Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(13):1769-1781. doi:10.7150/ijbs.29242 This issue Cite
Research Paper
Inhibition of AKT suppresses the initiation and progression of BRCA1-associated mammary tumors
Hye Jung Baek1*, Sun Eui Kim1*, Jong Kwang Kim1*, Dong Hoon Shin1, Tae Hyun Kim1, Kwang Gi Kim2, Chu-Xia Deng3, Sang Soo Kim1 ![]()
1. Research Institute, National Cancer Center, Goyang, 10408, Korea,
2. Department of Biomedical Engineering, Gachon University College of Medicine, Incheon, 21565, Korea,
3. Cancer Centre, Faculty of Health Sciences, University of Macau, Macau SAR 999078, China.
*These authors contributed equally.
Received 2018-8-15; Accepted 2018-9-7; Published 2018-10-3
Abstract

Despite the high incidence of BRCA1-mutant breast cancer, few substantial improvements in preventing or treating such cancers have been made. Using a Brca1-mutant mouse model, we examined the contribution of AKT to the incidence and growth of Brca1-mutated mammary tumors. A haploinsufficiency of Akt1 in Brca1-mutant mouse model significantly decreased mammary tumor formation from 54% in Brca1co/coMMTV-Cre mice to 22% in Brca1 co/coMMTV-Cre Akt1+/- mice. Notably, treatment of tumor-bearing Brca1-mutant mice with the AKT-inhibitor, MK-2206, yielded partial response or stable disease up to 91% of mice in maximum response. MK-2206 treatment also significantly reduced tumor volume and delayed recurrence in allograft and adjuvant studies, respectively. A correlation analysis of MK-2206 responses with gene expression profiles of tumors at baseline identified seven genes that were differentially expressed between tumors that did and did not respond to MK-2206 treatment. Our findings enhance our understanding of the involvement of AKT signaling in BRCA1-deficient mammary tumors and provide preclinical evidence that targeted AKT inhibition is a potential strategy for the prevention and therapeutic management of BRCA1-associated breast cancer.
Keywords: BRCA1, AKT, MK-2206, precision medicine
Introduction
The BRCA1 protein is a tumor suppressor that plays a critical role in maintaining genomic integrity through regulation of important cellular processes, including genetic stability, DNA damage repair, centrosome duplication, apoptosis, and cell-cycle control [1] [2] [3]. Germline mutation of BRCA1 is responsible for a considerable portion of hereditary breast and ovarian cancers [4]. Women with germline mutations in BRCA1 have a 57% (95% confidence interval [CI], 47%-66%) risk of developing breast cancer and a 40% (95% CI, 35%-46%) risk of developing ovarian cancer by the age of 70 [5]. Gene and protein expression profiling has revealed that cancers arising as a result of BRCA1 deficiency show triple-negative and basal-like properties, tend to be aggressive, and typically have a poor prognosis [6] [7].
The National Comprehensive Cancer Network (NCCN) recommends that BRCA1 mutation-positive women undergo periodic breast screening and consider mastectomy and salpingo-oophorectomy to reduce their cancer risk (NCCN guideline Ver. 2.2017). There are currently no effective therapies for preventing tumor initiation in this patient population, and the only available treatment option when tumors occur is resection of tumors followed by adjuvant chemotherapy. However, resection and chemotherapy may not be applicable and effective for all patients. Thus, although a means for improving the treatment of BRCA1-deficient breast cancer is urgently needed, efforts to develop and validate suitable therapies in clinical trials have encountered numerous hurdles.
Recently, two promising therapeutic agents were reported for the targeted therapy of BRCA1-related breast cancer. Olaparib (AZD2281) is an inhibitor of poly (ADP-ribose) polymerase (PARP), which senses DNA single-strand breaks and is essential for base-excision repair [8]. BRCA1 contributes to DNA-damage repair via homologous recombination. Treatment of Brca1-mutated tumor-bearing mice with olaparib was found to induce synthetic lethality by disrupting homologous recombination and inhibiting base-excision repair [9] [10]. A subsequent clinical trial showed that progression-free survival at a median follow-up of 14 months was 2.8 months longer and the risk for disease progression or death was 42% lower with olaparib monotherapy compared with conventional chemotherapy [11]. The second promising therapeutic candidate is NVP-BKM120, a pan-class IA phosphatidylinositol 3-kinase (PI3K) inhibitor that was shown to delay the tumor-doubling time from 5 to 26 days in the MMTV-Cre Brca1f/f Trp53+/- mouse model [12]. Notably, combined treatment with NVP-BKM120 and olaparib delayed tumor doubling to more than 70 days in the above-mentioned mouse model and to 50 days in mice xenotransplanted with human BRCA1-mutated tumors.
Here, we examined the contribution of AKT signaling in BRCA1-deficient breast cancer. Activation of AKT kinase activity is one of the most common molecular alterations associated with human malignancy, especially breast cancers [13, 14]. BRCA1 deficiency has been shown to activate the AKT oncogenic pathway by increasing the phosphorylation and kinase activity of AKT [15, 16], [17]. In the present study, we tested the ability of the AKT inhibitor MK-2206 to suppress the activity of AKT in Brca1co/coMMTV-Cre mice, which develop tumors that mimic human BRCA1 mutation-related mammary tumors, and evaluated the clinical applicability of these agents by examining tumor initiation and progression in vivo.
Materials and Methods
Animal experiments
Brca1 exon 11-deleted (Brca1-Δ11), Brca1 conditional-knockout (Brca1-co), and MMTV-Cre transgenic mice were provided by the National Cancer Institute (NCI, USA) mouse repository. Akt1-mutant mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA). All procedures involving animals and their care were approved by the Institutional Animal Care and Use Committee of the National Cancer Center (Goyang, Korea). Female Brca1-mutant mice were generated by intercrossing Brca1 conditional-knockout mice, MMTV-Cre mice and/or Akt1-knockout mice, originally generated by Drs. Deng, Hennighausen, and Birnbaum, respectively [18] [19] [20]. To generate the cohorts of Brca1/Akt1 mutant mice, we crossed male Brca1co/coMMTV-Cre Akt1+/- mice with female Brca1co/coAkt1+/- mice. This cross yielded mice with the following six genotypes: (1) Brca1co/co (12.5%), (2) Brca1co/coMMTV-Cre (12.5%), (3) Brca1co/coAkt1+/- (25%), (4) Brca1co/coAkt1-/- (12.5%), (5) Brca1co/coMMTV-Cre Akt1+/- (25%), and (6) Brca1co/coMMTV-Cre Akt1-/- (12.5%). Of these, Brca1co/co, Brca1co/coMMTV-Cre, Brca1co/coAkt1+/-, and Brca1co/coMMTV-Cre Akt1+/- mice were used in the tumorigenesis study, whereas Brca1co/co, Brca1co/coMMTV-Cre, Brca1co/coAkt1-/-, and Brca1co/coMMTV-Cre Akt1-/- were used for assessing mammary gland development. Mice carrying mutant alleles were genotyped by polymerase chain reaction (PCR) using the primers shown in Supplementary Table 5 and the following thermocycling conditions: 30 cycles of 94°C for 15 seconds, 60°C for 15 seconds, and 72°C for 1 minute.
MK-2206 and AZD2281 (olaparib) were purchased from Abmole Bioscience (Houston, TX, USA). MK-2206 was resuspended in 30% Captisol (Ligand Pharmaceuticals, San Diego, CA, USA), and olaparib was prepared as a stock solution (100 mg/ml) in dimethyl sulfoxide (DMSO) and diluted to 10 mg/ml with 10% 2-hydroxypropyl-cyclodextrin in phosphate-buffered saline (PBS).
