and a secondary antibody, the tissue sections were counterstained with DAPI. For IHC staining, the tissue sections were deparaffinized following standard procedures, the antigens were unmasked, and the sections were incubated with the anti-IKK2 (rabbit, Abcam, USA), anti-TNFR1 (rabbit, Abcam, USA), anti-E2F3a (rabbit, Abcam, USA) or anti-p65 antibody and the appropriate secondary antibody. Images were captured using a Leica DM 500B fluorescence microscope.
Western blot analysis
Cells and tissue samples were lysed in Pierce RIPA buffer (Thermo Scientific, USA) supplemented with 1 mM Na3VO4, 50 mM NaF, and 1× complete protease cocktail inhibitor (Roche, USA) to obtain the total proteins. After denaturation at 95°C for 5 min, equal amounts of proteins were loaded onto a 12% SDS-PAGE gel for separation. The specific primary antibodies used for Western blots were anti-p65 (rabbit, Abcam, USA), anti-TNFR1 (rabbit, Abcam, USA), anti-IKK2 (rabbit, Abcam, USA), and anti-GAPDH (mouse, Sigma, USA). The Western blot signals were recorded using a ChemiDoc MP Imaging System (Bio-Rad, USA).
Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated using the Qiagen Mini Kit (for gene expression analysis, Qiagen, USA) or the miRNeasy Mini Kit (for miRNA expression analysis, Qiagen, USA) following the manufacturer's guidelines. After the generation of cDNA with the M-MLV reverse transcriptase (for gene expression analysis, Promega, USA) or TaqMan MicroRNA Reverse Transcription kit (for miRNA expression analysis, Thermo Fisher Scientific, USA), qRT-PCR analysis was performed on a Bio-Rad CFX96 Real-Time PCR system (Bio-Rad, USA) using the specific primers listed in Supplementary Table 2. Actin and RNU6B were chosen as internal controls to normalize the gene and miRNA expression, respectively.
Microarray analyses
Microarray analyses of genes and miRNAs were performed as previously described [29, 30]. Briefly, the total RNAs were isolated from cells and clinical samples using the Qiagen Mini Kit (Qiagen, USA) or the miRNeasy Mini Kit (Qiagen, USA), respectively. Then, 1 μg RNA of each sample was labeled with the GeneChip 3' In Vitro Transcription (IVT) Express Kit (Affymetrix Inc., USA) or the miRCURY™ Hy3/Hy5 Power kit (Exiqon, Vedbaek, Denmark) following the manufacturers' guidelines. This process was followed by hybridization on an array chip at 45°C for 16 h. The data were obtained by subjecting slides and signals to scanning in the Axon GenePix 4000B microarray machine (Axon Instruments, USA).
Luciferase assay
HFLS-RA cells were transfected with the following combinations of plasmids: (1) miR-7-5p mimic + pGL4.26-Pp65-WT-Luc (firefly luciferase reporter vector) + pRL-TK (Renilla luciferase vector); (2) miR-NC (negative control) + pGL4.26-Pp65-WT-Luc + pRL-TK; (3) miR-7-5p mimic + pGL4.26-Pp65-Mutant-Luc + pRL-TK; and (4) miR-NC + pGL4.26-Pp65-Mutant-Luc + pRL-TK. After incubation at 37°C for 48 h, the cells were lysed and subjected to a luciferase assay using the Dual Luciferase Reporter Assay System (Promega, USA).
Small molecule screening
The pGADT7-TNF-α and pGBKT7-TNFR1 plasmids were co-transfected into wild-type yeast cells (BY4741). The yeast colonies expressing TNF-α and TNFR1 were picked and subjected to growth analysis in amino acid-deficient medium. After the determination of growth conditions, yeast cells were grown to mid-log phase (OD600=1.0) at 28°C for 24 h. The cells were then diluted 10-fold to OD600=0.1 with the same fresh medium, and 49 μl cells was added into individual wells of 96-well plates. Equal volumes of small molecules (1.0 μl) were then independently added into each well at a final concentration of 20 μM. The 96-well plates were placed in a 28°C incubator to grow the cells for another 18 h. Then, the cell density was determined at 600 nm using the Synergy HTX Multi-Mode Reader (BioTek, USA). The candidate compounds were assessed with a standard OD600<0.2 and subjected to a secondary round of screening under the same conditions to determine the reproducibility of growth inhibition.
Mice treatment with NSM00191 and assessment of arthritis
Six-week-old female DBA/1 mice (The Jackson Laboratory, USA) and TNF-α-overexpressing transgenic mice (Taconic Biosciences, USA) were maintained in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Xi'an Jiao Tong University. The mice were intraperitoneally injected with 20 μM NSM00191 (100 μl) or control DMSO (100 μl) for seven days, and then, plasma samples and joint tissue specimens were collected and subjected to the required experiments.
For macroscopic assessment of NSM00191 effect on improving arthritis, six-week-old female DBA/1 mice and TNF-α mice were intraperitoneally injected with 20 μM NSM00191 (100 μl) or control DMSO (100 μl) every two days for a total of 14 days. At the same time, the thickness of hind paws of mice was measured with microcalipers (Kroeplin GmbH, Germany) every two days. In addition, we also scored the clinical signs of mice with the standards as described previously [31, 32]. The normal mice were scored as 0, and 1, 2, 3 and 4 represented mild, moderate, severe and complete erythema, or swelling of the wrist (or ankle), respectively. The arthritic score of individual mouse was obtained by summing the scores recorded for each limb. To maintain the impartiality, this experiment was performed by three investigators in a blinded manner and the mean scores were calculated.
Statistical analysis
The data are presented as the means ± SE. All experiments in this study were independently replicated at least three times. The data were analyzed using the independent t-test (SPSS, USA). P < 0.05 was considered statistically significant.
Results
Elevated levels of pro-inflammatory cytokines in RA plasma samples
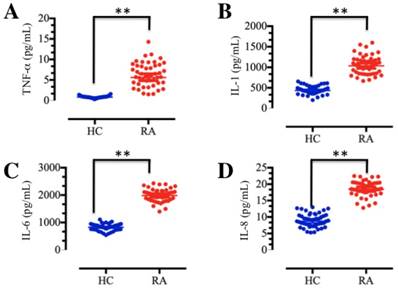
TNF-α and IL-1 are the major macrophage-derived cytokines, and they play important roles in the rheumatoid joint by inducing the synthesis and secretion of IL-6, IL-8 and granulocyte-macrophage colony-stimulating factor (GM-CSF) from synovial fibroblasts [7, 11]. To determine the concentrations of these soluble pro-inflammatory cytokines in foot and ankle RA patients, we collected a total of 48 blood samples from both foot and ankle RA patients and healthy volunteer participants without inflammation. The isolated plasma was subjected to ELISA, which indicated that TNF-α, IL-1, IL-6 and IL-8 levels were significantly elevated in foot and ankle RA patients compared to those in healthy controls (Figure 1). These results are consistent with previous publications [9-11], and further indicate that inflammation is very common in RA pathogenesis.
