Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Birth-and-death balance at all...
Individuals at different levels...
Animal cells have three...
Apoptosis is irrelevant to...
Cancer cells may lack SD and...
Most studies professed to be on...
There are four basic SICD...
Concluding remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(13):1800-1812. doi:10.7150/ijbs.26962 This issue Cite
Review
Evaluating the Remote Control of Programmed Cell Death, with or without a Compensatory Cell Proliferation
Xixi Dou1,2 ![]() , Lichan Chen3
, Lichan Chen3 ![]() , Mingjuan Lei4, Lucas Zellmer5, Qingwen Jia1, Peixue Ling1,2, Yan He6, Wenxiu Yang7, Dezhong Joshua Liao6,7
, Mingjuan Lei4, Lucas Zellmer5, Qingwen Jia1, Peixue Ling1,2, Yan He6, Wenxiu Yang7, Dezhong Joshua Liao6,7 ![]()
1. Key Laboratory of Biopharmaceuticals, Shandong Academy of Pharmaceutical Sciences, Jinan 250101, Shandong Province, P.R. China
2. Technology Center, Shandong Freda Pharmaceutical Group, Jinan 250101, Shandong Province, P.R. China
3. College of Chemical Engineering, Huaqiao University, Xiamen 361021, Fujian Province, P.R. China
4. Hormel Institute, University of Minnesota, Austin, MN 55912, USA
5. Masonic Cancer Center, University of Minnesota, 435 E. River Road, Minneapolis, MN 55455, USA
6. Key Lab of Endemic and Ethnic Diseases of the Ministry of Education of China in Guizhou Medical University, Guiyang 550004, Guizhou Province, P.R. China
7. Department of Pathology, Guizhou Medical University Hospital, Guiyang 550004, Guizhou province, P.R. China
Received 2018-4-30; Accepted 2018-8-24; Published 2018-10-19
Abstract

Organisms and their different component levels, whether organelle, cellular or other, come by birth and go by death, and the deaths are often balanced by new births. Evolution on the one hand has built demise program(s) in cells of organisms but on the other hand has established external controls on the program(s). For instance, evolution has established death program(s) in animal cells so that the cells can, when it is needed, commit apoptosis or senescent death (SD) in physiological situations and stress-induced cell death (SICD) in pathological situations. However, these programmed cell deaths are not predominantly regulated by the cells that do the dying but, instead, are controlled externally and remotely by the cells' superior(s), i.e. their host tissue or organ or even the animal's body. Currently, it is still unclear whether a cell has only one death program or has several programs respectively controlling SD, apoptosis and SICD. In animals, apoptosis exterminates, in a physiological manner, healthy but no-longer needed cells to avoid cell redundancy, whereas suicidal SD and SICD, like homicidal necrosis, terminate ill but useful cells, which may be followed by regeneration of the live cells and by scar formation to heal the damaged organ or tissue. Therefore, “who dies” clearly differentiates apoptosis from SD, SICD and necrosis. In animals, apoptosis can occur only in those cell types that retain a lifelong ability of proliferation and never occurs in those cell types that can no longer replicate in adulthood. In cancer cells, SICD is strengthened, apoptosis is dramatically weakened while SD has been lost. Most published studies professed to be about apoptosis are actually about SICD, which has four basic and well-articulated pathways involving caspases or involving pathological alterations in the mitochondria, endoplasmic reticula, or lysosomes.
Keywords: Apoptosis, Stress-induced cell death, Senescent death, Necrosis, Cancer, Evolution, Regeneration
Introduction
In the culture of China and quite a few other east Asian countries, “Yin” (meaning negative, female, etc.) and “Yang” (meaning positive, male, etc.) are often used to describe two opposite extremes or situations, like black vs white, night vs day, life vs death, etc. This Yin-Yang contrast has, in the recent decades, been borrowed to describe different balances between two extremes in the biomedical sphere, with exemplary references cited here [1-5]. For instance, in an animal's body, cells may die via a predetermined procedure, which are coined as programmed cell deaths, with ensuing proliferation of the live cells to compensate for the cell loss [6], together constituting a Yin-Yang balance. Cells can die via a predetermined procedure because evolution has built death program(s) in the genome of each animal species. However, in the meantime evolution has also built mechanisms to allow the cells' host tissue, organ and even the entire body of the animal to control the death program(s) for the animal's ultimate interest, although this systemic regulation has not been sufficiently addressed in the literature. Because of the evolutionary establishment of this systemic control, both the programmed cell death and the death-and-birth balance are not predominantly regulated by the cells themselves, but are mainly regulated by the cells' superior(s), i.e. the host organ or tissue or even the animal's body [7-9]. Actually, this superior and external control of deaths and death-birth balances is a common rule of the earth's ecosystem and occurs at all levels of life, in our opinion. We infer that, because the death program(s) are controlled superiorly, an individual at any level has to be loyal to its superior as a condition for its survival, with organismal species controlled by the earth's ecosystem, which is the paramount “superior” and consists of the earth's environment and the interactions among different organismal species. In this essay we describe our musings on the control and coordination of cell deaths and births by host tissues or organs, and in turn by the animal's body, in physiological and pathological situations, as these external and superior regulations of different modes of programmed cell death have not been sufficiently addressed in the literature.
Birth-and-death balance at all levels of life is regulated externally and from above
Organisms of all kinds constantly come by birth and go by death. Actually, here on earth, the birth-and-death relationship overarches, and is the pivot of, life at all levels, i.e. at the levels of organelle, cell, organ/tissue, organism, and species, as stratified and adumbrated below:
- Many organismal species have reached extinction or are becoming extinct [10-13]. The ecocide does not occur as the wish of the extinct species themselves but, instead, is largely due to environmental changes [12, 14-19]. For instance, the dinosaur's extinction was not due to collective suicide of the dinosaurs but was because the environment had changed to a situation that was no longer suitable for their survival. In the meantime, environmental change also prods organisms into adaptation that leads to evolution either to new species (Fig 1) or to the development of new mechanisms for the organisms to survive in the new environment. As an example of the latter case, throughout evolution, a variety of microbes have equipped themselves with an ability to produce antibiotics, such as ampicillin, to kill their foes [20-25]. Similarly, many plants have also evolutionarily established ability to produce certain chemicals to fend off their enemies [26-33]. For example, many plants have evolutionarily equipped themselves with a mechanism to produce phytoestrogens that can interfere with animals' reproductive function [34-38]. We speculate that this can be a self-defensive mechanism, as those animals who eat too much of the plants will have their fertility inhibited and thus their population decreased, leading to the preservation of the plants (Fig 1).
- Within each species, individual organisms will die when they reach the end of their lifespans [39-41], but the species as a whole will survive via reproduction. The eventual death of individuals is a condition of the earth's ecosystem to allow the species to continue to exist, as the ecosystem would not allow any species to limitlessly expand its population to dominate the earth. To warrant this control by the ecosystem, aging-caused death of organisms is evolutionarily programmed in the genome of the species to make its individuals allegiant to, and controlled by, the species itself [39, 42-45]. In our opinion, this built-in program is a condition of the earth's ecosystem to allow the species to survive and, in turn, is a condition of the species to allow its individuals to survive, which again shows an ecosystem's basic rule that an individual has the built-in death program but its superior has the “remote control” of the program. In addition, because many species of plants and animals often die from various natural reasons, such as predation by their enemies, we surmise that evolution has also balanced their fecundity against their deaths caused by natural causes. Cannibalism is another mechanism evolutionarily conferred on many animal species, which allows these animal species to regulate their populations, to survive certain adverse situations, or to gain other advantages [46-48].