After 8 months of age, mice were examined weekly for the occurrence of tumors. When a mammary tumor was detected, the mouse was assigned to one of the following treatment groups: control (30% Captisol and 10% 2-hydroxypropyl-cyclodextrin), MK-2206 (240 mg/kg, oral, 3 times/wk) with 10% 2-hydroxypropyl-cyclodextrin, olaparib (100 mg/kg, IP, 3 times/wk) with 30% Captisol, and MK-2206+olaparib. To examine the progress of tumorigenesis, we anesthetized mice once a week and acquired tumor images using a 7-T MR imager (Bruker BioSpec, Rheinstetten, Germany). Tumor volume was estimated according to the following formula: V = L × W × D × π/6, where V is volume, L is length, W is width, and D is depth. For tumor allografts, spontaneously formed primary tumors obtained from Brca1co/coMMTV-Cre mice were orthotopically implanted into 5-week-old female HsdCpb:NMRI-Foxn1nu mice (Orient-Harlan Laboratories, Sungnam, Korea). After each grafted tumor reached a volume of ~1 cm3, the tumor tissue was excised, trimmed with a tissue slicer, and reimplanted into recipient mice. After implantation, the recipient mice were left untreated or were treated with MK-2206 (120 mg/kg) or olaparib, as indicated above. Tumor growth was assessed as the ratio of tumor volume at a given time to that recorded at the initiation of treatment. For experiments testing MK-2206 adjuvant therapy, mammary tumors from Brca1co/coMMTV-Cre mice were surgically resected under halothane anesthesia upon reaching a volume of ~1 cm3. MK-2206 treatment was initiated 1 week after surgery; during this 1-week interval, mice were pretreated with a course of antibiotics (0.001% enrofloxacin; Baytril, Leverkusen, Germany) provided in the drinking water. For tumors identified after surgery, the duration between surgery and relapse was taken as the time (days) to recurrence. Tumor progression in response to drug treatment was accessed according to RECIST (Response Evaluation Criteria In Solid Tumors, version 1.1) guideline.
Cell culture
MCF7 cells and BRCA1-mutant MDA-MB-436 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). The authenticity of human cell lines was confirmed by short tandem repeat (STR) analysis performed by the Omics Core of the National Cancer Center. Brca1Δ11/Δ11Tp53+/- and Brca1Δ11/Δ1153BP1-/- mouse mammary tumor cell lines were generated from the corresponding tumors as described previously [21] [22]. In brief, portions of mammary tumors that developed spontaneously in mice were dissected, briefly washed in PBS, and minced in DMEM containing 10 μg/ml of collagenase type III (Worthington, Lakewood, NJ, USA). After digestion, released cells and finely minced tissues were pelleted, washed in PBS, and cultured in DMEM supplemented with 10% FBS. Cultures were monitored for epithelial cell growth, and the media were changed every 3 days. After fibroblastic cells had died, leaving adherent epithelial cells, the cultures were trypsinized and passaged in the same medium. For growth assays, cells were plated at 2 × 104 cells per well in 4-well plates in quadruplicate, with or without the indicated treatments, and cell viability was determined using an In Vitro Toxicology Assay Kit (Sigma, St. Louis, MO, USA) according to the manufacturer's instructions. BRCA1 expression in MCF7 cells was knocked down by transfecting cells with a pool of three BRCA1-targeting small interfering RNAs (siRNAs; Santa Cruz, Dallas, TX, USA) or scrambled siRNA controls (Dharmacon, Lafayette, CO, USA) using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) according to the manufacturer's protocol. For experiments involving estrogen treatment, cells were maintained in hormone-depleted, phenol red-free Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% dextran/charcoal-treated fetal bovine serum (FBS; Gemini, West Sacramento, CA, USA). AKT inhibitor IV was obtained from Merck-Millipore (Darmstadt, Germany).
Immunoblotting
Western blot analysis was carried out according to standard procedures using enhanced-chemiluminescence detection (GE Life Science, Chicago, IL, USA). Tumor tissue lysates were prepared using an electric homogenizer for 30 seconds after the addition of lysis buffer. The following antibodies were used: anti-β-actin, anti-acetyl-CoA carboxylase (ACC), anti-phospho-ACC, anti-phospho-AKT, anti-phospho-BRCA1, anti-caspase-3, anti-caspase-7, anti-β-catenin, anti-phospho-cyclin D1, anti-phospho-GSKα/β, anti-MAPK, anti-phospho-MAPK, anti-PARP, anti-PDK1, anti-phospho-PDK1, anti-PI3K, anti-phospho-PI3K, anti-phospho-S6, anti-phospho-mTOR, anti-phospho-Rb, anti-VEGFR, anti-phospho-VEGFR (all from Cell Signaling Technology, Danvers, MA, USA); anti-β-actin, anti-AKT, anti-AKT1, anti-BRCA1, anti-cyclin D1, anti-Rb, anti-S6, anti-mTOR, anti-α-tubulin (all from Santa Cruz, Dallas, TX, USA); and anti-PCNA (Atlas Antibodies, Bromma, Sweden). Horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse antibodies (Jackson Immuno Research, West Grove, PA, USA) were used as secondary antibodies as appropriate.
Histology and immunohistochemical staining
For histology, tissues were fixed in 10% (v/v) formalin, embedded in paraffin, sectioned, stained with hematoxylin and eosin (H&E), and examined by light microscopy. Immunoreactive proteins were detected using anti-phospho-AKT (Cell Signaling Technology) and anti-phospho-histone H3 (Merck-Millipore, Darmstadt, Germany) primary antibodies and a Zymed Histostain kit (Invitrogen, Waltham, MA, USA) according to the manufacturer's instructions. The accumulation of collagen in tissues was monitored using a Masson's trichrome staining kit (Sigma). Proportions of phospho-histone H3-positive cells in embryonic tissues were estimated using HistoQuest 4.0 software (TissueGnostics, Vienna, Austria).
Expression analysis
Tumor tissues were dissected free of surrounding normal tissues and immediately frozen in a pre-chilled aluminum block. Total RNA was extracted from tumor tissues using an RNeasy mini kit (Qiagen, Venlo, Netherlands) and subjected to genome-wide RNAseq analyses, performed by eBiogene (Seoul, Korea). Raw data obtained from duplicate analyses of two sets of 28 samples were normalized using Cufflinks RNAseq workflow [23]. Two-sample t-tests (P < 0.05) were performed for each set to identify genes that were differentially expressed between tumors that did and did not respond to MK-2206 treatment. The differentially expressed genes (DEGs) that overlapped between the sets were selected as markers, and a heat map was generated using the z-scores of their normalized expression in fragments per kilobase per million mapped fragments (FPKM). Sorted samples showed a good correlation between the ratio of tumor volume (RTV) and gene expression patterns. Results were cross-validated by generating a heat map using normalized FPKMs of the genes in each adjuvant-based experiment as input to the heat map function of the Superheat R open source package. DEGs in the above-described RNA samples were further validated by quantitative reverse transcription-PCR (RT-qPCR) using the primers shown in Supplementary Table 2.
Functional network analysis
All known interactions of proteins with our markers were analyzed using STRING, a database of known and predicted protein-protein interactions. Direct and indirect associations determined by computational predictions and interactions obtained from other interaction-analysis databases (https://www. string-db.org) were included [24] [25]. The STRING score is calculated from the combination of all predictions (range, 0 to 1) and classified into four categories: highest confidence (0.900), high confidence (0.700), medium confidence (0.400), and low confidence (0.150). For network analysis, confidence was set at high (STRING score = 0.7). Only experimentally determined interactions and known interactions from curated databases were included in our analysis. Nodes in the network were grouped according to enriched KEGG pathways (false discovery rate [FDR] < 0.05).