The levels of plasma cytokines were upregulated in RA patients. Blood samples were obtained from RA patients (RA, n=48) and healthy volunteer donors (HC, n=48). The samples were subjected to ELISAs to measure the levels of cytokines, including TNF-α (A), IL-1 (B), IL-6 (C) and IL-8 (D). ** P <0.001.
Activation of the TNF-α/NF-κB signaling pathway in RA patients
Given that TNF-α is a typical molecule that stimulates the activation of transcription factor NF-κB [17], we next sought to determine if elevated TNF-α expression in RA patients was able to cause the activation of NF-κB. Accordingly, we randomly selected paired tissue samples from 4 RA patients and 4 healthy controls to measure the levels of critical members of the TNF-α/NF-κB signaling pathway. As shown in Figure 2A, the protein levels of TNFR1, IKK2 and p65 were significantly induced in RA patients compared to the levels in the healthy controls. We also examined the histological changes of TNFR1, IKK2 and p65 using IHC staining in these 4 paired tissue samples. Consistent with the above results, the TNFR1, IKK2 and p65 stains were much more obvious in RA patient tissues than in healthy control tissues, but not E2F3a, a known target of NF-κB (Figure 2B).
The TNF-α/NF-κB signaling pathway was activated in RA patients. Four paired joint tissue specimens from RA patients (RA) and healthy volunteer donors (HC) were subjected to Western blot (A) and IHC staining assays (B) to examine the TNFR1, IKK2, p65 and E2F3a levels. Bars = 100 µm. (C) Two paired samples used in B (HC-1/RA-1, and HC-3/RA-3) were used for IMF staining with the anti-p65 antibody. The localization and levels of p65 are shown in green (left panel). The nuclei were counterstained with DAPI (blue, middle panel), and the merged images are in the right panel. Bars = 25 µm.
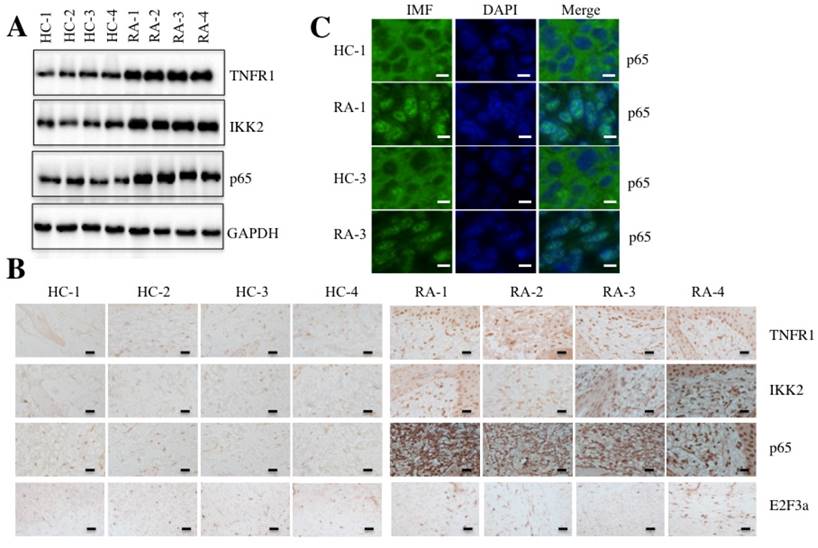
NF-κB translocates from the cytoplasm to the nucleus with TNF-α stimulation [33]. To determine if this also happens in RA patients, we performed an IMF staining assay to evaluate the native protein levels of p65 in paired RA patient samples from two patients (1 and 3). As shown in Figure 2C, the IMF results indicated that p65 translocated to the nucleus in RA patient samples significantly more than in healthy control tissues.
The inflammatory targets of NF-κB were universally activated in RA patients
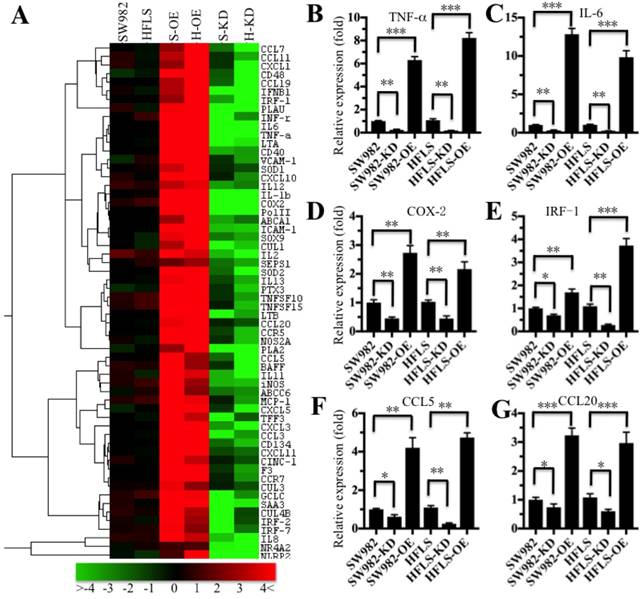
The translocation of NF-κB from the cytoplasm to the nucleus is critical for coupling extracellular stimuli to the transcriptional activation of specific target genes [33]. To determine which NF-κB target gene expression was changed in RA patients, we primarily evaluated the candidate target genes in both HFLS-RA and SW982 cells expressing different p65 levels. Accordingly, we constructed and obtained p65 overexpression (H-OE and S-OE) and knockdown (H-KD and S-KD) cells in both HFLS-RA and SW982 backgrounds. After verification of the p65 levels in these cells (Supplementary Figure 1), we subjected the mRNAs from p65-overexpressing and downregulated cells, as well as their parental cells, to microarray analysis. The hierarchical clustering analysis indicated distinctive mRNA expression patterns between the p65-overexpressing cells, p65 downregulated cells and their parental cells (Figure 3A). Overall, 134 genes and 98 genes were identified to have consistently aberrant expression in p65-overexpressing and downregulated cells, respectively, in comparison to their parental cells. By comparing gene expression levels in these three different p65-background cells, we found 61 genes were consistently upregulated in p65-overexpressing cells but downregulated in p65 knockdown cells in comparison to their parental cells (Figure 3A). Of these 61 genes, over 80% were identified as downstream targets of NF-κB. For instance, several cytokines (e.g., IL-1β, TNF-α, IL-6 and INF-γ), chemokines (e.g., CCL5, CCL7, CCL11, CCL20 and MCP-1), immunoreceptors (e.g., CD40, CD48 and CCR5), cell adhesion molecules (e.g., ICAM-1 and VACM-1), and stress response genes (e.g., COX-2, SOD1 and SOD2) were upregulated in p65-overexpressing cells but downregulated in p65-knockdown cells (Figure 3A). To verify the accuracy of the microarray results, we randomly selected 6 genes, TNF-α, IL-6, COX-2, IRF-1, CCL5 and CCL20, as representatives to examine their expression in different p65-background cells using qRT-PCR. Consistent with the microarray results, the levels of TNF-α, IL-6, COX-2, IRF-1, CCL5 and CCL20 were upregulated by 2.5- to 14-fold in SW982-OE and HFLS-OE cells, whereas they were downregulated by 2- to 5-fold in SW982-KD and HFLS-KD cells compared with the levels in their parental cells (Figure 3B).