- Within an individual with multiple organs, such as a human being, some organs or tissues “die” after a certain embryonic stage or certain age, such as the disappearance of the pronephroi and then the mesonephroi during the embryonic development [49-54], and the regression of the thymus after puberty [55-59]. On the other hand, some other tissue structures “are born” after a certain embryonic stage or certain age, such as the appearance of the mesonephroi and then the permanent kidneys during the embryonic development [49-54]. The development of the mammary glands during the pregnant and lactating periods in women is another example of “new tissue birth” (women before pregnancy only have the breasts but not the mammary glands) [60-62]. Although usually considered as a disease, degeneration as a manifestation of aging is a natural event we all will experience sooner or later, which in most cases is due to cell demise in those cell types that can no longer regenerate in adulthood [63, 64], and sometimes is associated with the “birth” of new histological structures. For instance, some new histological entities may appear in the brains of certain neurodegenerative patients, such as the senile plaques and neurofibrillary tangles in the patients with Alzheimer's Disease [65, 66] and the Lewy bodies in the patients with Parkinson's Disease [67, 68].
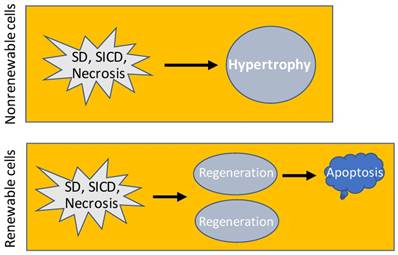
- Cells of an animal may be killed by external hazards such as infectious bacteria. Sometimes, external hazards may activate a death program of the cells, as explained before [6] and later in this essay. Even if without encountering an external hazard, cells will still die of aging when they reach the end of their lifespans [69-74]. Cells in a multi-cellular animal can be dichotomized based on whether they are renewable, i.e. whether they still retain a regenerative ability when they come to a certain age [75]. One group, exemplified by the cardiac myocytes [76-79] and neurons [75, 80], experiences proliferation during the periods of development and growth but loses the replicative ability in the adulthood [64]. When this nonrenewable group of cells experiences a massive death, such as from a bacterial infection or lack of blood supply, the remaining cells will undergo hypertrophy, i.e. increase in their cellular size, with the purpose of restoring the function of the organ or tissue (Fig 2), as often seen in cardiac myocytes [81-84], although hypertrophy appears in neurons only occasionally for unaddressed reasons [85-91]. The other group, such as white blood cells and epidermal keratinocytes, retains a lifelong ability to proliferate, although some cell types actually do not replicate themselves but are derived from another cell type, like most white blood cells that are constantly derived from the bone marrow. This renewable group not only has a short lifespan but also is often requested by the animal's body to defend the nonrenewable group, and in turn the body itself, by fighting against various external hazards, often causing the cells to die quickly and massively in adverse situations. For example, white blood cells are required to fight against infectious bacteria and are often killed by the bacteria. Therefore, this renewable group of cells usually has a teeming loss but, in the meantime, also regenerates robustly, ending with a high rate of cell turnover. In either of the two cell groups, when cell death abounds, the connective tissue may step in to form a granulation and an ensuing scar as new tissues to help in healing the damaged tissue or organ [92-94]. For instance, a myocardial infarction may cause the death of many cardiac myocytes with ensuing fibrosis in the affected areas of the heart [95, 96], and a chronic infection by hepatitis viruses may cause constant death of hepatocytes followed by the development of liver fibrosis and cirrhosis [97, 98]. The death-and-hypertrophy balance or the death-and-regeneration balance, with or without formation of granulation and scar as new tissues, is controlled, or, more correctly, coordinated, by the animal's body.
- Within individual cells, various organelles are often increased or decreased in their number or size in various situations, often discerned in the mitochondria [99], lysosomes [100] and endoplasmic reticula (ER) [101-103], for example. Skeletal muscle cells even have many nuclei and may gain more with additional muscle growth [104]. In a stressful situation, organelles may be requested to “die” so that the cell as a whole can survive, which is often manifested as autophagy that can be regarded as cannibalism at the organelle level or as self-cannibalism [6, 105]. In our opinion, neither the increase nor the decrease of these organelles is their own wish, but rather is willed and controlled by their superior, i.e. the cell.
Interrelationships among the death, birth and evolution of organisms. Left panel: The death and birth of organisms are balanced by the ecosystem, so that the earth will not be over-crowded by organisms and that there will not be any particular species dominating the earth. Deaths often occur when the environment is changed, whereas individual species, organisms, organs, etc., are not willing to die and will find ways to survive, resulting in evolution to new species and, sometimes, also to neoplasms that can be considered as “new species” at the tissue/organ level. Right panel: Phytoestrogens are, hypothetically, developed during evolution as a mechanism for plants to protect themselves from animals: The more an animal (such as a rabbit) eats a phytoestrogen-containing plant (such as a sort of grass), the more severely its reproductive function will be interfered with by the phytoestrogens. The plant is thus preserved by the decrease in the fertility of the animals.

Consequence of SD, SICD and necrosis. In nonrenewable cell types, SD, SICD or necrosis may cause hypertrophy of the remaining cells to restore the cell-death-caused functional loss. In renewable cell types, SD, SICD or necrosis triggers regeneration that usually results in over regeneration and ensuing apoptosis of the over-regenerated cells, which is a one-way procedure because apoptosis will not trigger regeneration.
Individuals at different levels of life are also egocentric, which drives evolution
As stratified above, individual deaths happen constantly at the levels of the organelle, cell, organ/tissue, organism, and species. Many of the deaths are evolutionarily programmed events, but the deaths are not actual suicides. Instead, the deaths are controlled by the superior, such as the animal's body, the species or the earth's ecosystem, and this control is the superior's condition to allow the individual to exist. This is, in our opinion, because evolution has built death program(s) inside a cell's genome but has handed the control of the program to their superior, e.g. the organ or tissue, the animal's body, up to the earth's ecosystem, to force individuals at all levels to be loyal to their superior and to obey its “orders”. However, individuals are also egocentric and, once they are endangered, their egoism will push them to look for ways to survive [8, 9, 106-109]. “Try to survive” is an impetus for organismal evolution that leads to the development of new species via mutations [107-109]. We are all familiar with the question “which comes first, the chicken or the egg?” The correct evolutionary answer for it is that neither one comes first. The first chicken evolved from a species that genetically is not chicken but is very close to chicken, with mutations occurring in the egg or the bird leading to its transformation, i.e. evolution, to the chicken.
Within an animal species, individuals also fight against each other to live better and longer, with cannibalism as an extreme phenomenon of this fight and filial cannibalism as an even more extreme example [110-113]. Similarly, within individual organs or tissues of an animal, individual cells fight against their fellow cells to live better or longer, which is commonly coined as “cell competition” [114-118]. We infer, with trepidation, that some cells may win the competition by mutating certain gene(s) to gain competence, and some of the mutations may convert the cells to a neoplastic version. This conjecture, which has not been explored and thus has so far not received material evidence, actually states that tumorigenesis can occur via an active mechanism, as it occurs due to a motivation of the parental normal cell to live longer or better. Of course, there are two additional possible mechanisms for neoplastic transformation [119]. One is that environmental change imposes survival pressure onto normal cells, such as when the cells encounter irradiation, genotoxic chemicals, etc. The cells need to evolve via efficacious mutations to survive the stressful situation, and some of the mutations beget neoplastic transformation [119]. This mechanism has received a rich vein of clinical evidence that chemo- or radio-therapy of tumors, which can cause mutations, increases the incidence of a second primary tumor [120-125]. As another mechanism, DNA replication during routine proliferation of normal cells may mistakenly result in mutations, and some of the mutations may be irreparable and lead to neoplastic transformation [126]. In this latter case, neoplasms as a sort of “new organism” are fortuitously developed without specific motivation, i.e. neither because the parental cells want to live longer or better nor because they want to survive in a stressful situation.