Results
Inhibition of AKT rescues abnormalities associated with estrogen treatment and BRCA1 mutation
Although mutation of BRCA1 ultimately contributes to tumorigenesis, cells at an initial stage of BRCA1 loss exhibit genetic instability, senescence, apoptosis and growth suppression, indicating that growth stimulation and additional mutations are required for tumor formation by BRCA1-mutant cells. Estrogen, which activates downstream targets including the phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway is a potent mitogen for breast epithelial cells [26]. Indeed, the survival defect of BRCA1-deficient mammary epithelial cells is rescued by increasing estradiol (E2) levels [27]. In addition, estrogen receptor-α (ER-α) is highly expressed in the premalignant mammary gland and during initiation stages of tumorigenesis in the Brca1-mutant mouse, although its expression is gradually diminished during mammary tumor progression [28].
To identify the contribution of estrogen signaling to the survival of BRCA1-deficient cells, we analyzed various regulatory proteins in BRCA1 knockdown MCF7 cells treated with or without E2 or irradiation (Fig. 1A). BRCA1 knockdown in MCF7 cells induced phosphorylation of AKT, glycogen synthase kinase 3 beta (GSK3β), cyclin D1 and the ribosomal protein S6, and E2 treatment induced phosphorylation of Rb (retinoblastoma) and AKT. Notably, AKT phosphorylation was synergistically induced by BRCA1 knockdown and E2 treatment, but was unaffected by irradiation-induced DNA damage. Next, we tested whether E2-induced survival required the activation of AKT, and whether inhibition of AKT reversed the cellular events induced by E2 treatment and/or BRCA1 deficiency. We found that G1/S progression induced by E2 treatment in BRCA1 knockdown MCF7 cells was significantly reduced by treatment with shRNA against AKT1, which is ubiquitously expressed and is the major AKT isoform in MCF7 cells (Supplementary Fig. 1A); a similar reduction in G1/S progression was observed following treatment with AKT inhibitor IV (Fig. 1B and 1C). Moreover, treatment with AKT inhibitor IV attenuated E2-induced phosphorylation of Rb, cyclin D1 and S6 (Fig. 1D) and significantly reduced E2-induced survival, regardless of BRCA1 status (Fig. 1E).
AKT is activated by estrogen treatment and BRCA1 downregulation, and its inhibition rescues the abnormalities associated with the loss of BRCA1. (A) MCF7 cells were transfected with control or BRCA1 siRNA, and then treated with E2 (100 nM) or irradiation (10 Gy). Protein expression patterns were analyzed by Western blotting, using β-actin as a control for equal loading. (B) Inhibition of AKT prevented E2-induced G1/S transition. MCF7 cells transfected with siRNA against BRCA1 were treated with E2 in the absence or presence of AKT inhibition (infection with AKT1-shRNA lentivirus or treatment with AKT inhibitor IV) and analyzed by flow cytometry with propidium iodide staining. (C) Percentage of cell distributions in different phases of the cell cycle. Results from three independent experiments are shown (**P < 0.01, *P < 0.05). (D) MCF7 cells transfected with scrambled or BRCA1 siRNA were treated with E2 or AKT inhibitor IV, after which protein expression patterns were analyzed by Western blotting. (E) E2-induced survival was measured in MCF7 cells transfected with scrambled or BRCA1 siRNA, treated with or without AKT inhibitor IV. Each number represents survival relative to that in the absence of E2 (**P < 0.01). (F) Phospho-histone H3 staining of cerebellar and liver tissues from WT, Brca1Δ11/Δ11, and Brca1Δ11/Δ11Akt1+/- embryos at 14.5 days post coitus (dpc). Proportions of phospho-histone H3-positive nuclei are indicated. Scale bars: 100 μm. (G) Hyperplastic mammary glands (upper panel) and tumor tissues (lower panel) from Brca1co/coMMTV-Cre mice were subjected to immunohistochemical staining with anti-phospho-AKT antibody. Scale bars: 50 μm. (H) Immortalized Brca1Δ11/Δ11 and Brca1+/+ MEFs were analyzed by Western blotting.
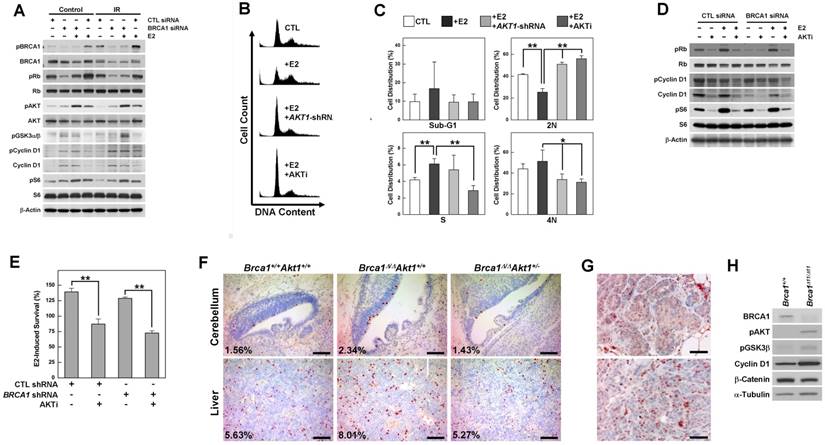
To determine whether downregulation of AKT is capable of rescuing the developmental abnormalities caused by the loss of Brca1 in mice, we crossed Akt1-mutant mice with Brca1-Δ11-mutant mice, which exhibit embryonic lethality. We found that the additional alterations in Brca1/Akt1-combined mutant mouse (Brca1Δ11/Δ11Akt1+/-) embryos were associated with even more severe embryonic lethality and abnormalities, with defects including exencephaly, failure of neural tube closure, and severe lymphedemas (Supplementary Fig. 2). However, the level of phospho-histone H3, which was increased in the cerebellum and liver of Brca1Δ11/Δ11 embryos, was restored in Akt1-haploinsufficient Brca1-mutant (Brca1Δ11/Δ11Akt1+/-) embryos (Fig. 1F). Together, our results suggest that AKT is a key molecule in the cellular effects caused by BRCA1 mutation and estrogen, and that its inhibition can rescue the abnormalities caused by estrogen treatment and BRCA1 mutation.
However, increases in AKT phosphorylation induced by the loss of BRCA1 were identified not only in hyperplastic mammary glands, but also in adenocarcinomas from Brca1-mutant mice and estrogen-insensitive mouse embryonic fibroblasts (MEFs) (Fig. 1G and 1H), suggesting differential contributions of ER-α and AKT to the initiation and progression of BRCA1-associated mammary tumor.
Downregulation of AKT1 in Brca1-mutant mice reduces mammary tumor formation
A previous study showed that Wap-Cre transgene-mediated deletion of Brca1 from mouse mammary epithelial cells results in aberrant mammary development [18]. Here, we used whole-mount preparations to assess the development of mammary glands in post-pubertal Brca1/ Akt1-mutant mice. The mammary glands of 2-month-old wild-type (WT) and Brca1co/coMMTV-Cre mice were filled with elongated ducts and exhibited branches in the fat pad. In contrast, 5 of 11 Brca1co/coAkt1-/- mice and 7 of 19 Brca1co/coMMTV-Cre Akt1-/- mice failed to form a proper mammary structure above the lymph node (Fig. 2A right panel). According to measurements of mammary gland density obtained using Branch software (ver. 1.1, [29]), mammary glands of AKT1-deficient mice exhibited a significantly diminished ductal network (Fig. 2B, and Supplementary Table 1). A further analysis of mammary gland development in these mice up to 6 months of age revealed that ductal expansion did not reach the tip of the fat pad (Supplementary Fig. 3). These findings suggest that AKT is required for the proliferation of mammary epithelial cells.