The inflammatory targets of NF-κB were activated in p65-overexpressing cells. The heat maps of the consistently altered genes in p65-overexpressing and knockdown cells are shown. The RNA from SW982 cells, SW982 cells overexpressing p65 (S-OE), SW982 cells with knockdown of p65 (S-KD), HFLS-RA cells (HFLS), HFLS-RA cells overexpressing p65 (H-OE), and HFLS-RA cells with knockdown of p65 (H-KD) were subjected to microarray analysis. The elevated genes are indicated in red, and the downregulated genes are shown in green. qRT-PCR was performed to verify the expression of TNF-α (B), IL-6 (C), COX-2 (D), IRF-1 (E), CCL5 (F), and CCL20 (G) in SW982, S-OE, S-KD, HFLS, H-OE and H-KD cells. * P <0.05; ** P < 0.001; *** P <0.0001.
Given that the microarray results were obtained from in vitro cell lines, we next examined if these candidate targets were also dysregulated in RA samples. Accordingly, we selected a total of 12 known NF-κB targets that play roles in different biological processes as representatives to examine their expression in paired tissues from 4 RA patients and 4 healthy controls (Samples were the same as those used in Figure 2). These genes included TNF-α, IL-6, MCP-1, CCL5, COX-2, VACM-1, CD40, MHC-1, two regulators of apoptosis (Bax and Bcl-2), and two transcription factors (c-Myc and E2F3a). Consistent with the results from the microarray results, TNF-α, IL-6, MCP-1, CCL5, COX-2, VACM-1, CD40 and MHC-1 expression was significantly upregulated in RA samples (Supplementary Figures 2A-2H), while no obvious change was identified in the expression of Bax, Bcl-2, c-Myc or E2F3a (Supplementary Figures 2I-2L). These results suggested that the inflammatory targets of NF-κB were universally activated in RA patients.
Identification of the miRNA profile involved in RA pathogenesis
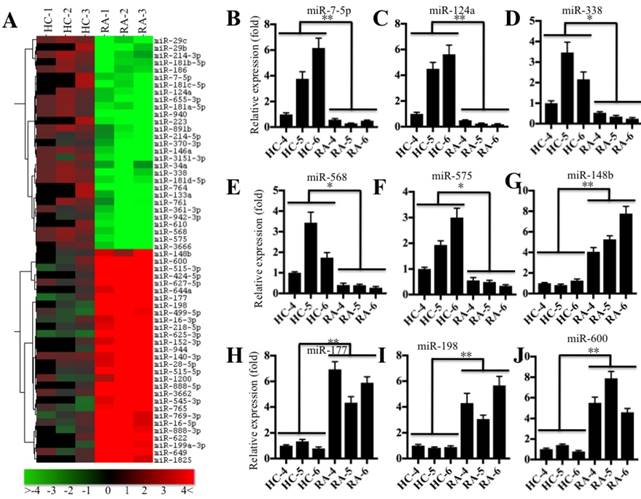
Based on the notion that miRNA expression is highly variable in different biological processes, even in the same disease but different samples [22], we next sought to investigate the miRNA expression profile in the pathogenesis of RA. Accordingly, we subjected RNA samples labeled with miRCURY™ Hy3/Hy5 to microarray analysis and then compared the miRNA expression in RA patients (RA-1 to -3) and healthy controls (HC-1 to -3). Hierarchical clustering analysis revealed distinctive miRNA expression patterns between RA patients and healthy controls (Figure 4A). Overall, 56 miRNAs had consistently aberrant expression in all RA patients compared to healthy controls. Of them, 29 miRNAs were downregulated, and the others were upregulated (Figure 4A). To verify the accuracy of the microarrays, we randomly selected 5 downregulated miRNAs, miR-7-5p, miR-124a, miR-338, miR-568 and miR-575, and 4 upregulated miRNAs, miR-148b, miR-177, miR-198 and miR-600, as representatives to examine their expression in RA patients (RA-4 to -6) and healthy controls (HC-4 to -6), which were different from the samples used in microarray analysis. Consistent with the microarray results, the qRT-PCR results indicated that the expression levels of miR-7-5p, miR-124a, miR-338, miR-568 and miR-575 were dramatically decreased (Figures 4B-4F), whereas the expression levels of miR-148b, miR-177, miR-198 and miR-600 were significantly upregulated in RA patients compared to the levels in healthy controls (Figures 4G-4J).
miR-7-5p was significantly downregulated in RA patients. (A) The heat maps of the consistently altered miRNAs in RA patients are shown. RNA samples from three paired joint tissue specimens (HC-4, -5, -6 and RA-4, 5, -6) were subjected to miRNA microarray analysis. The elevated miRNAs are indicated in red, and the downregulated miRNAs are shown in green. qRT-PCR was performed to verify the expression of miR-7-5p (B), miR-124a (C), miR-338 (D), miR-568 (E), miR-575 (F), miR-148b (G), miR-177 (H), miR-198 (I) and miR-600 (J) in the RNA samples used in A. * P <0.05; ** P < 0.001.