Animal cells have three programmed death modes
As abovementioned, all cells in an animal have a lifespan and have, beginning at birth, started their journey of aging towards death [41, 127, 128], but the lifespans of different cell types vary drastically. We define cell death via aging as “senescent death” (SD) [6], because aging and senescence are highly interrelated [129-137], although senescence itself is often defined as permanent growth arrest that does not necessarily lead to the death of the cell [131, 135-138]. Obviously, SD has been programmed in the cells [39], likely in the nuclear genome although it remains possible that part of the program is allocated to the mitochondrial genome [139, 140]. The fact that all cells of an animal have the same genome but may differ greatly in lifespan insinuates that the same SD program may be regulated quite differently in different cell types. A longer lifespan not only connotes a weaker SD ability, i.e. having more difficulty in dying of SD, but also is associated with a weaker regenerative ability, and vice versa, in our opinion [6]. For instance, cardiac myocytes and neurons have the longest lifespan but have the weakest SD mechanism and the weakest regenerative ability [64, 75, 76, 78, 80, 141-146], whereas the epidermal keratinocytes show the opposite properties [147, 148].
Any cell in an animal may encounter a host of stressors in its lifetime. A stressor may be an external one such as a toxic chemical or an infection by micropathogens. A stressor may also be an internal one such as a genetic mutation occurring spontaneously during DNA replication, but often an internal stressor may initially be created by an external one. For example, a genotoxic chemical often causes DNA mutations, and a change in the microenvironmental temperature (such as a fever) can alter the cellular metabolic rate and in turn the cellular pH. A very severe stress will directly kill the cell, which is dubbed as “necrosis” [6, 149]. However, when a stress is not severe enough to kill instantly, it may elicit cell death via the cell's own demise program, which is coined by us as “stress-induced cell death”, or “SICD” [6, 7, 149, 150]. For instance, when a cell cannot repair a mutation, caused by irradiation or a genotoxic chemical for example, it will likely turn on a death program to commit suicide, so that the mutation will not be passed to the filial cells during cell replication and become inherited [6, 7, 149]. Actually, causing mutation-driven SICD has become a major mechanism for irradiation and many chemotherapeutic agents in eliciting their therapeutic effects on cancer cells [119, 149, 151, 152], although this mechanism is widely misconstrued as apoptosis [149, 151, 152], as discussed below. As another commonly seen example, when white blood cells are infected by bacteria or viruses but cannot kill the micropathogens, the cells may also commit suicide by turning on a demise program, so that they will not carry the micropathogens to other body sites and spread the infection [153-157]. These exemplary cell deaths are the iron will of the animal's body for its ultimate interest [6, 149].
In animals, many cells will no longer be useful and thus need to be eliminated after certain developmental or physiological stages or certain ages [158, 159]. Examples include digit individualization during the human embryonic development which is associated with the death of the interdigital cells to avoid hand-webbing [131, 160], thymus regression after puberty which is actually a cell dooming procedure [55-59], post-partum involution of the uterus [161-164] and post-weaning involution of the mammary glands [165-168] featuring massive cell death, and atrophy of the gonads in older men and women which is also associated with death of certain gonadal cells [169-174]. These physiological deaths occur in part because each organ or tissue has a physiological total number of the described cells, and, to keep the cell number within the physiological range, jettison of the excessive cells is a must [175]. In the exemplary physiological situations mentioned above, the jettisoned cells are those no-longer needed or, in Savill's word [176-180], “unwanted”, by the animal's body, thus being redundant and useless, and the dooming procedure has been evolutionarily programmed inside the cells. This program needs to ensure 1) that the decease remains physiological without eliciting immune reactions that damage the host organ or tissue, and 2) that the decease occurs in a cannibalistic manner so that the materials of the dead cell can be recycled. These two requirements are met by a swift engulfment of the dooming or doomed cell by a macrophage or another phagocytic cell [6, 7, 149, 150, 181]. Since the engulfment functions as the scavenging of the cell corpse [177-180, 182], these cells are collectively tagged by us as “scavengers” [6, 7, 149, 150]. Because of the requirement of scavengers, this dooming mechanism becomes well established only when an animal's lymphatic and blood circulation systems have been evolutionarily developed to allow macrophages to travel distantly to the apoptotic cells [6, 7, 149, 150, 181]. We define this particular death program that aims at purging away useless and redundant cells from a tissue or organ as “apoptosis”, as we construe that this is the cell death mode described by Kerr et al in their seminal study in which the word “apoptosis” was created [183]. As summarized by Savill [176], Kerr et al and a professor of ancient Greek created the word “apoptosis” to liken the cell death to “the dropping off as of leaves from a tree”, which emphasizes that the death is physiological, occurs to individual cells (or leaves) via an endogenous program, and can be triggered through a program regulated by external stimuli (or autumn). We emphasize the “external regulation” of this “endogenous program” because it is insufficiently addressed in the literature.
If we are familiar with how some companies are so wary of causing themselves troubles when they want to get rid of their redundant employees, we will be able to fathom why and how evolution equips animal cells with an apoptotic mechanism for the animal's body to expunge its redundant cells, in a manner not only harmless but also wholesome, in order to lower the total cell number of each tissue or organ to the physiological range. As a caveat that needs to be given, although probably all of us describe apoptosis as a suicidal event, this is only partially correct. On the one hand we can indeed regard it as a suicide because the cell dies via a program contained in itself. However, on the other hand we can also consider the death as a homicide, not only because it is not the cell's own wish but also because there is a killer, who is the cell's host, and the killing has a motivation, which is that the host wants to get rid of no-longer needed cells. Restated, the death is remotely controlled.
Apoptosis is irrelevant to nonrenewable cell types
During the developmental and growth stages, all cell types, including the nonrenewable ones like cardiac myocytes and neurons, proliferate to meet the body's growth needs, but this growth is not regeneration. After cessation of the body growth, nonrenewable cell types become well-differentiated and post-mitotic, and thus can no longer proliferate [64]. These facts lead us to a conclusion that in the whole lifespan the body never has excessive cells of a nonrenewable type to scrap via apoptosis. In other words, nonrenewable cell types do not have a chance to undergo apoptosis, if apoptosis is defined as a physiological mechanism to remove excessive cells. However, nonrenewable cell types may die of SD, SICD or necrosis. Actually, if these cells experience a massive SICD or necrosis during the developmental or growth period, such as when the cells are severely infected by bacteria, regeneration may still occur in these early stages of the life. Therefore, although apoptosis, a form of cell death, and regeneration, a form of cell birth, have a Yin-Yang contract, they share a property, i.e. being skipped by nonrenewable cell types. This property has never been pointed out before in the literature, to our knowledge.
Cancer cells may lack SD and have strengthened SICD but weakened apoptosis
It has been known to all pathologists for a century that cancer cells are weaker and have a higher death toll than their normal counterparts [119, 149, 152]. As the Nobel laureate Peyton Rous had pointed out in 1941, “that cancer cells are often sick cells and die young is known to every pathologist” [184]. In our opinion, these myriad deaths occur via necrosis and SICD, but not apoptosis. We opine that the SICD program in cancer cells has been strengthened, compared to that in their parental normal cells (Table 1), which justifies the use of SICD-causing irradiation or genotoxic chemicals to treat cancer. In contrast, the apoptotic mechanism may have been weakened or even lost in cancer cells (Table 1), since a neoplasm is autonomous and thus is generally acknowledged as a “new organism” [149, 185-188], somewhat similar to a new bacterial strain, because both of them are maintained by constant replication of their cells and have no predetermined physiological total number of cells and thus no redundant cells [149]. Indeed, tumor lumps continue enlarging their sizes, proving their lack of cell redundancy. This conjecture that the apoptotic mechanism in cancer cells has been greatly weakened still lacks substantiation but deserves testing, as it nixes the popular strategy of cancer therapy by enhancing cancer cells' apoptosis [189-192], although in most cases this so-called “apoptosis” is actually SICD, as discussed before [6] and further enlarged below.
Differences between normal cells and cancer cells in the four cell death modes
| Apoptosis | SD | SICD | Necrosis | |
|---|---|---|---|---|
| Normal | + | + | + | + |
| Cancer | - | - | ++ | ++ |
Note: "++" means more severe than "+", and "-' means lacking or weakened.