Downregulation of AKT suppresses mammary gland development and mammary tumor formation caused by the loss of BRCA1. (A) Whole-mount staining of mammary glands from 2-month-old Brca1co/coMMTV-Cre and Brca1co/coMMTV-Cre Akt1-/- mice. Arrows indicate where sprouting ducts are appeared in the mammary glands. (B) Branch software was used to estimate the total length (open) and branch numbers (filled) of ducts between the lymph node and end tip in mammary glands of 2-month-old mice with the indicated genotypes. The density of mammary gland ducts was significantly lower in mice with Akt1-mutant (**P < 0.01). (C) Kaplan-Meier curves of tumor-free survival. WT (N = 30) and Brca1co/coAkt1+/- (N = 30) mice were normal and did not develop tumors. By contrast, 54% (15 of 28) of Brca1co/coMMTV-Cre (N = 28) mice spontaneously developed mammary tumors by 16 months of age, whereas 22% (6 of 27) of Brca1co/coMMTV-Cre Akt1+/- mice developed mammary tumors by the same age. Log-rank tests revealed a significant difference (P = 0.007) in the tumor-free survival of Brca1co/coMMTV-Cre mice compared with Brca1co/coMMTV-Cre Akt1+/- mice. (D and E) Protein expression patterns in tumors from Brca1co/coMMTV-Cre and Brca1co/coMMTV-Cre Akt1+/- mice. (F) Histological analysis (H&E and trichrome staining) of two tumor cases from Brca1co/coMMTV-Cre and Brca1co/coMMTV-Cre Akt1+/- mice. The second set of two panels of H&E staining in tumors from Brca1co/coMMTV-Cre Akt1+/- mice represent magnifications of the boxed areas in adjacent images. Scale bars: 100 μm.
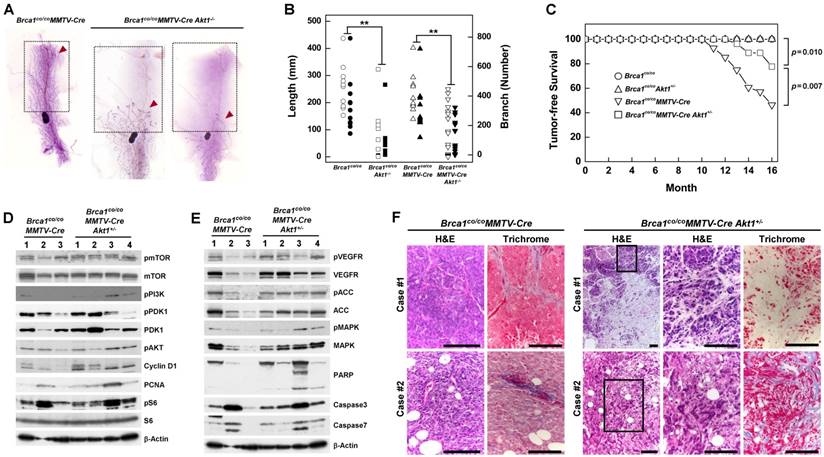
To determine whether AKT contributes to BRCA1-deficient mammary tumor formation, we generated cohorts of Brca1co/co, Brca1co/coAkt1+/-, Brca1co/coMMTV-Cre and Brca1co/coMMTV-Cre Akt1+/- mice, and examined tumor formation. Mice in each genotype were healthy and were not readily distinguishable from one another, and no mice exhibited any externally visible abnormalities until 10 months of age. Beginning at 10 months of age, Brca1co/coMMTV-Cre mutant mice developed breast cancer, and by 16 months of age showed a high incidence of mammary tumors (54%; 15 of 28). No Brca1co/co or Brca1co/coAkt1+/- mice showed any signs of neoplasia, including mammary tumors, up to 16 months. During the same period, Brca1co/coMMTV-Cre Akt1+/- mice exhibited a mammary tumor incidence of 22% (6 of 27) and showed significantly better tumor-free survival than Brca1co/coMMTV-Cre mice (P = 0.007) (Fig. 2C). These findings suggest that downregulation of AKT1 in Brca1co/coMMTV-Cre mice efficiently reduces breast cancer formation.
A further analysis of tumor proteins revealed that mammary tumors from Brca1co/coMMTV-Cre Akt1+/- mice showed similar patterns in the PI3K/AKT/mTOR pathway (Fig. 2D), but showed increased levels of phosphorylated VEGFR (vascular endothelial growth factor receptor), ACC (acetyl-CoA carboxylase) and MAPK (mitogen-activated protein kinase), and enhanced apoptosis compared with tumors from Brca1co/coMMTV-Cre- mice (Fig. 2E). In addition, tumors from Brca1co/coMMTV-Cre Akt1+/- mice displayed large stromal areas with lower-grade properties, including epithelial cells with glandular/tubular structures and regular outlines, and uniform nuclear chromatin, whereas tumors from Brca1co/coMMTV-Cre mice exhibited frequent nuclear pleomorphisms (Fig. 2F), suggesting that AKT status contributes to the aggressiveness of Brca1-mutant tumors.
Inhibition of AKT attenuates the growth of BRCA1-mutant tumor cells
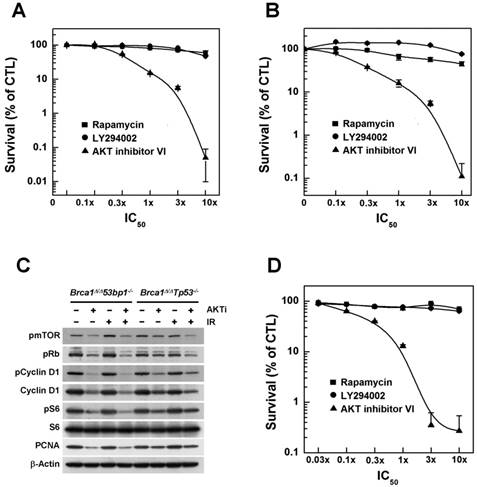
Next, we further analyzed whether inhibition of AKT was capable of exerting anticancer efficacy against tumor cells in addition to its apparent ability to prevent BRCA1-related breast cancer. Accordingly, we treated breast cancer cell lines derived from tumors obtained from Brca1△11/△1153bp1-/- and Brca1△11/△11Tp53-/- mice with increasing concentrations of AKT inhibitor IV. We found that this agent caused a concentration-dependent reduction in cell growth (Fig. 3A and 3B). We also treated these cells with AKT inhibitor IV in the absence or presence of irradiation, and analyzed the expression of proliferation-related proteins (Fig. 3C). Inhibition of AKT decreased the levels of phosphorylated mTOR, Rb, cyclin D1 and S6, as well as the levels of cyclin D1 and PCNA (proliferating cell nuclear antigen) in both cell lines, suggesting that suppression of AKT activity reduced the activities/levels of various proteins related to cell-cycle progression and proliferation in BRCA1-deficient breast cancer cells. In addition, we examined the effects of AKT inhibition on a human cancer cell line with BRCA1 mutation. Notably, treatment with AKT inhibitor IV reduced the survival of BRCA1 mutant MDA-MB436 cells (Fig. 3D).