MicroRNA-7-5p directly targeted the 3'-UTR of p65
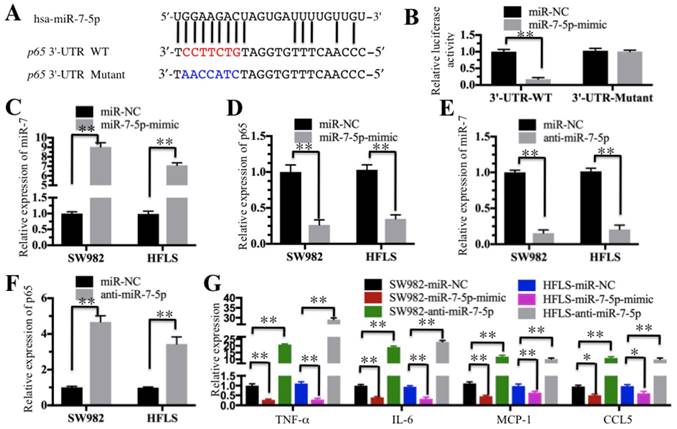
Our above results indicated the TNF-α/NF-κB signaling pathway is activated and a number of miRNAs are downregulated in RA patients. We next determined if these dysregulated miRNAs could target critical members of the TNF-α/NF-κB signaling pathway. By searching possible miRNA targets in an online database (miRDB, http://www.mirdb.org), we found that miR-7-5p was predicted to bind to the 3'-UTR of the p65 gene (Figure 5A). To verify this prediction, we constructed luciferase reporter vectors containing either wild-type (WT) or mutant 3′-UTR of p65 and then co-transfected them with miR-7-5p-mimic into HFLS-RA cells. The luciferase assay results indicated that miR-7-5p significantly decreased the luciferase activity in cells containing p65-3′-UTR-WT but failed to repress the luciferase activity in cells containing 3′-UTR-mutant (Figure 5B). These results suggest that miR-7-5p might direct bind to the p65 3′-UTR. To further verify this conclusion, we also transfected a miR-7-5p-mimic into SW982 and HFLS-RA cells. After confirmation of the miR-7-5p expression using qRT-PCR in these two cell lines (Figure 5C), we examined the p65 level and found that p65 expression was significantly downregulated with the overexpression of miR-7-5p (Figure 5D). Similarly, we evaluated the effect of a miR-7-5p inhibitor (anti-miR-7-5p) on p65 expression in SW982 and HFLS-RA cells. With the downregulation of miR-7-5p in these two cell lines (Figure 5E), p65 expression was significantly increased (Figure 5F). These results further demonstrate that miR-7-5p was able to repress p65 expression. We next sought to determine if the changes in miR-7-5p level might affect the expression of NF-κB downstream targets. Using the cells in Figures 5C and 5E, we examined the expression of TNF-α, IL-6, MCP-1 and CCL5. As shown in Figure 5G, with the overexpression of miR-7-5p, these four genes were dramatically downregulated. Conversely, with the downregulation of miR-7-5p, these four genes were significantly induced.
miR-7-5p specifically targeted p65. (A) The 3′-UTR of p65 contains a putative miR-7-5p binding site. The seed binding sequence of miR-7-5p in the WT 3′-UTR of p65 is indicated in red, while it is indicated in blue for mutant 3′-UTR. (B) The mutant of p65 3′-UTR failed to bind miR-7-5p. The miR-7-5p-mimic or its negative control (miR-NC) was co-transfected with pMIR-p65-3′-UTR-WT or pMIR-p65-3′-UTR-mutant into HFLS-RA cells for 24 h. The luciferase activity was quantified using the dual-luciferase assay reporter system by normalizing the signal to the Renilla activity. (C-F) The expression of miR-7-5p was negatively correlated with the p65 level. The miR-7-5p-mimic, anti-miR-5p or miR-NC was transfected into SW982 or HFLS-RA (HFLS) cells. The expression of miR-7-5p and p65 was determined in these cells. (G) The overexpression or downregulation of miR-7-5p affected the expression of NF-κB downstream targets. The cells used in C-F were subjected to qRT-PCR to examine the expression of NF-κB downstream targets, including TNF-α, IL-6, MCP-1 and CCL5. ** P < 0.001.
NSM00191 specifically disrupted the TRADD-TRAF2 interaction
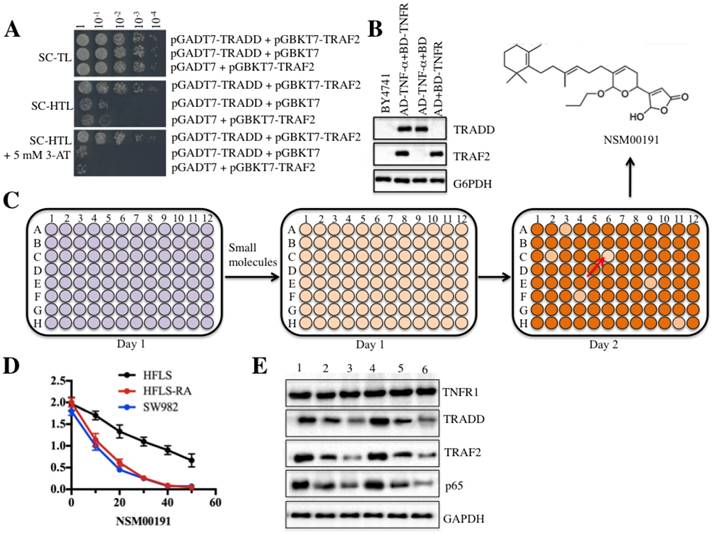
As mentioned earlier, current medications for RA treatment have serious side effects [26]. To identify some small molecules that specifically target critical members involved in RA pathogenesis, we set up a small molecule pool containing 50,000 chemicals derived from plants and marine species. We then developed an in vitro HTS method in yeast cells through disrupting the interaction between TRADD and TRAF2. After verification of the growth of yeast cells harboring pGADT7-TRADD and pGBKT7-TRAF2 in nutrient-deficient medium and the protein levels of TRADD and TRAF2 (Figures 6A and 6B), we placed equal volumes of yeast cells into 96-well plates supplemented with an individual small molecule in each well. The candidate small molecules were assessed based on cell density (OD600<0.2) after 18 h of incubation (Figure 6C). Fortunately, we identified a series of small molecules that exhibited different abilities to inhibit yeast cell growth. One small molecule, known as NSM00191, showed the strongest ability to inhibit cell growth. Its chemical structure is shown in Figure 6C. To verify its cytotoxic activity, we utilized different concentrations of NSM00191 (0, 10, 20, 30, 40 and 50 μM) in the treatment of the HFLS, HFLS-RA and SW982 cells to evaluate cell proliferation. As shown in Figure 6D, NSM00191 significantly reduced the cell proliferation of HFLS-RA and SW982 cells, with an IC50 growth-inhibitory concentration of nearly 10 µM. In addition, the protein levels of TRADD, TRAF2 and p65 but not TNFR1 were dramatically downregulated with NSM00191 treatment in HFLS-RA and SW982 cells (Figure 6E). These results demonstrated that NSM00191 might specifically function as an inhibitor to disrupt the interaction between TRADD and TRAF2 but not affect the upstream members of TRADD (e.g., TNFR1) in the TNF-α/NF-κB signaling pathway.