In our opinion, cancer cells cannot undergo aging and thus will not die of SD (Table 1), because cancer cells by their definition in all pathology textbooks are immortal, making cancers somewhat resemble bacterial strains as abovementioned. The concept of “cancer cell senescence” and its enhancement as cancer therapy, which has recently become popular [193-197], is unfathomable to us, as it hints slyly that “immortal cells still undergo aging and die of it”, and thus is illogical [6]. Those cells that can undergo aging and die of SD should not be called neoplastic, no matter how their morphology and/or behavior resembles that of neoplastic cells, if we all agree to stick with the definition of neoplasm in pathology textbooks that put immortality as an indispensable criterion for neoplastic cells, including the benign ones. However, a caveat that needs to be given is that certain treatments may be able to induce differentiation of certain, but probably not all, types of cancer, and thus reestablish an SD mechanism. This reestablished SD via induction of differentiation (probably, re-differentiation) is actually a “reprogramming of a reprogrammed program” and thus is not the same thing as a simple enhancement of an existing SD mechanism.
An enchanting question that has thus far not been answered is whether SD, apoptosis and SICD, or any two of the three (such as apoptosis and SD), share the same death program. Reiterated, it remains unknown whether animal cells have only one suicidal program or, instead, have two or three different suicidal programs responsible for apoptosis, SD and/or SICD. Since building more programs will likely enlarge the genome and become extravagant, we intend to believe that evolution has likely followed a more efficient path and built only one single program in a cell but allows it to be regulated differently to control SD, apoptosis or SICD. Despite that cancer cells may have lost SD and even apoptosis, the fact that irradiation and many chemotherapeutic agents can cause SICD of cancer cells, as described above, evinces that these immortal cells still retain a suicidal program (Table 1). One possibility is that normal cells have three different death programs controlling apoptosis, SD and SICD, respectively, and their immortalization deletes the one for SD and weakens the one for apoptosis but leaves the one for SICD intact. Alternatively, normal cells have only one single but flexible death program that can be directed to apoptosis, SD or SICD in different situations, and immortalization blocks the pathway to SD and dramatically neutralizes that to apoptosis without affecting that to SICD. Although neither of these two possibilities has hitherto been supported by material evidence, it is clear that the death program(s) in cancer cells differ, in one way or another, from the one(s) in their parental normal cells.
Most studies professed to be on apoptosis are actually on SICD
As we have repeatedly pointed out before [6, 7, 149, 198], SICD has been extensively studied under the name of apoptosis, whereas authentic apoptosis has hitherto received insufficient attention. In case readers do not want to leaf through our previous perspective articles, herein we adumbrate our rationales for why most relevant studies have misconstrued SICD as apoptosis: First, in apoptosis, it is those healthy but no longer needed cells that die [176-179], which is a black-white demarcation distinguishing apoptosis from SD, SICD and necrosis wherein it is those ill but useful cells that die [6, 7, 149]. Therefore, apoptosis does not trigger regeneration, because the animal's body will not annihilate redundant cells via apoptosis and simultaneously produces more cells to increase the redundancy [6, 7, 149]. In contrast, SD, SICD or necrosis of renewable cells triggers regeneration to restore the physiological cell number. The other way around may be correct, i.e. a regeneration triggered by SD, SICD or necrosis usually produces more cells than what is needed, and thus apoptosis usually ensues to eliminate the over-regenerated cells. Indeed, fresh granulations and scars usually experience such over regeneration and thus usually shrink (regress) later via apoptosis of the over-regenerated cells (Fig 2) [199-202]. Second, almost all studies claimed to be on apoptosis involve a stressor, such as a chemotherapeutic agent, to trigger the death, making it pathological and homicidal with the stressor as the killer, whereas apoptosis is a physiological event initiated by the animal's body and not by an external agent [6, 7, 150, 176, 178]. Indeed, in most of these studies, the stressor causes damage to the mitochondria and/or lysosomes, typically increasing the permeability of their membranes, which then results in leakage of their proteins into the cytoplasm [203-211] and thus is unadulteratedly pathological. Third, most studies professed to be on apoptosis involve cell lines in cell culture systems, making the studies irrelevant to apoptosis for at least three reasons [6, 7]: 1) Cell lines are immortalized with their apoptotic mechanism drastically weakened or even lost, as we have expounded above for immortal cancer cells. Even if they still retain an apoptotic program, it is reprogrammed from, and thus is not the same as, the program in their normal parental cells, whereas our purpose in studying apoptosis is to know the original death program, but not the reprogrammed program. In other words, we are dealing with two different programs, i.e. the original, intact one and its altered version, and we should only name one of the two as apoptosis. 2) Unlike their normal counterparts in animals, immortal cell lines growing in culture dishes no longer have a physiological total number and cannot be split into the “useful” and “useless” categories, although they may behave differently and may die when their growth becomes confluent [212-218]. Therefore, in a culture dish there is no way of deciding how many cells, and which ones, should be extinguished. Actually, no cell within the dish has any interest in, and qualifications for, making these decisions. 3) Apoptosis requires two players, i.e. a scavenger cell, the predator, and the dying or dead cell, the prey [178, 179]. The later part of the apoptotic procedure occurs inside, and is performed by, the scavenger cell [177-180, 182]. This means that apoptosis also involves proteins, mostly lysosomal enzymes, of the scavenger origin [6, 7]. In contrast, in most cell culture studies there is only one player, i.e. the dying cell itself, with the whole procedure occurring inside it only and involving its proteins only; therefore, the cell deaths have no way of being a true apoptosis [6, 7]. There are other iconic features of authentic apoptosis that differ from SD, SICD and necrosis and have been detailed by us before [6, 7, 149, 198].
There are four basic SICD pathways that have been well delineated
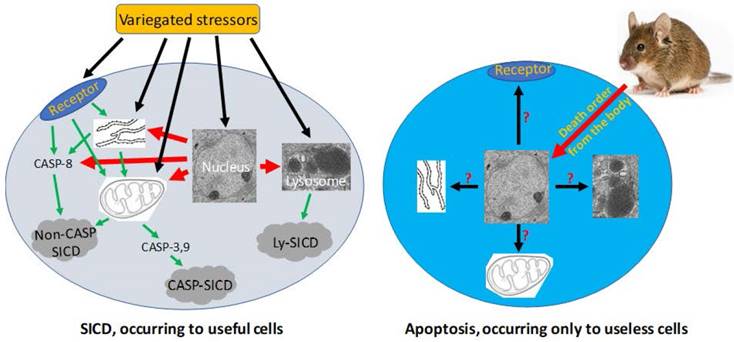
Since SICD has been extensively studied under the name of apoptosis, we can search its mechanisms in the literature simply by changing the keyword from apoptosis to SICD. Because stressors are variegated, SICD has numerous circumstances and thus multitudinous ad hoc modes [6]. A stressor, no matter whether it is an external one like a chemotherapeutic agent or an internal one like a change in the cellular pH, may trigger SICD via one of four basic pathways (Fig 3), as summarized from a rich vein of the literature that is actually overbearing [203-211, 219-223]: 1) The stressor may bind to a death receptor on the cell membrane, in turn activating caspase-8 and then the so-called “extrinsic apoptosis pathway”, which does not involve mitochondria [209, 224-231] and, in our opinion, should be renamed as “mitochondria-independent SICD” as it is not apoptosis at all. 2) The stressor may occur within, or directly act on, the mitochondria, resulting in its leakage of cytochrome c and other proteins to the cytoplasm and then the activation of the so-called “caspase-dependent apoptosis” or “caspase-independent apoptosis” [204, 207, 224, 232, 233], which in our opinion should be redefined as “caspase-dependent or -independent SICD”. We remain the only ones who are intrepid enough to remind peers that the permeability change of the mitochondrial membrane is caused by a stressor, is an unadulteratedly pathological event, and thus cannot be part of physiological apoptosis [6, 198]. 3) The stressor may occur within, or directly act on, the ER [234], in turn triggering a mitochondria-independent death via caspase-8 [229, 230] or triggering a mitochondria-dependent death that may be caspase-dependent or -independent [206, 229, 230, 235-238], as described above. 4) The stressor may occur within, or act directly on, lysosomes, leading to the leakage of its enzymes into the cytoplasm to kill the cells by hydrolysis of various cellular components [219-223]. Obviously, the lysosomal damage is also pathological. In addition, a stressor may also directly occur within or act on the nuclear genome (Fig 3), such as a lethal mutation occurring spontaneously, which may elicit an SICD via a pathway involving mitochondria and caspases described above.