Inhibition of AKT decreases the survival of BRCA1-deficient tumor cells. The survival of (A) Brca1Δ11/Δ1153bp1-/- and (B) Brca1Δ11/Δ11Tp53-/- (closed) mammary tumor cell lines was estimated in the presence of rapamycin, LY294002, or AKT inhibitor IV. (C) Brca1Δ11/Δ1153bp1-/- and Brca1Δ11/Δ11Tp53-/- mammary tumor cells were exposed to irradiation or treated with the AKT inhibitor IV. Protein expression patterns were analyzed by Western blotting, using β-actin as a control for equal loading. (D) The survival of BRCA1-deficient MDA-MB436 tumor cells was estimated in the presence of rapamycin, LY294002, or AKT inhibitor IV. The IC50 values for rapamycin, LY294002, and AKT inhibitor IV were calculated to be 50 pM, 1.4 μM, and 1.25 μM, respectively.
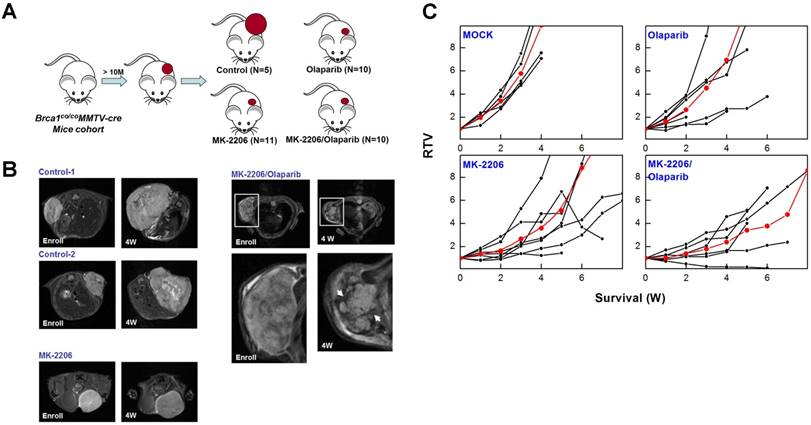
To determine whether targeting AKT could suppress BRCA1-mutant breast cancer, we tested the efficacy of MK-2206 (the AKT inhibitor) with olaparib (the PARP inhibitor) in Brca1co/coMMTV-Cre mice, which spontaneously develop mammary tumors beginning at 10 months of age. Mammary tumors in Brca1co/coMMTV-Cre mice could be detected by observation and palpation during periodic examinations upon reaching a size of ~0.5 cm3. Tumor-bearing mice were randomized to receive the following: vehicle control (N = 5); MK-2206, via oral gavage (N = 11); olaparib, via intraperitoneal administration (N = 10); or MK-2206 plus olaparib (N = 10) (Fig. 4A). Treatments were applied three times a week for the duration of the experiment. Baseline and progressed tumor volumes were measured by scanning the mice once a week by magnetic resonance (MR) imaging until the tumor volume reached ~3 cm3 (Fig. 4B). Three weeks after the initiation of treatment, the volume of tumors in vehicle-treated mice had increased ~5.8-fold relative to baseline, whereas tumors in MK-2206-treated mice were significantly smaller, exhibiting only an approximate 2.8-fold increase. In mice co-treated with olaparib and MK-2206, tumor volume was only increased ~1.8-fold at this same time, indicating that these agents exerted an additive effect on tumor growth. Moreover, MR images of these tumors revealed a partial response with necrosis that was absent in other groups (Table 1 and Fig. 4C). The maximum percentage of mice responding to treatment (disease control rate) was 91% (10 of 11) for MK-2206, 30% (3 of 10) for olaparib, and 82% (9 of 11) for co-treatment. Obviously, MK-2206 monotherapy significantly extended survival compared with that in the non-treatment group (7.8 vs. 3.8 weeks, respectively). Taken together, our results indicate that AKT inhibition decreases mammary tumor growth and increases survival in mice with BRCA1-associated mammary tumors. Notably, not all individual mice with the same Brca1 mutation showed the same response to a given treatment, suggesting a degree of variability between individuals in the effect of the studied therapy.
Summarized results of a preclinical experiment testing the effect of MK-2206 treatment on Brca1-mutant tumors
| Treatment | RTV2 at 3 W | Survival (W) | SAE3 | DCR4 |
|---|---|---|---|---|
| Not Treated | 5.77±1.22 | 3.80±0.45 | 0/5 | 0/5 |
| MK-2206 | 2.79±1.80** | 7.77±2.68** | 2/11 | 10/11 |
| Olaparib | 3.98±3.04 | 4.65±3.87 | 5/10 | 3/10 |
| Combination | 1.79±1.06** | 4.00±2.91 | 8/11 | 9/11 |
1The numbers represent mean±SD, with significant differences indicated by asterisks (**P<0.01); 2RTV, ratio of tumor volume after treatment versus baseline (at inception of treatment); 3SAE, serious adverse effect; 4DCR, disease control rate
Therapeutic effects of the AKT inhibitor MK-2206 on mammary tumors in Brca1co/coMMTV-Cre mice. (A) Brca1co/coMMTV-Cre female mice spontaneously developed mammary tumors beginning at 10 months of age. Upon tumor appearance, tumor-bearing mice were randomized into the following treatment groups: no treatment; the PARP inhibitor, olaparib (100 mg/kg, IP, 3 times/week); the AKT inhibitor, MK-2206 (240 mg/kg, oral, 3 times/week); or olaparib plus MK-2206. Tumor growth progression was monitored by weekly MRI scans. (B) Representative MRI scans of tumor-bearing mice at baseline (left) and after 4 weeks of the indicated treatments. The arrows indicate partial responses with necrosis in the tumors of mice co-treated with MK-2206 and olaparib. (C) Graphs show RTVs (ratios of tumor volumes) between post-treatment and baseline (at the start of treatment). Red lines represent the averages of cohorts.
Inhibition of AKT suppresses the growth of BRCA1-deficient tumors in an allograft model and as an adjuvant therapy after surgery
To further confirm the therapeutic effect of AKT inhibition, we investigated prognostic biomarkers of MK-2206 treatment outcome in tumors obtained at baseline. Previous studies have reported that mammary tumors spontaneously generated from Brca1-alterd mice can be orthotopically transplanted into female mice without losing their original phenotype, gene expression profile, or sensitivity to anticancer agents [9, 30]. Accordingly, we collected 18 tumor samples from spontaneously developed mammary tumors in Brca1co/coMMTV-Cre mice and orthotopically transplanted them into HsdCpb: NMRI-Foxn1nu (nude) female mice. We then used this model to test the efficacy of MK-2206 by comparing treated and non-treated tumors with the same origin (Fig. 5A). An analysis of tumors in individual mice revealed that 44.4% (8 of 18) and 38.9% (7 of 18) of mice treated with MK-2206 or olaparib, respectively, had ratios of tumor volume (RTVs) < 50%; 27.8% (5 of 18) exhibited at least a 50% reduction in response to both treatments; and 33.3% (6 of 18) failed to respond to either treatment (Fig. 5B). The overall RTVs (treated vs. non-treated tumors) for mice bearing Brca1-mutant tumor allografts were 60.8% and 62.6% for mice treated with MK-2206 and olaparib, respectively (Fig. 5C).