The small molecule NSM00191 specifically disrupted the interaction between TRADD and TRAF2. TRADD interacted with TRAF2 in yeast cells. Different combinations of plasmids, including pGADT7-TRADD + pGBKT7-TRAF2; pGADT7-TRADD + pGBKT7; and pGADT7 + pGBKT7-TRAF2, were co-transformed into BY4741 cells. Cell growth was determined in media with different nutrient deficiencies, including the lack of Trp or Leu (SC-TL) (top panel), the lack of Trp, Leu and His (SC-HTL) (middle panel), and SC-HTL supplemented with 5 mM 3-amino-1,2,4-trizole (3-AT). Columns in each panel represent the serial decimal dilutions. (B) The protein levels of TRADD and TRAF2 in yeast cells. A Western blot was performed to examine the protein levels of TRADD and TRAF2 in yeast cells used in (A). (C) Schematic representations of screening small molecules that disrupt the TRADD-TRAF2 interaction. The yeast cells harboring pGADT7-TRADD + pGBKT7-TRAF2 were subjected to HTS in the SC-HTL+5 mM 3'-AT medium supplemented with an individual compound in each well. Small molecules that significantly inhibited cell growth (OD600<0.2) were selected. The chemical structure of NSM00191 is shown. (D) NSM00191 significantly inhibited SW982 and HFLS-RA cell proliferation. The HFLS, HFLS-RA and SW982 cells were treated with different concentrations (0, 10, 20, 30, 40 and 50 μM) of NSM00191 for 48 hr. Cell viability was determined at 490 nm. (E) NSM00191 specifically disrupted the TRADD-TRAF2 interaction in HFLS-RA and SW982 cells. The HFLS-RA and SW982 cells were first treated with DMSO (lanes 1 and 4), 10 μM NSM00191 (lanes 2 and 5), or 20 μM NSM00191 (lanes 3 and 6), followed by the detection of TNFR1, TRADD, TRAF2 and p65 by Western blot. GAPDH was used as a loading control.
NSM00191 greatly improved inflammation status in RA mouse models
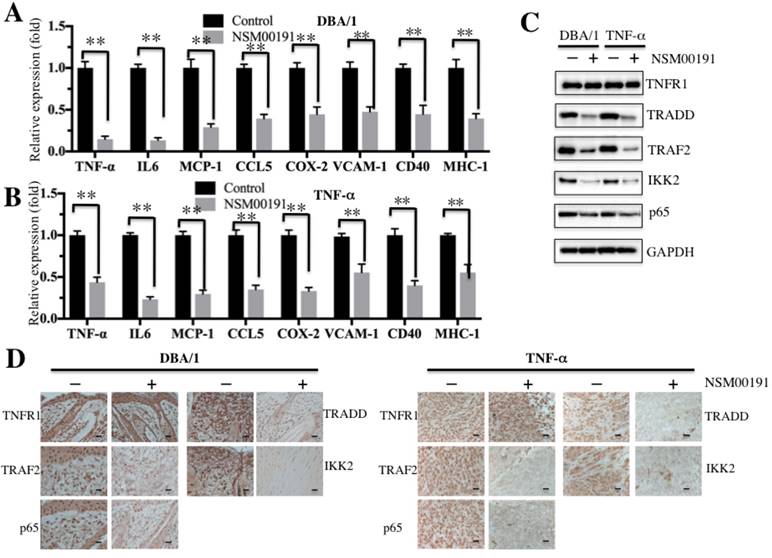
Next, we selected a collagen-induced arthritis mouse model, DBA/1, and a TNF-α-overexpression transgenic mouse model, which have been widely used in the investigation of RA pathogenesis [34], to further evaluate the in vivo effect of NSM00191. The mice were intraperitoneally injected with 20 μM NSM00191 or control DMSO for 7 days, and then, plasma samples and joint tissue specimens were collected. The ELISA results indicated that NSM00191 treatment significantly decreased TNF-α, IL-1, IL-6 and IL-8 expression compared to DMSO treatment in mice from both backgrounds (Figures 7A and 7B). Moreover, we also evaluated the effect of NSM00191 on clinical progression of arthritis. The paw thickness and arthritis scores were significantly reduced in mice treated with NSM00191 compared to those untreated mice (Supplementary Figure 4). In addition, we also detected the protein levels of TNFR1, TRADD, TRAF2, IKK2 and p65 in joint tissues. As shown in Figure 7C, NSM00191 treatment significantly decreased the protein levels of TRADD, TRAF2, IKK2 and p65 but not TNFR1. Consistent with the Western blot results, the IHC staining results also indicated a decrease in TRADD, TRAF2, IKK2 and p65 expression in mice treated with NSM00191 (Figure 7D). These results clearly demonstrate the in vivo efficiency of NSM0091, which might provide a new possibility for the development of a therapeutic reagent disrupting the interaction between TRADD and TRAF2.
NSM00191 significantly improved the inflammatory response in vivo. (A-B) NSM00191 significantly inhibited the expression of NF-κB downstream targets. The DBA/1 mice (A, n=12) and TNF-α-overexpressing mice (B, n=12) were divided into two groups. One group of mice was treated with DMSO; the other group mice were treated with 20 μM NSM00191 for 7 days. Then, the joint tissues were collected, and their RNAs were isolated. The RNAs from each treatment mice were equally combined and subjected into qRT-PCR analyses to examine the expression of TNF-α, IL-6, MCP-1, CCL5, COX-2, VCAM-1, CD40 and MHC-1. (C) NSM00191 inhibited the interaction between TRADD and TRAF2 in vivo. The joint samples used in A and B were subjected to Western blot analysis to determine the protein levels of TNFR1, TRADD, TRAF2, IKK2 and p65. GAPDH was used as a loading control. (D) NSM00191 decreased the p65 levels in vivo. One pair of joint samples used in A and B were subjected to IHC staining to determine the protein levels of TNFR1, TRADD, TRAF2, IKK and p65.