Pathological SICD vs physiological apoptosis. Left-panel: SICD occurs to useful cells and is often caused by an external stress directly or indirectly via causing an internal stress (such as a genomic DNA damage). The external or internal stressor acts on a death receptor at the cell membrane, the endoplasmic reticulum (ER), the mitochondria, or the lysosomes, executing a suicidal procedure via a caspase-dependent (CASP-SICD) or -independent (Non-CASP SICD) pathway or via a lysosome (Ly) mediated pathway. Right-panel: Apoptosis occurs as an order from the animal's body to eliminate no-longer useful (thus redundant) cells. Because few in vivo studies on authentic apoptosis without involving a stressor as the death inducer have been documented, we really know little about apoptosis, but it cannot be ruled out, or ruled in, that the apoptotic mechanism is the same as, or similar to, that used in SD and/or SICD.
The fact that mitochondria, lysosomes and ER play roles in programmed cell death, besides their primary functions in physiological situations, reflects another fact that most, if not all, components and genes in our cells have dual functions, i.e. can be good or bad to the cells [239]. Indeed, our genes and organelles often function oppositely in physiological and pathological situations [239]. What function a gene or an organelle should elicit depends on our body's decision regarding its ultimate interest in that particular situation, which usually is an attempt to convert a pathological situation to a normal one [127, 239]. For instance, mitochondria normally power the cell but kill the cell in many pathological situations [240], as described above. Cytochrome c normally serves as the most potent oncoprotein as it sustains our cells' life by powering them, but, once released from the mitochondria to the cytoplasm, it serves as the most potent tumor suppressor to kill our cells via SICD [6, 198, 241]. Somewhat oppositely, the P53 protein normally arrests progression of the cell cycle to gain time for the cell to repair mutations and in turn to return to its normal life trajectory [242-245], for which it should not be considered as a tumor suppressor, in our opinion [239, 246]. However, if the mutations are irreparable, P53 goads the cell into SICD and thus functions as a tumor suppressor [131, 239].
Concluding remarks
There has been a prodigious amount of research on animal cell death modes. However, most of these studies have been focused on the death procedures and on the underlying mechanisms within the dying cell only, with insufficient attention on how the animal's body regulates the death and coordinates it with the responses of the live cells in the involved organ or tissue and in other body sites, including cells of the connective tissue that form granulations and scars. We sort cell demise modes into four basic types, i.e. physiological SD and apoptosis as well as pathological SICD and necrosis [6, 149]. SD, apoptosis and SICD are programmed events, with the program(s) evolutionarily built into the cells that do the dying, but largely with the animal's body maintaining remote control of the program(s). Apoptosis expunges those useless yet normal cells while SD and SICD, like necrosis, exterminate those useful but ill cells, warning us that uselessness is always fatal and illness is dangerous in many cases. In cancer cells, SICD may have been strengthened while SD has been lost and apoptosis has been dramatically weakened. Apoptosis involves phagocytosis of the dooming or doomed cell by a macrophage or another scavenger-type cell, with the later part of the procedure occurring inside, and performed by, the scavenger cell. This insinuates that apoptosis has a cannibalistic property at the cellular level. SICD as a programmed pathological event resides between, and overlaps with, apoptosis and necrosis [6, 149]. This unique trait makes it often misconstrued as apoptosis or necrosis. Admixing a physiological death, like apoptosis, with a pathological one, like SICD involving a stressor and at least mitochondrial or lysosomal damage, is a major source of confusion in the literature. Because stressors are multifarious, SICD has a sheer number of ad hoc modes that in the literature are categorized into four basic pathways that involve death receptors on the cell membrane or involve pathologically altered mitochondria, ER or lysosomes, with some of the pathways using caspases as downstream effectors. Although these SICD pathways have been well articulated, they are often mistakenly put under the umbrella of apoptosis.
Acknowledgements
We would like to thank Dr. Fred Bogott at the Austin Medical Center, Mayo Clinic in Austin, Minnesota, USA, for his excellent English editing of this manuscript. This work was supported by a grant from Chinese Natural Science Foundation to D. Joshua Liao (grant No. 81660501).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jezek J, Cooper KF, Strich R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants (Basel). 2018:7 (1):-pii: E13. doi: 10.3390/antiox7010013
2. Kean LS, Turka LA, Blazar BR. Advances in targeting co-inhibitory and co-stimulatory pathways in transplantation settings: the Yin to the Yang of cancer immunotherapy. Immunol Rev. 2017;276(1):192-212
3. Faas MM, Saez T, de VP. Extracellular ATP and adenosine: The Yin and Yang in immune responses? Mol Aspects Med. 2017;55:9-19
4. Komar AA. The Yin and Yang of codon usage. Hum Mol Genet. 2016;25(R2):R77-R85
5. Li Y, Yamane D, Masaki T, Lemon SM. The yin and yang of hepatitis C: synthesis and decay of hepatitis C virus RNA. Nat Rev Microbiol. 2015;13(9):544-558
6. Liu X, Yang W, Guan Z, Yu W, Fan B, Xu N. et al. There are only four basic modes of cell death, although there are many ad-hoc variants adapted to different situations. Cell Biosci. 2018;8:6-doi 10.1186/s13578-018-0206-6
7. Liu B, Xu N, Man Y, Shen H, Avital I, Stojadinovic A. et al. Apoptosis in Living Animals Is Assisted by Scavenger Cells and Thus May Not Mainly Go through the Cytochrome C-Caspase Pathway. J Cancer. 2013;4(9):716-723
8. West SA, Griffin AS, Gardner A. Evolutionary explanations for cooperation. Curr Biol. 2007;17(16):R661-R672
9. Nowak MA. Five rules for the evolution of cooperation. Science. 2006;314(5805):1560-1563
10. Burgess MG, Costello C, Fredston-Hermann A, Pinsky ML, Gaines SD, Tilman D. et al. Range contraction enables harvesting to extinction. Proc Natl Acad Sci U S A. 2017;114(15):3945-3950
11. Costello MJ, May RM, Stork NE. Can we name Earth's species before they go extinct? Science. 2013;339(6118):413-416
12. Grant PR, Grant BR, Huey RB, Johnson MTJ, Knoll AH, Schmitt J. Evolution caused by extreme events. Philos Trans R Soc Lond B Biol Sci 2017; 372(1723): -pii. 2016 0146. doi: 10.1098/rstb.2016.0146
13. Tedesco PA, Bigorne R, Bogan AE, Giam X, Jezequel C, Hugueny B. Estimating how many undescribed species have gone extinct. Conserv Biol. 2014;28(5):1360-1370