Therapeutic effects of the AKT inhibitor MK-2206 on BRCA1-deficient tumor transplantation and adjuvant models. (A) Overview of the allograft model and drug treatments. Eighteen spontaneously developed mammary tumors were collected from Brca1co/coMMTV-Cre mice and transplanted into nude mice. Growth of the corresponding tumors in sham-treated mice versus mice treated with olaparib (100 mg/kg, IP, 3 times/week) or MK-2206 (120 mg/kg, oral, 3 times/week) was tested. When the tumor of any mouse implanted with the same original tumor reached ~3 cm3, all mice implanted with that tumor were sacrificed and examined. (B) Graph shows calculated RTVs (RTV of treated tumor/RTV of control tumor × 100) for tumors treated with olaparib and MK-2206. (C) Summary of the allograft experiment. The numbers represent means ± SE (**P < 0.01). (D) Overview of adjuvant therapy applied after surgery. Upon tumor appearance in Brca1co/coMMTV-Cre mutant mice, the tumors were surgically removed. One week post-surgery, mice were randomized into a control group (N = 10) or MK-2206 treatment group (N =10, 240 mg/kg, oral, 3 times/week). (E) Kaplan-Meier curves for recurrence-free survival. The overall recurrence-free periods were 29.3 days and 49.4 days in control and MK-2206-treated groups, respectively. Log-rank tests showed that tumor-free survival was significantly longer (P = 0.041) in the MK-2206 treatment group.
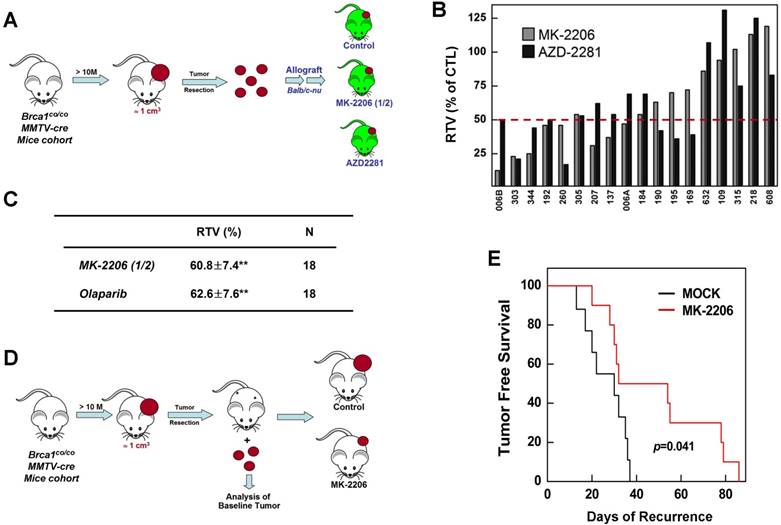
We next compared the efficacy of MK-2206 as an adjuvant therapy after resection of BRCA1-deficient mammary tumors. After female Brca1co/coMMTV-Cre mice formed palpable tumors, the tumors were surgically removed and the mice were randomized to treatment with vehicle control (N = 10) or MK-2206 (N = 10), and monitored until recurrence (Fig. 5D). Tumor-free survival periods were 29.3 days (95% CI, 16.6-42.0) and 49.4 days (95% CI, 36.8-62.1) in control and MK-2206 groups, respectively, a difference that was statistically significant (P = 0.041, log-rank test) (Fig. 5E). Taken together, these results suggest that inhibition of AKT activity in Brca1-mutant mammary tumors significantly suppresses tumor progression.
Analysis of MK-2206 response-associated biomarkers
Although the overall results of MK-2206 treatment showed significant improvement in cohorts of mice harboring Brca1-mutant tumors, some individuals in each group failed to respond to this AKT-inhibition strategy. To increase the potential clinical efficacy of AKT inhibition against BRCA1-associated breast cancer, it would be helpful to distinguish potential responders from non-responders before initiation of treatment. In an effort to identify candidate prognostic markers, we classified the cases based on their responsiveness to MK-2206 treatment, and further analyzed protein patterns in tumor tissue. To determine whether responsiveness to MK-2206 treatment was distinguishable at the protein level, we then analyzed tumors from allograft and adjuvant models (Fig. 6A and 6B). However, this approach failed to identify substantial differences in protein expression patterns in baseline tumors between responders and non-responders.
Analysis of MK-2206 response-associated biomarkers. (A) Protein expression patterns in tumors from the allograft model are shown. 'Responder' indicates that the tumor responded to MK-2206 with at least a 50% reduction in growth. C, controls (vehicle treatment); T, MK-2206 treatment. (B) Tumors from adjuvant therapy were analyzed by Western blotting. The numbers indicate the day of recurrence after surgery/MK-2206 treatment. (C) MK-2206 response-related genes were identified by screening for genes that were differentially expressed in baseline tumors of MK-2206-sensitive versus -insensitive tumors in allograft and adjuvant experiments. Red and blue numbers represent the numbers of genes that were expressed at higher and lower levels, respectively, in responders. (D) Heat map shows upregulation of seven selected genes in the MK-2206-responder group compared with the non-responder group in the allograft model (P < 0.05). Tumor samples are sorted with respect to their RTV to highlight the correlation with gene expression. These sensitive markers were also upregulated in the delayed-recurrence population in the adjuvant model (P < 0.05). All genes were validated by RT-qPCR (fold change > 1.5). (E) Summary of enriched gene ontology (GO) terms of the seven genes associated with tumor volumes (FDR < 0.05). (F) Integrated functional network analysis of the selected genes using the STRING protein interaction network and KEGG pathway.
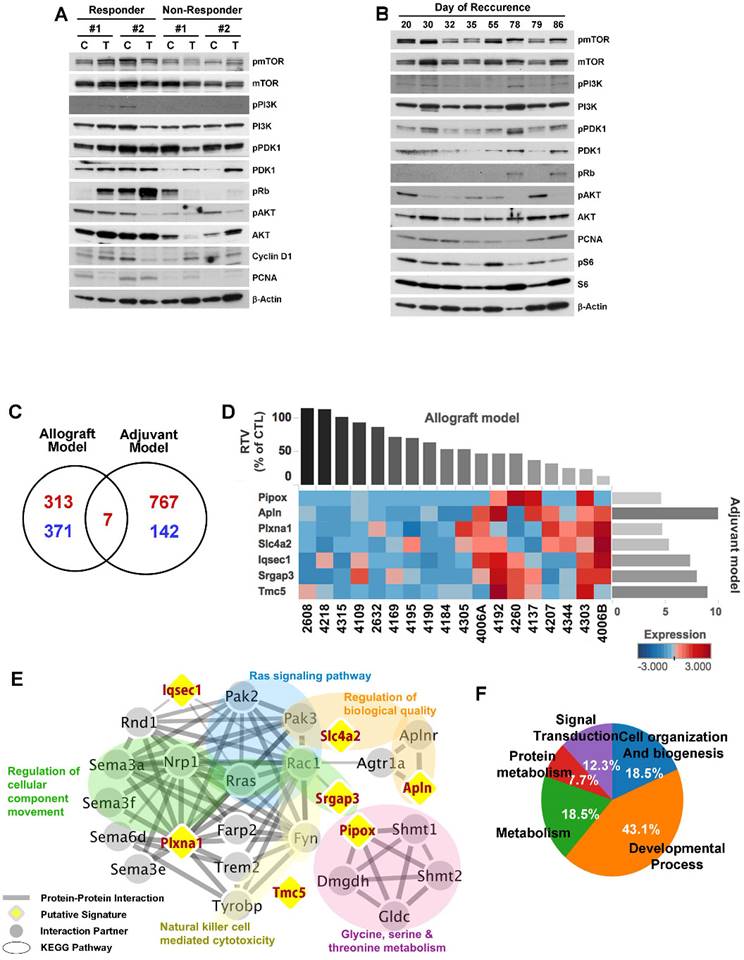
As an alternative strategy, we examined gene expression patterns in the corresponding baseline tumor tissue samples based on their responsiveness to MK-2206 treatment. We collected 18 non-treatment allograft mammary tumors, and screened the whole transcriptome using mRNA sequencing data and the Cufflinks computational pipeline to predict which genes were differentially expressed following MK-2206 treatment of Brca1-deleted tumors. We identified 691 genes that appeared to be differentially expressed between robust and poor responders at a significance level of 0.05 (two-sample t-test). We then used the same data-processing procedure on 10 tumor samples obtained from resection of the adjuvant model. We identified 916 genes that were differentially expressed between early- and delayed-recurrence populations in the adjuvant model. Of these, seven overlapped with the 691 putative predictors identified in the allograft model (Fig. 6C, 6D, and Supplementary Table 2). Express of the seven DEGs were further confirmed higher in the responder (P < 0.05), as validated using RTqPCR (fold change >1.5) (Supplementary Fig. 4).