Discussion
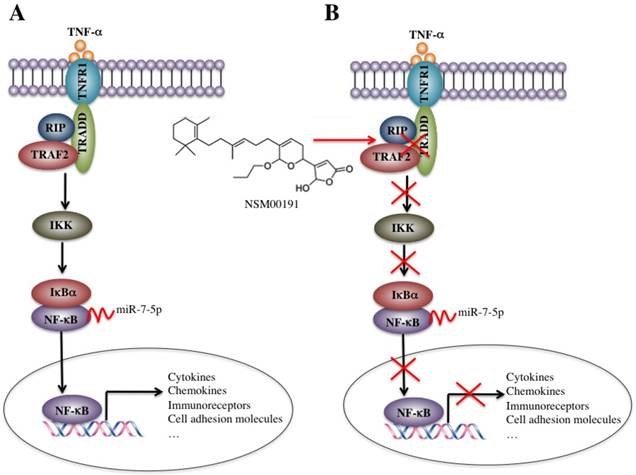
In the rural areas of northwestern China, RA, especially the foot and ankle RA, is very common in people over 50 years old, which causes a heavy burden on families and society. However, the traditional medications employed in the medical therapy of RA, including NSAIDs, DMARDs, cannot always effectively prevent the development of foot and ankle RA in our clinical observations, which motivated us to conduct more in-depth studies on the pathogenic mechanisms of foot and ankle RA, identify more targets and develop new therapeutic medications. In the present study, we found that the plasma levels of TNF-α, IL-1, IL-6 and IL-8 were significantly elevated in foot and ankle RA patients. The activated TNF-α/NF-κB signaling pathway caused the universal activation of NF-κB downstream targets, such as cytokines, chemokines, stress response genes, cell adhesion molecules and immunoreceptors. Interestingly, a number of miRNAs were dysregulated in foot and ankle RA patients, and one miRNA, known as miR-7-5p, specifically bound to the 3'-UTR of the p65 gene and negatively regulated its expression. Using an in vitro HTS assay, we discovered a small molecule known as NSM00191 that was able to specifically disrupt the interaction between TRADD and TRAF2 and thus greatly inhibit inflammation status in vitro and in vivo (Figure 8).
Schematic diagrams of the activation of TNF-α/NF-κB and NSM00191 function in RA patients. (A) The elevated levels of TNF-α initiate signaling transduction by interacting with the membrane receptor TNFR1. TNFR1 further interacts with TRADD and recruits TRAF2 and RIP to form a complex. This complex activates IKK2, enabling the phosphorylation of IκBα, eventually causing the release of NF-κB. The increased level of NF-κB in the cytoplasm then translocates to the nucleus and induces the transcription of a number of genes, such as cytokines, chemokines, immunoreceptors and cell adhesion molecules. At the transcriptional level, a microRNA known as miR-7-5p can specifically bind to the 3'-UTR of p65, a subunit of NF-κB. (B) A small molecule called NSM00191 specifically disrupts the interaction between TRADD and TRAF2, causing the inhibition of TRAF2 downstream signaling transduction, which eventually results in the transcriptional inhibition of NF-κB downstream targets.
TNF-α is the prime cytokine that mediates inflammation in RA [35]. The current understanding of TNF-α function in RA mainly focuses on its roles in autoimmune response and mediating downstream signaling [10, 35]. In local inflammatory tissues, the costimulation-dependent interactions between dendritic cells, T-cells, and B-cells lead to T-cell activation, which in turn activates NF-κB signaling and upregulates a wide range of pro-inflammatory cytokines, chemokines, and metabolic proteins [7, 36]. Consistent with previous studies, we found elevated TNF-α expression in the foot and ankle RA samples (Figure 1) and the activation of NF-κB signaling, as well as the upregulation of a variety of NF-κB targets, such as TNF-α, IL-6, MCP-1, CCL5, COX-2, VACM-1, CD40 and MHC-1 (Figure 3 and Supplementary Figure 2). Due to the limitations of our laboratory, we did not isolate the dendritic cells, T-cells or B-cells in foot and ankle RA samples. Our experimental results come from a sum of all cell types in inflammatory tissues, which limit our understanding of how TNF-α induces inflammation and whether the activation of NF-κB was cell type-specific. In addition to TNF-α, various stimuli, including IL-1, cytokine receptors (TNFR1 and 2), and pattern-recognition receptors (PRRs) (e.g., Toll-like receptor 7 and 9), can activate NF-κB signaling [16, 37, 38]. Although we also found elevated IL-1 levels in the serum of foot and ankle RA patients (Figure 1B) and observed upregulated IL-1β expression in p65-overexpressing cells but repressed IL-1β expression in p65 knockdown cells (Figure 3A), we did not investigate IL-1-mediated signaling in the current study, further limiting our understanding of the pathogenesis of foot and ankle RA because we do not know whether the TNF-α-mediated signaling pathway is the most important pathway leading to inflammation in the absence of IL-1- or other stimulus-mediated activation.
Although numerous studies have identified aberrant miRNA expression profiles in RA patients, only a few of these miRNAs have been well studied and have known targets [21-25, 39-41]. For instance, miR-22 can target the 3'-UTR of cysteine-rich angiogenic inducer 61 (CYR61), regulating inflammation response in the synovial cells from RA patients [39]. MiR-23b can bind to the 3'-UTRs of three genes, including TGF-β-activated kinase 1/MAP3K7- binding protein 2 (TAB2), TAB3 and IKK-α, resulting in the suppression of IL-17-, TNF-α- or IL-1β-mediated activation of NF-κB [40]. In this study, we found a total of 56 miRNAs with aberrant levels in foot and ankle RA patients, only a few of which have been reported to be involved in RA pathogenesis. For instance, miR-124a acts as a negative regulator of MCP-1 in fibroblast-like synoviocytes from RA patients [42]. The DNA methylation-mediated downregulation of miR-34a in RA synovial fibroblasts can promote resistance to apoptosis by targeting X-linked inhibitor of apoptosis protein (XIAP) [43]. In addition, miR-146a is able to target tumor necrosis factor receptor-associated factor 6 (TRAF6) and IL-1 receptor-associated kinase 1 (IRAK-1) [44]. Although we only investigated the role of miR-7-5p here, we also found a few miRNAs that might target other TNF-α/NF-кB signaling members and NF-кB downstream targets by predicting their candidate targets in the miRNA database. For instance, miR-214-5p might bind to the 3'-UTR of TRAF2 (Supplementary Figure 3A), and a cluster of miRNAs, including miR-181a-5p, miR-181b-5p, miR-181c-5p and miR-181d-5p, might target the 3'-UTR of TNF-α (Supplementary Figure 3B). We are currently verifying and investigating the roles of these miRNAs in the pathogenesis of RA but do not stress their importance in the present study. In addition, among these TNF-α/NF-кB signaling members, only IκBα was downregulated in RA patients [45, 46]. We found an upregulated miRNA, miR-16-5p, was predicted to bind to the 3'-UTR of IκBα (Supplementary Figure 3C). These results indicated that not only the downregulated miRNAs but also the upregulated miRNAs might contribute to the pathogenesis of RA. Moreover, cytokines such as TNF-α, IL-1β and IL-17 have been shown to play roles in the regulation of miRNA expression [47-49]. For instance, both TNF-α and IL-1β can induce miR-146a expression [50], while IL-17 can suppress miR-23b expression in RA [40]. We did not examine whether TNF-α or IL-1β can regulate the miR-7-5p level in this study, and we will conduct these experiments in the future.