14. Borer ET, Grace JB, Harpole WS, MacDougall AS, Seabloom EW. A decade of insights into grassland ecosystem responses to global environmental change. Nat Ecol Evol. 2017;1(5):118-doi 10.1038/s41559-017-0118
15. Carey C. Climate change, extinction risks, and reproduction of terrestrial vertebrates. Adv Exp Med Biol. 2014;753:35-54
16. Estrada A, Garber PA, Rylands AB, Roos C, Fernandez-Duque E, Di FA. et al. Impending extinction crisis of the world's primates: Why primates matter. Sci Adv. 2017;3(1):e1600946
17. Hull P. Life in the Aftermath of Mass Extinctions. Curr Biol. 2015;25(19):R941-R952
18. Pimm SL, Jenkins CN, Abell R, Brooks TM, Gittleman JL, Joppa LN. et al. The biodiversity of species and their rates of extinction, distribution, and protection. Science. 2014;344(6187):1246752-doi 10.1126/science.1246752
19. Tilman D, Clark M, Williams DR, Kimmel K, Polasky S, Packer C. Future threats to biodiversity and pathways to their prevention. Nature. 2017;546(7656):73-81
20. Larsson DG. Antibiotics in the environment. Ups J Med Sci. 2014;119(2):108-112
21. van der Meij A, Worsley SF, Hutchings MI, van Wezel GP. Chemical ecology of antibiotic production by actinomycetes. FEMS Microbiol Rev. 2017;41(3):392-416
22. Metsa-Ketela M. Evolution inspired engineering of antibiotic biosynthesis enzymes. Org Biomol Chem. 2017;15(19):4036-4041
23. Perry J, Waglechner N, Wright G. The Prehistory of Antibiotic Resistance. Cold Spring Harb Perspect Med. 2016:6 (6):- pii: a025197. doi: 10.1101/cshperspect.a025197
24. Rahman H, Austin B, Mitchell WJ, Morris PC, Jamieson DJ, Adams DR. et al. Novel anti-infective compounds from marine bacteria. Mar Drugs. 2010;8(3):498-518
25. Mohr KI. History of Antibiotics Research. Curr Top Microbiol Immunol. 2016;398:237-272
26. Chen X, Mukwaya E, Wong MS, Zhang Y. A systematic review on biological activities of prenylated flavonoids. Pharm Biol. 2014;52(5):655-660
27. Beck JJ, Alborn HT, Block AK, Christensen SA, Hunter CT, Rering CC. et al. Interactions Among Plants, Insects, and Microbes: Elucidation of Inter-Organismal Chemical Communications in Agricultural Ecology. J Agric Food Chem. 2018;66(26):6663-6674
28. Zagrobelny M, de Castro ECP, Moller BL, Bak S. Cyanogenesis in Arthropods: From Chemical Warfare to Nuptial Gifts. Insects. 2018:9 (2):-pii: E51. doi: 10.3390/insects9020051
29. Velasques J, Cardoso MH, Abrantes G, Frihling BE, Franco OL, Migliolo L. The rescue of botanical insecticides: A bioinspiration for new niches and needs. Pestic Biochem Physiol. 2017;143:14-25
30. Fonseca S, Radhakrishnan D, Prasad K, Chini A. Fungal Production and Manipulation of Plant Hormones. Curr Med Chem. 2018;25(2):253-267
31. LoPresti EF. Chemicals on plant surfaces as a heretofore unrecognized, but ecologically informative, class for investigations into plant defence. Biol Rev Camb Philos Soc. 2016;91(4):1102-1117
32. Pedras MS, Yaya EE. Plant chemical defenses: are all constitutive antimicrobial metabolites phytoanticipins? Nat Prod Commun. 2015;10(1):209-218
33. Mithofer A, Boland W. Plant defense against herbivores: chemical aspects. Annu Rev Plant Biol. 2012;63:431-450
34. Sirotkin AV, Harrath AH. Phytoestrogens and their effects. Eur J Pharmacol. 2014;741:230-236
35. Bennetau-Pelissero C. Risks and benefits of phytoestrogens: where are we now? Curr Opin Clin Nutr Metab Care. 2016;19(6):477-483
36. Magnusson U, Persson S. Endocrine Disruptors in Domestic Animal Reproduction: A Clinical Issue? Reprod Domest Anim. 2015;50(Suppl 3):15-19
37. Woclawek-Potocka I, Mannelli C, Boruszewska D, Kowalczyk-Zieba I, Wasniewski T, Skarzynski DJ. Diverse effects of phytoestrogens on the reproductive performance: cow as a model. Int J Endocrinol. 2013;2013:650984-doi 10.1155/2013/650984
38. Dusza L, Ciereszko R, Skarzynski DJ, Nogowski L, Opalka M, Kaminska B. et al. Mechanism of phytoestrogens action in reproductive processes of mammals and birds. Reprod Biol. 2006;6(Suppl 1):151-174
39. Khokhlov AN. Does aging need its own program, or is the program of development quite sufficient for it? Stationary cell cultures as a tool to search for anti-aging factors. Curr Aging Sci. 2013;6(1):14-20
40. Maklakov AA, Lummaa V. Evolution of sex differences in lifespan and aging: causes and constraints. Bioessays. 2013;35(8):717-724
41. Maklakov AA, Rowe L, Friberg U. Why organisms age: Evolution of senescence under positive pleiotropy? Bioessays. 2015;37(7):802-807
42. Allocati N, Masulli M, Di IC, De L, V. Die for the community: an overview of programmed cell death in bacteria. Cell Death Dis. 2015;6:e1609-doi 10.1038/cddis.2014.570
43. Buttner S, Eisenberg T, Herker E, Carmona-Gutierrez D, Kroemer G, Madeo F. Why yeast cells can undergo apoptosis: death in times of peace, love, and war. J Cell Biol. 2006;175(4):521-525
44. Ameisen JC. Looking for death at the core of life in the light of evolution. Cell Death Differ. 2004;11(1):4-10
45. Ameisen JC. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ. 2002;9(4):367-393
46. Corcos D. Food-Nonfood Discrimination in Ancestral Vertebrates: Gamete Cannibalism and the Origin of the Adaptive Immune System. Scand J Immunol. 2015;82(5):409-417
47. Wise DH. Cannibalism, food limitation, intraspecific competition, and the regulation of spider populations. Annu Rev Entomol. 2006;51:441-465
48. Kinchen JM, Hengartner MO. Tales of cannibalism, suicide, and murder: Programmed cell death in C. elegans. Curr Top Dev Biol. 2005;65:1-45
49. Upadhyay KK, Silverstein DM. Renal development: a complex process dependent on inductive interaction. Curr Pediatr Rev. 2014;10(2):107-114
50. Solhaug MJ, Bolger PM, Jose PA. The developing kidney and environmental toxins. Pediatrics. 2004;113(4 Suppl):1084-1091
51. Drummond I. The pronephros. Results Probl Cell Differ. 2002;40:322-345
52. Kuure S, Vuolteenaho R, Vainio S. Kidney morphogenesis: cellular and molecular regulation. Mech Dev. 2000;92(1):31-45
53. Aterman K. Pronephros and mesonephros-Cohnheim revisited. Pediatr Pathol. 1990;10(6):1021-1032
54. McCrory WW. The normal embryologic development of the kidney: a basis for understanding structural abnormalities. Birth Defects Orig Artic Ser. 1974;10(4):3-11
55. Boehm T, Swann JB. Thymus involution and regeneration: two sides of the same coin? Nat Rev Immunol. 2013;13(11):831-838
56. Gui J, Mustachio LM, Su DM, Craig RW. Thymus Size and Age-related Thymic Involution: Early Programming, Sexual Dimorphism, Progenitors and Stroma. Aging Dis. 2012;3(3):280-290
57. Aw D, Palmer DB. The origin and implication of thymic involution. Aging Dis. 2011;2(5):437-443
58. Calder AE, Hince MN, Dudakov JA, Chidgey AP, Boyd RL. Thymic involution: where endocrinology meets immunology. Neuroimmunomodulation. 2011;18(5):281-289
59. Hince M, Sakkal S, Vlahos K, Dudakov J, Boyd R, Chidgey A. The role of sex steroids and gonadectomy in the control of thymic involution. Cell Immunol. 2008;252(1-2):122-138
60. Macias H, Hinck L. Mammary gland development. Wiley Interdiscip Rev Dev Biol. 2012;1(4):533-557
61. Inman JL, Robertson C, Mott JD, Bissell MJ. Mammary gland development: cell fate specification, stem cells and the microenvironment. Development. 2015;142(6):1028-1042