To assess the downstream effects of the seven putative markers following MK-2206 treatment, we identified downstream pathways that might specifically affect the regulation of MK-2206 resistance or sensitivity. To this end, we constructed a protein-protein interaction (PPI) network of these seven genes and their interacting molecules using the STRING database [24] [25] and KEGG pathway. This analysis showed that the 27 proteins were connected to various pathways (Supplementary Table 4) through a previously annotated PPI in the constructed network (Fig. 6E). It also revealed five clusters of proteins associated with various biological functions and cellular component (FDR < 0.05]. Among the five clusters, one was associated with the function “Ras signaling pathway”. Notably in this context, acquired resistance to AKT inhibition is associated with upregulation of the Ras pathway in breast cancer cell lines [31]. In this cluster, R‑ras, Rac1, and Pak2/3 directly interacted with Plxna1 (Plexin A1), Iqsec1 (IQ motif and Sec7 domain 1), and Srgap3 (SLIT-ROBO Rho GTPase activating protein 3). Other modules, including “Natural killer cell mediated cytotoxicity” and “Glycine, serine and threonine metabolism”, might also be associated with the response to MK-2206. Additionally, the network facilitated identification of Plexin A1 as potentially being responsible for the crosstalk between the “Ras signaling pathway” and other categories.
Gene ontology (GO) analyses also revealed that the 27 proteins in the constructed network were involved in signal transduction (GO:0007165), cellular response to stimulus (GO:0051716), negative regulation of cellular component movement (GO:0051271), and regulation of cell morphogenesis (GO:0022604) (Supplementary Table 3). These results are summarized in Figure 6F, which highlights five categories of affected biological processes.
Discussion
BRCA1-deficient breast cancer is a very well-known hereditary cancer. However, except for resection of normal mammary glands and ovaries, there is no accepted measure for proactively preventing tumor development. Mutations of BRCA1 are known to confer an increased risk for breast, ovarian and prostatic cancers, but it is unclear why these hormone-responsive tissues are particularly sensitive to such genetic alterations. Previously, we showed that estrogen signaling contributes to BRCA1-deficient mammary tumors and demonstrated that its inhibition may be useful for the prevention of BRCA1-mutated breast cancer [32]. In addition, tamoxifen might be an effective adjuvant treatment for BRCA1-related breast cancer to prevent the recurrence or development of a contralateral mammary tumor, regardless of the estrogen receptor (ER) status of the tumor [33].
We herein show that AKT plays an important role in the tumorigenesis caused by the loss of BRCA1, and provide evidence suggesting that targeting AKT could be therapeutically relevant for preventing and treating BRCA1-associated breast cancer. Yang and colleagues previously showed that loss of BRCA1 leads to AKT activation, and AKT depletion rescues cells from the abnormalities associated with BRCA1 mutation [15, 17]. Here, we tested whether reducing AKT function reversed the abnormalities associated with the loss of BRCA1 in a mouse model. Brca1Δ11/Δ11 embryos exhibited an increased number of phospho-histone H3-positive cells in the cerebellum and liver during development. By contrast, the level of phospho-histone H3-positive cells in Brca1Δ11/Δ11Akt1+/- embryos resembled that in BRCA1-proficient embryos, but more severely abnormal neural development phenotypes were observed in these double-mutant embryos. Our analysis of mammary tissues from pubertal mice showed that the loss of AKT1, the major isoform of AKT in mammary gland epithelial cells, significantly reduced ductal length and branching, suggesting that suppression of AKT activity effectively reduces the proliferation of mammary epithelial cells. At 16 months of age, significantly fewer Brca1co/coMMTV-Cre mice with a haploinsufficiency of Akt1 exhibited mammary tumor formation (22%; 6 of 27) compared to Brca1co/coMMTV-Cre mice with WT Akt1 (54%; 15 of 28), suggesting that AKT activity is important for the formation of Brca1-mutant mammary tumors. Our in vivo studies using tumor-bearing Brca1-mutant mice showed that monotherapy with the AKT inhibitor, MK-2206, significantly extended survival (>2-fold), reduced tumor growth (48.5% and 60.8% control of naturally formed and transplanted tumors, respectively), and delayed the recurrence of tumors after resection (~1.7-fold).
Of particular interest was our observation that, although the AKT pathway is downstream of estrogen signaling, inhibition of AKT not only suppressed tumor initiation, it also reduced the progression of Brca1 mutation-associated tumors. Interestingly, we found that extent of G1/S progression, reduced the levels of proliferation-associated proteins, and suppressed cell survival by inhibition of AKT were more than reversing the stimulatory effects of estrogen treatment, suggesting that AKT is able to mediate proliferation signals other than those of the ER-PI3K-AKT pathway in the context of a BRCA1 deficiency. Indeed, it is known that phosphorylation of AKT is modulated by insulin-like growth factor signaling in the absence of BRCA1 [34], indicating that AKT can be activated through pathways other than estrogen signaling. Thus, targeting of AKT could be a more effective strategy for preventing tumorigenesis compared with suppression of estrogen signaling.
Importantly, our in vivo results show that tumor-bearing mice harboring the same Brca1 mutation (Brca1-Δ11) were not uniform in their responses to MK-2206. Although MK-2206 treatment triggered significant responses in three different preclinical trials, tumor progression in some mice resembled that observed in the corresponding untreated control mice. In our allograft experiment, tumors in nine of 18 mice treated with MK-2206 were more than 50% smaller than those of their control counterparts, but the tumors in three mice were larger than those of their control counterparts. In experiments involving adjuvant therapy after resection, the average time to recurrence was 29.3 days in the control group (N = 10; 95% CI, 16.2-42.0), indicating that tumors recurred in half of mice over a similar period, despite MK-2206 treatment. Classification of MK-2206-treated mice based on whether their time to recurrence was inside or outside the CI of untreated mice (non-responders and responders, respectively) revealed that average recurrence time in MK-2206 non-responders (N = 5) was 28.4 days (95% CI, 17.0-39.8). Moreover, tumor-free survival in responders (N = 5) was 2.4- and 2.5-times longer than that in untreated and non-responder groups, respectively (mean, 70.4 days; 95% CI, 59.0-81.8). Precision (or personalized) medicine, in which an individual's drug response is predicted based on a genetic analysis of their baseline tumor, is the next horizon of efficient tumor therapy and patient safety. Although we do not yet understand the mechanisms underlying the variability in the response to a given drug, recent findings have suggested that genetic markers can be closely associated with sensitivity to a specific drug. Here, we compared the gene expression profiles of two sets of baseline tumors with the responsiveness of these tumors to MK-2206. This analysis identified seven DEGs as MK-2206 response-associated genes that have previously been reported to have a role in regulating AKT activity (APLN, PIPOX, SLC4A, and PLXNA1) [35] [36] [37] [38] or are related to PI3K activation (TMC, SRGAP3, and IQSEC1) [39] [40] [41]. These genes encode proteins involved in developmental process, metabolism, cell organization and biogenesis, signal transduction, and protein metabolism. A further analysis of PPI networks of these genes and their interacting proteins revealed that a number of protein clusters are associated with MK-2206 responsiveness. These include the Ras signaling pathway; regulation of cellular component movement; natural killer cell-mediated cytotoxicity; glycine, serine, and threonine metabolism; and regulation of biological quality.