With the well-known molecular mechanisms of RA pathogenesis, a variety of small molecules have been identified and have shown better specificity but fewer side effects in comparison to traditional medications in clinical trials for RA treatment [10, 51-53]. The basic strategy to develop these small molecules is to block the activity of most of the critical pro-inflammatory cytokines and kinases involved in the pathogenesis of RA [10, 51-53]. A number of TNF-α antagonists, such as infliximab, etanercept, adalimumab, golimumab and certolizumab pegol, have been developed and used in the treatment of RA [27]. The inhibitors of JAKs (e.g., tofacitinib and baricitinib) and spleen tyrosine kinase (SYK) (e.g., fostamatinib), have shown advantages in the interruption of kinase activities, thereby affecting intracellular signaling through kinase inhibition [54]. In this study, we identified a naturally sourced small molecule, NSM00191, that specifically disrupted the interaction between TRADD and TRAF2 (Figures 6A, 6B and 6E), significantly reduced cell proliferation in vitro (Figure 6D), and greatly improved inflammation status in RA mouse models (Figure 7). On the other hand, one limitation regarding the inhibition efficiency of NSM00191 in this study is that we did not compare it with other well-established small molecules, such as TNF-α inhibitors and JAK inhibitors. We will extensively compare the specificity, toxicity, and inhibitory ability of NSM00191 in future studies.
In summary, our findings verified that the activation of TNF-α/NF-κB signaling in foot and ankle RA patients resulted in the universal upregulation of NF-κB downstream targets. We found new miRNAs that might directly target various members of the TNF-α/NF-κB signaling pathway. More importantly, we found that the small molecule NSM00191 was able to significantly decrease the inflammation status in vitro and in vivo. Our findings will benefit the understanding of RA pathogenesis and therapy.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the following fundings: National Natural Science Foundation of China (81301604), Shaanxi Province Natural Science Foundation (2014JQ4164), China Postdoctoral Science Foundation (2017M613178) and Shaanxi Province Postdoctoral Science Foundation (2017BSHQYXMZZ13).
Authors' contributions
J. L and X. S designed the research and experiments. X. W performed most of the experiments. X. C, X. L, H. Z and Y. L performed some parts of the experiments. X. W, J. L and X. S analyzed data, performed statistical analyses and wrote the manuscript. All authors reviewed and approved the manuscript.
Ethical approval and informed consent
The clinical samples were acquired with written informed consent from all of the participants following protocols approved by the ethical board of Xi'an Jiaotong University College of Medicine. All experimental procedures used in this study were carried out in accordance with the approved guidelines of the ethical board of Xi'an Jiaotong University College of Medicine.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Heidari B. Rheumatoid Arthritis: Early diagnosis and treatment outcomes. Caspian J Intern Med. 2011;2:161-70
2. Davis JM 3rd, Matteson EL. My treatment approach to rheumatoid arthritis. Mayo Clin Proc. 2012;87:659-73
3. Grassi W, De Angelis R, Lamanna G, Cervini C. The clinical features of rheumatoid arthritis. Eur J Radiol. 1998;27(Suppl 1):S18-24
4. Burska A, Boissinot M, Ponchel F. Cytokines as biomarkers in rheumatoid arthritis. Mediators Inflamm. 2014;2014:545493
5. Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537-45
6. Piotr Wojdasiewicz, Lukasz A. Poniatowski, Dariusz Szukiewicz. The Role of Inflammatory and Anti-Inflammatory Cytokines in the Pathogenesis of Osteoarthritis. Mediators Inflamm. 2014;2014:561459
7. Arango Duque G, Descoteaux A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front Immunol. 2014;5:491
8. Lavagno L, Gunella G, Bardelli C, Spina S, Fresu LG, Viano I, Brunelleschi S. Anti-inflammatory drugs and tumor necrosis factor-alpha production from monocytes: role of transcription factor NF-kappa B and implication for rheumatoid arthritis therapy. Eur J Pharmacol. 2004;501:199-208
9. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205-19
10. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6:15
11. Kinne RW, Bräuer R, Stuhlmüller B, Palombo-Kinne E, Burmester GR. Macrophages in rheumatoid arthritis. Arthritis Res. 2000;2:189-202
12. Chadwick W, Magnus T, Martin B, Keselman A, Mattson MP, Maudsley S. Targeting TNF-alpha receptors for neurotherapeutics. Trends Neurosci. 2008;31:504-11
13. Al-Lamki RS, Sadler TJ, Wang J, Reid MJ, Warren AY, Movassagh M. et al. Tumor necrosis factor receptor expression and signaling in renal cell carcinoma. Am J Pathol. 2010;177:943-54
14. Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853-66
15. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651
16. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023
17. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86
18. Takeuchi T, Miyasaka N, Tatsuki Y, Yano T, Yoshinari T, Abe T, Koike T. Baseline tumour necrosis factor alpha levels predict the necessity for dose escalation of infliximab therapy in patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70:1208-15
19. Wei ST, Sun YH, Zong SH, Xiang YB. Serum Levels of IL-6 and TNF-α May Correlate with Activity and Severity of Rheumatoid Arthritis. Med Sci Monit. 2015;21:4030-8
20. Ceribelli A, Nahid MA, Satoh M, Chan EK. MicroRNAs in rheumatoid arthritis. FEBS Lett. 2011;585:3667-74