62. Capuco AV, Ellis SE. Comparative aspects of mammary gland development and homeostasis. Annu Rev Anim Biosci. 2013;1:179-202
63. Vijg J, Dong X, Milholland B, Zhang L. Genome instability: a conserved mechanism of ageing? Essays Biochem. 2017;61(3):305-315
64. Warner HR, Hodes RJ, Pocinki K. What does cell death have to do with aging? J Am Geriatr Soc. 1997;45(9):1140-1146
65. Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018;14:450-464
66. Hernandez F, Llorens-Martin M, Bolos M, Perez M, Cuadros R, Pallas-Bazarra N. et al. New Beginnings in Alzheimer's Disease: The Most Prevalent Tauopathy. J Alzheimers Dis. 2018 -doi: 10.3233/JAD-179916
67. Foguem C, Manckoundia P. Lewy Body Disease: Clinical and Pathological "Overlap Syndrome" Between Synucleinopathies (Parkinson Disease) and Tauopathies (Alzheimer Disease). Curr Neurol Neurosci Rep. 2018;18(5):24-doi 10.1007/s11910-018-0835-5
68. Jellinger KA. Dementia with Lewy bodies and Parkinson's disease-dementia: current concepts and controversies. J Neural Transm (Vienna ). 2018;125(4):615-650
69. Newbold RF, Overell RW, Connell JR. Induction of immortality is an early event in malignant transformation of mammalian cells by carcinogens. Nature. 1982;299(5884):633-635
70. Newbold RF, Overell RW. Fibroblast immortality is a prerequisite for transformation by EJ c-Ha-ras oncogene. Nature. 1983;304(5927):648-651
71. Newbold RF. Multistep malignant transformation of mammalian cells by carcinogens: induction of immortality as a key event. Carcinog Compr Surv. 1985;9:17-28
72. Newbold RF. Malignant transformation of mammalian cells in culture: delineation of stages and role of cellular oncogene activation. IARC Sci Publ. 1985;67:31-53
73. Newbold RF, Cuthbert AP, Themis M, Trott DA, Blair AL, Li W. Cell immortalization as a key, rate-limiting event in malignant transformation: approaches toward a molecular genetic analysis. Toxicol Lett. 1993;67(1-3):211-230
74. Thomas F, Nesse RM, Gatenby R, Gidoin C, Renaud F, Roche B. et al. Evolutionary Ecology of Organs: A Missing Link in Cancer Development? Trends Cancer. 2016;2(8):409-415
75. Martin GM. Cellular aging-postreplicative cells. A review (Part II). Am J Pathol. 1977;89(2):513-530
76. Tam SK, Gu W, Mahdavi V, Nadal-Ginard B. Cardiac myocyte terminal differentiation. Potential for cardiac regeneration. Ann N Y Acad Sci. 1995;752:72-79
77. Rumyantsev PP. Interrelations of the proliferation and differentiation processes during cardiact myogenesis and regeneration. Int Rev Cytol. 1977;51:186-273
78. Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res. 2003;92(2):139-150
79. Anversa P, Leri A, Kajstura J, Nadal-Ginard B. Myocyte growth and cardiac repair. J Mol Cell Cardiol. 2002;34(2):91-105
80. Frade JM, Ovejero-Benito MC. Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle. 2015;14(5):712-720
81. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018 -doi: 10.1038/s41569-018-0007-y
82. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245-262
83. Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89(9):1401-1438
84. Lyon RC, Zanella F, Omens JH, Sheikh F. Mechanotransduction in cardiac hypertrophy and failure. Circ Res. 2015;116(8):1462-1476
85. O'Ferrall EK, Gendron D, Guiot MC, Hall J, Sinnreich M. Lower motor neuron syndrome due to cauda equina hypertrophy with onion bulbs. Muscle Nerve. 2013;48(2):301-305
86. Ladd AA, Ladd FV, da Silva AA, Oliveira MF, de Souza RR, Coppi AA. SCG postnatal remodelling-hypertrophy and neuron number stability-in Spix's yellow-toothed cavies (Galea spixii). Int J Dev Neurosci. 2012;30(2):129-137
87. PANNESE E. NUMBER AND STRUCTURE OF PERISOMATIC SATELLITE CELLS OF SPINAL GANGLIA UNDER NORMAL CONDITIONS OR DURING AXON REGENERATION AND NEURONAL HYPERTROPHY. Z Zellforsch Mikrosk Anat. 1964;63:568-592
88. EDDS MV Jr. Hypertrophy of nerve fibers to functionally overloaded muscles. J Comp Neurol. 1950;93(2):259-275
89. Manz HJ, Phillips TM, Rowden G, McCullough DC. Unilateral megalencephaly, cerebral cortical dysplasia, neuronal hypertrophy, and heterotopia: cytomorphometric, fluorometric cytochemical, and biochemical analyses. Acta Neuropathol. 1979;45(2):97-103
90. Fricano CJ, Despenza T Jr, Frazel PW, Li M, O'Malley AJ, Westbrook GL. et al. Fatty acids increase neuronal hypertrophy of Pten knockdown neurons. Front Mol Neurosci. 2014;7:30-doi 10.3389/fnmol.2014.00030
91. Yamauchi H, Sakurai S, Tsukagoshi R, Suzuki M, Tabe Y, Fukasawa T. et al. A case of diffuse neuronal hypertrophy in acute appendicitis. Int Surg. 2012;97(3):249-253
92. Martin P, Nunan R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br J Dermatol. 2015;173(2):370-378
93. Ud-Din S, Volk SW, Bayat A. Regenerative healing, scar-free healing and scar formation across the species: current concepts and future perspectives. Exp Dermatol. 2014;23(9):615-619
94. Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res. 2012;49(1):35-43
95. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71(4):549-574
96. Schirone L, Forte M, Palmerio S, Yee D, Nocella C, Angelini F. et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxid Med Cell Longev. 2017;2017:3920195-doi 10.1155/2017/3920195
97. Weiskirchen R, Tacke F. Liver Fibrosis: From Pathogenesis to Novel Therapies. Dig Dis. 2016;34(4):410-422
98. Friedman SL. Hepatic fibrosis - overview. Toxicology. 2008;254(3):120-129
99. Jazwinski SM, Jiang JC, Kim S. Adaptation to metabolic dysfunction during aging: Making the best of a bad situation. Exp Gerontol. 2018;107:87-90
100. Hipolito VEB, Ospina-Escobar E, Botelho RJ. Lysosome remodelling and adaptation during phagocyte activation. Cell Microbiol. 2018:20 (4):-doi: 10.1111/cmi.12824
101. Diederich M, Cerella C. Non-canonical programmed cell death mechanisms triggered by natural compounds. Semin Cancer Biol. 2016;40-41:4-34
102. Winkle CC, Gupton SL. Membrane Trafficking in Neuronal Development: Ins and Outs of Neural Connectivity. Int Rev Cell Mol Biol. 2016;322:247-280
103. Griffing LR, Lin C, Perico C, White RR, Sparkes I. Plant ER geometry and dynamics: biophysical and cytoskeletal control during growth and biotic response. Protoplasma. 2017;254(1):43-56
104. Van der Meer SF, Jaspers RT, Degens H. Is the myonuclear domain size fixed? J Musculoskelet Neuronal Interact. 2011;11(4):286-297
105. Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci. 2013;70(14):2425-2441
106. Rizvi SMA, Prajapati HK, Ghosh SK. The 2 micron plasmid: a selfish genetic element with an optimized survival strategy within Saccharomyces cerevisiae. Curr Genet. 2018;64(1):25-42
107. McLaughlin RN Jr, Malik HS. Genetic conflicts: the usual suspects and beyond. J Exp Biol. 2017;220(Pt 1):6-17
108. Penny D. Cooperation and selfishness both occur during molecular evolution. Biol Direct. 2014;10:26-doi 10.1186/s13062-014-0026-5
109. Holmquist GP. Cell-selfish modes of evolution and mutations directed after transcriptional bypass. Mutat Res. 2002;510(1-2):141-152
110. Deal ND, Wong BB. HOW MATE AVAILABILITY INFLUENCES FILIAL CANNIBALISM. Q Rev Biol. 2016;91(1):47-67