Prevention and treatment options for BRCA1-associated breast cancer have been limited by many clinical trial-related hurdles. Thus, preclinical simulation in a mouse model, such as our approach using Brca1-mutant mice bearing endogenous tumors, is a useful strategy for testing treatment efficacy. Results of the current study based on this mouse simulation system suggest that inhibition of AKT could be a useful strategy for both preventing and treating BRCA1-associated breast cancer. Additional studies are needed to confirm the potential of this strategy and the utility of these markers for future clinical applications of MK-2206 responsiveness in BRCA1-associated breast cancer.
Supplementary Material
Supplementary figures and tables.
Abbreviations
BRCA1, breast cancer type 1 susceptibility protein; DEG, differentially expressed gene; ER, estrogen receptor; FPKM, fragments per kilobase per million mapped fragments; MMTV, mouse mammary tumor virus; PARP, inhibitor of poly (ADP-ribose) polymerase; PI3K, phosphatidylinositol 3-kinase; RTV, ratio of tumor volume.
Acknowledgements
This work was supported by the National Cancer Center of Korea (NCC-1610030/1710900), and the National Research Foundation of Korea (2014R1A2A1A11049935/2018R1A2B6001216). We would like to thank Omics, Animal Imaging, and Bioinformatics Cores at National Cancer Center Korea for supporting analysis.
Ethics Approval
All procedures involving animals and their care were approved by the Institutional Animal Care and Use Committee of the National Cancer Center of Korea.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416-26
2. Caestecker KW, Van de Walle GR. The role of BRCA1 in DNA double-strand repair: past and present. Exp Cell Res. 2013;319:575-87
3. Takaoka M, Miki Y. BRCA1 gene: function and deficiency. Int J Clin Oncol. 2018;23:36-44
4. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66-71
5. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329-33
6. Venkitaraman R. Triple-negative/basal-like breast cancer: clinical, pathologic and molecular features. Expert Rev Anticancer Ther. 2010;10:199-207
7. Nanda R. "Targeting" triple-negative breast cancer: the lessons learned from BRCA1-associated breast cancers. Semin Oncol. 2011;38:254-62
8. Glendenning J, Tutt A. PARP inhibitors-current status and the walk towards early breast cancer. Breast. 2011;20(Suppl 3):S12-9
9. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA. et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079-84
10. Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152-8
11. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N. et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017
12. Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J. et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2:1048-63
13. Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29-86
14. Liu W, Bagaitkar J, Watabe K. Roles of AKT signal in breast cancer. Front Biosci. 2007;12:4011-9
15. Xiang T, Ohashi A, Huang Y, Pandita TK, Ludwig T, Powell SN. et al. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008;68:10040-4
16. Xiang T, Jia Y, Sherris D, Li S, Wang H, Lu D. et al. Targeting the Akt/mTOR pathway in Brca1-deficient cancers. Oncogene. 2011;30:2443-50
17. Jia Y, Song W, Zhang F, Yan J, Yang Q. Akt1 inhibits homologous recombination in Brca1-deficient cells by blocking the Chk1-Rad51 pathway. Oncogene. 2013;32:1943-9
18. Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T. et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37-43
19. Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L. et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323-30
20. Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349-52
21. Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA. et al. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001;28:266-71
22. Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL. et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009;35:534-41
23. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562-78
24. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447-52
25. Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000;28:3442-4
26. Ciruelos Gil EM. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat Rev. 2014;40:862-71
27. Gorrini C, Gang BP, Bassi C, Wakeham A, Baniasadi SP, Hao Z. et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc Natl Acad Sci U S A. 2014;111:4472-7
28. Li W, Xiao C, Vonderhaar BK, Deng CX. A role of estrogen/ERalpha signaling in BRCA1-associated tissue-specific tumor formation. Oncogene. 2007;26:7204-12
29. Baek HJ, Kim SE, Choi EK, Kim JK, Shin DH, Park EJ, Kim TH, Kim JY, Kim KG, Deng CX, Kim SS. Inhibition of Estrogen Signaling Reduces the Incidence of BRCA1-associated Mammary Tumor Formation. Submitted.
30. Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T. et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell. 2008;32:11-20
31. Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W. et al. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics. 2010;3:26
32. Bachelier R, Xu X, Li C, Qiao W, Furth PA, Lubet RA. et al. Effect of bilateral oophorectomy on mammary tumor formation in BRCA1 mutant mice. Oncol Rep. 2005;14:1117-20
33. Foulkes WD, Goffin J, Brunet JS, Begin LR, Wong N, Chappuis PO. Tamoxifen may be an effective adjuvant treatment for BRCA1-related breast cancer irrespective of estrogen receptor status. J Natl Cancer Inst. 2002;94:1504-6
34. Shukla V, Coumoul X, Cao L, Wang RH, Xiao C, Xu X. et al. Absence of the full-length breast cancer-associated gene-1 leads to increased expression of insulin-like growth factor signaling axis members. Cancer Res. 2006;66:7151-7
35. Tang SY, Xie H, Yuan LQ, Luo XH, Huang J, Cui RR. et al. Apelin stimulates proliferation and suppresses apoptosis of mouse osteoblastic cell line MC3T3-E1 via JNK and PI3-K/Akt signaling pathways. Peptides. 2007;28:708-18
36. Natarajan SK, Muthukrishnan E, Khalimonchuk O, Mott JL, Becker DF. Evidence for Pipecolate Oxidase in Mediating Protection Against Hydrogen Peroxide Stress. J Cell Biochem. 2017;118:1678-88
37. Liu X, Zhou X, Xu H, He Z, Shi X, Wu S. SLC34A2 Regulates the Proliferation, Migration, and Invasion of Human Osteosarcoma Cells Through PTEN/PI3K/AKT Signaling. DNA Cell Biol. 2017;36:775-80
38. Castro-Rivera E, Ran S, Brekken RA, Minna JD. Semaphorin 3B inhibits the phosphatidylinositol 3-kinase/Akt pathway through neuropilin-1 in lung and breast cancer cells. Cancer Res. 2008;68:8295-303
39. Cizkova M, Cizeron-Clairac G, Vacher S, Susini A, Andrieu C, Lidereau R. et al. Gene expression profiling reveals new aspects of PIK3CA mutation in ERalpha-positive breast cancer: major implication of the Wnt signaling pathway. PLoS One. 2010;5:e15647
40. Jain P, Fierst TM, Han HJ, Smith TE, Vakil A, Storm PB. et al. CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene. 2017;36:6348-58
41. Sakurai A, Jian X, Lee CJ, Manavski Y, Chavakis E, Donaldson J. et al. Phosphatidylinositol-4-phosphate 5-kinase and GEP100/Brag2 protein mediate antiangiogenic signaling by semaphorin 3E-plexin-D1 through Arf6 protein. J Biol Chem. 2011;286:34335-45
Author contact
![]() Corresponding author: Sang Soo Kim, Phone: (8231) 920-2491, Fax: (8231) 920-2494, Email: sangsookimre.kr
Corresponding author: Sang Soo Kim, Phone: (8231) 920-2491, Fax: (8231) 920-2494, Email: sangsookimre.kr