21. Filkova M, Jungel A, Gay RE, Gay S. MicroRNAs in rheumatoid arthritis: potential role in diagnosis and therapy. BioDrugs. 2012;26:131-41
22. Ludwig N, Leidinger P, Becker K, Backes C, Fehlmann T, Pallasch C. et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016;44:3865-77
23. Alsaleh G, Francois A, Philippe L, Gong YZ, Bahram S, Cetin S. et al. MiR-30a-3p negatively regulates BAFF synthesis in systemic sclerosis and rheumatoid arthritis fibroblasts. PLoS One. 2014;9:e111266
24. Kawano S, Nakamachi Y. miR-124a as a key regulator of proliferation and MCP-1 secretion in synoviocytes from patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70(Suppl 1):i88-91
25. Churov AV, Oleinik EK, Knip M. MicroRNAs in rheumatoid arthritis: altered expression and diagnostic potential. Autoimmun Rev. 2015;14:1029-37
26. Kumar P, Banik S. Pharmacotherapy options in rheumatoid arthritis. Clin Med Insights Arthritis Musculoskelet Disord. 2013;6:35-43
27. Ma X, Xu S. TNF inhibitor therapy for rheumatoid arthritis. Biomed Rep. 2013;1:177-84
28. Rivellese F, Lobasso A, Barbieri L, Liccardo B, De Paulis A, Rossi FW. Novel therapeutic approaches in Rheumatoid Arthritis: Role of Janus Kinases Inhibitors. Curr Med Chem. 2018 doi: 10.2174/0929867325666180209145243
29. Liu CG, Calin GA, Volinia S, Croce CM. MicroRNA expression profiling using microarrays. Nat Protoc. 2008;3:563-78
30. Jiang K, Zhang C, Yu B, Chen B, Liu Z, Hou C. et al. Autophagic degradation of FOXO3a represses the expression of PUMA to block cell apoptosis in cisplatin-resistant osteosarcoma cells. Am J Cancer Res. 2017;7:1407-22
31. Mukai T, Gallant R, Ishida S, Yoshitaka T, Kittaka M, Nishida K. et al. SH3BP2 gain-of-function mutation exacerbates inflammation and bone loss in a murine collagen-induced arthritis model. PLoS One. 2014;9:e105518
32. Inglis JJ, Criado G, Medghalchi M, Andrews M, Sandison A, Feldmann M. et al. Collagen-induced arthritis in C57BL/6 mice is associated with a robust and sustained T-cell response to type II collagen. Arthritis Res Ther. 2007;9:R113
33. Trask OJ Jr. Nuclear Factor Kappa B (NF-kappaB) Translocation Assay Development and Validation for High Content Screening. 2004.
34. Asquith DL, Miller AM, McInnes IB, Liew FY. Animal models of rheumatoid arthritis. Eur J Immunol. 2009;39:2040-44
35. Alam J, Jantan I, Bukhari SNA. Rheumatoid arthritis: Recent advances on its etiology, role of cytokines and pharmacotherapy. Biomed Pharmacother. 2017;92:615-33
36. Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771-84
37. Mogensen TH. Pathogen recognition and inflammatory signaling in the innate immune defenses. Clin Microbio Rev. 2009;22:240-73
38. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805-20
39. Lin J, Huo R, Xiao L, Zhu X, Xie J, Sun S. et al. A novel p53/microRNA-22/Cyr61 axis in synovial cells regulates inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014;66:49-59
40. Zhu S, Pan W, Song X, Liu Y, Shao X, Tang Y. et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat Med. 2012;18:1077-86
41. Chen XM, Huang QC, Yang SL, Chu YL, Yan YH, Han L. et al. Role of Micro RNAs in the Pathogenesis of Rheumatoid Arthritis: Novel Perspectives Based on Review of the Literature. Medicine (Baltimore). 2015;94:e1326
42. Kawano S, Nakamachi Y. miR-124a as a key regulator of proliferation and MCP-1 secretion in synoviocytes from patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70:Suppl 1:i88-91
43. Niederer F, Trenkmann M, Ospelt C, Karouzakis E, Neidhart M, Stanczyk J. et al. Down-regulation of microRNA-34a in rheumatoid arthritis synovial fibroblasts promotes apoptosis resistance. Arthritis Rheum. 2012;64:1771-9
44. Pauley KM, Satoh M, Chan AL, Bubb MR, Reeves WH, Chan EK. Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther. 2008;10:R101
45. Zhang B, Wang LS, Zhou YH. Elevated microRNA-125b promotes inflammation in rheumatoid arthritis by activation of NF-κB pathway. Biomed Pharmacother. 2017;93:1151-7
46. Hruskova V, Jandova R, Vernerova L, Mann H, Pecha O, Prajzlerova K. et al. MicroRNA-125b: association with disease activity and the treatment response of patients with early rheumatoid arthritis. Arthritis Res Ther. 2016;18:124
47. Asirvatham AJ, Magner WJ, Tomasi TB. miRNA regulation of cytokine genes. Cytokine. 2009;45:58-69
48. Liu X, He F, Pang R, Zhao D, Qiu W, Shan K. et al. Interleukin-17 (IL-17)-induced microRNA 873 (miR-873) contributes to the pathogenesis of experimental autoimmune encephalomyelitis by targeting A20 ubiquitin-editing enzyme. J Biol Chem. 2014;289:28971-86
49. Sharma AR, Sharma G, Lee SS, Chakraborty C. miRNA-Regulated Key Components of Cytokine Signaling Pathways and Inflammation in Rheumatoid Arthritis. Med Res Rev. 2016;36:425-39
50. Nahid MA, Satoh M, Chan EK. Interleukin 1β-Responsive MicroRNA-146a Is Critical for the Cytokine-Induced Tolerance and Cross-Tolerance to Toll-Like Receptor Ligands. J Innate Immun. 2015;7:428-40
51. Ursini F, Russo E, Ruscitti P, Giacomelli R, De Sarro G. The effect of non-TNF-targeted biologics and small molecules on insulin resistance in inflammatory arthritis. Autoimmun Rev. 2018;17:399-404
52. Mócsai A, Kovács L, Gergely P. What is the future of targeted therapy in rheumatology: biologics or small molecules? BMC Med. 2014;12:43
53. Li P, Zheng Y, Chen X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front Pharmacol. 2017;8:460
54. Yamaoka K, Tanaka Y. Targeting the Janus kinases in rheumatoid arthritis: focus on tofacitinib. Expert Opin Pharmacother. 2014;15:103-13
Author contact
![]() Corresponding authors: zeltasuncom; lujun23com
Corresponding authors: zeltasuncom; lujun23com