111. Schausberger P. Cannibalism among phytoseiid mites: a review. Exp Appl Acarol. 2003;29(3-4):173-191
112. Manica A. Filial cannibalism in teleost fish. Biol Rev Camb Philos Soc. 2002;77(2):261-277
113. Fitzgerald GJ. Filial cannibalism in fishes: Why do parents eat their offspring? Trends Ecol Evol. 1992;7(1):7-10
114. Wang C, Tai Y, Lisanti MP, Liao DJ. c-Myc induction of programmed cell death may contribute to carcinogenesis: a perspective inspired by several concepts of chemical carcinogenesis. Cancer Biol Ther. 2011;11(7):615-626
115. Maruyama T, Fujita Y. Cell competition in mammals - novel homeostatic machinery for embryonic development and cancer prevention. Curr Opin Cell Biol. 2017;48:106-112
116. Di GS, Sollazzo M, Paglia S, Grifoni D. MYC, Cell Competition, and Cell Death in Cancer: The Inseparable Triad. Genes (Basel). 2017:8 (4):-doi: 10.3390/genes8040120
117. Claveria C, Torres M. Cell Competition: Mechanisms and Physiological Roles. Annu Rev Cell Dev Biol. 2016;32:411-439
118. Johnston LA. Socializing with MYC: cell competition in development and as a model for premalignant cancer. Cold Spring Harb Perspect Med. 2014;4(4):a014274
119. Zhang J, Lou X, Zellmer L, Liu S, Xu N, Liao DJ. Just like the rest of evolution in Mother Nature, the evolution of cancers may be driven by natural selection, and not by haphazard mutations. Oncoscience. 2014;1(9):580-590
120. Oeffinger KC, Baxi SS, Novetsky FD, Moskowitz CS. Solid tumor second primary neoplasms: who is at risk, what can we do? Semin Oncol. 2013;40(6):676-689
121. Marcu LG, Santos A, Bezak E. Risk of second primary cancer after breast cancer treatment. Eur J Cancer Care (Engl ). 2014;23(1):51-64
122. Kaiser HE, Nasir A, Groger AM, Link CJ Jr. The etiology of second primary neoplasms. In Vivo. 1998;12(1):89-93
123. Mahmood S, Vu K, Tai P, Joseph K, Koul R, Dubey A. et al. Radiation-induced second malignancies. Anticancer Res. 2015;35(4):2431-2434
124. Marta GN, Murphy E, Chao S, Yu JS, Suh JH. The incidence of second brain tumors related to cranial irradiation. Expert Rev Anticancer Ther. 2015;15(3):295-304
125. Wang Z, Luo D, Xiao H, Joshua LD. Case report: metastases from thyroid and nasopharyngeal carcinomas in the same lymph node following chemotherapy for mantle cell lymphoma. Head Neck Pathol. 2015;9(1):123-126
126. Rozhok AI, DeGregori J. The evolution of lifespan and age-dependent cancer risk. Trends Cancer. 2016;2(10):552-560
127. Jia QW, Chen XH, Jia YP, Dou XX, Ezeogu L, Xu NZ.. et al. is type 2 diabetes one of such aging phenomena that lack an irreversible structural change? J Diabetes Metab 2105; 6(5):543-doi: 10.4172/2155-6156.1000543.
128. Rubin H. Cell aging in vivo and in vitro. Mech Ageing Dev. 1997;98(1):1-35
129. Passaro F, Testa G. Implications of Cellular Aging in Cardiac Reprogramming. Front Cardiovasc Med. 2018;5:43-doi 10.3389/fcvm.2018.00043
130. Shakeri H, Lemmens K, Gevaert AB, De Meyer GRY, Segers V. Cellular senescence links aging and diabetes in cardiovascular disease. Am J Physiol Heart Circ Physiol. 2018 -doi: 10.1152/ajpheart.00287.2018
131. Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15(11):1139-1153
132. Falandry C, Bonnefoy M, Freyer G, Gilson E. Biology of cancer and aging: a complex association with cellular senescence. J Clin Oncol. 2014;32(24):2604-2610
133. Sikora E, Bielak-Zmijewska A, Mosieniak G. Cellular senescence in ageing, age-related disease and longevity. Curr Vasc Pharmacol. 2014;12(5):698-706
134. Tan FC, Hutchison ER, Eitan E, Mattson MP. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology. 2014;15(6):643-660
135. van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509(7501):439-446
136. Vicencio JM, Galluzzi L, Tajeddine N, Ortiz C, Criollo A, Tasdemir E. et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path-a mini-review. Gerontology. 2008;54(2):92-99
137. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194-1217
138. Sapieha P, Mallette FA. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018 -pii: S0962-8924(18)30059-X. doi: 10.1016/j.tcb.2018.03.003
139. Lauri A, Pompilio G, Capogrossi MC. The mitochondrial genome in aging and senescence. Ageing Res Rev. 2014;18:1-15
140. Greiner S, Sobanski J, Bock R. Why are most organelle genomes transmitted maternally? Bioessays. 2015;37(1):80-94
141. Tan FC, Hutchison ER, Eitan E, Mattson MP. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology. 2014;15(6):643-660
142. Fielder E, von ZT, Jurk D. The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J Alzheimers Dis. 2017;60(s1):S107-S131
143. Vicencio JM, Galluzzi L, Tajeddine N, Ortiz C, Criollo A, Tasdemir E. et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path-a mini-review. Gerontology. 2008;54(2):92-99
144. Sapolsky RM. Second generation questions about senescent neuron loss. Neurobiol Aging. 1987;8(6):547-548
145. Bernecker OY, Huq F, Heist EK, Podesser BK, Hajjar RJ. Apoptosis in heart failure and the senescent heart. Cardiovasc Toxicol. 2003;3(3):183-190
146. Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C. et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res. 2003;93(7):604-613
147. Choi M, Lee C. Immortalization of Primary Keratinocytes and Its Application to Skin Research. Biomol Ther (Seoul ). 2015;23(5):391-399
148. Rubin H. The disparity between human cell senescence in vitro and lifelong replication in vivo. Nat Biotechnol. 2002;20(7):675-681
149. Zhang J, Lou XM, Jin LY, Zhou RJ, Liu SQ, Xu NZ. et al. Necrosis, and then stress induced necrosis-like cell death, but not apoptosis, should be the preferred cell death mode for chemotherapy: clearance of a few misconceptions. Oncoscience. 2014;1(6):407-422
150. Liao DJ. The scavenger cell hypothesis of apoptosis: apoptosis redefined as a process by which a cell in living tissue is destroyed by phagocytosis. Med Hypotheses. 2005;65(1):23-28
151. Liu B, Ezeogu L, Zellmer L, Yu B, Xu N, Liao DJ. Protecting the normal in order to better kill the cancer. Cancer Med. 2015;4(9):1394-1403
152. Wang G, Chen L, Yu B, Zellmer L, Xu N, Liao DJ. Learning about the Importance of Mutation Prevention from Curable Cancers and Benign Tumors. J Cancer. 2016;7(4):436-445
153. Kobayashi SD, Malachowa N, DeLeo FR. Influence of Microbes on Neutrophil Life and Death. Front Cell Infect Microbiol. 2017;7:159-doi 10.3389/fcimb.2017.00159
154. Upton JW, Chan FK. Staying alive: cell death in antiviral immunity. Mol Cell. 2014;54(2):273-280
155. Badley AD, Sainski A, Wightman F, Lewin SR. Altering cell death pathways as an approach to cure HIV infection. Cell Death Dis. 2013;4:e718-doi 10.1038/cddis.2013.248
156. LeGrand EK. Implications of early apoptosis of infected cells as an important host defense. Med Hypotheses. 2000;54(4):591-596
157. LeGrand EK. Genetic conflict and apoptosis. Perspect Biol Med. 2001;44(4):509-521
158. Zakeri Z, Lockshin RA. Cell death during development. J Immunol Methods. 2002;265(1-2):3-20
159. Mondello C, Scovassi AI. Apoptosis: a way to maintain healthy individuals. Subcell Biochem. 2010;50:307-323
160. Hernandez-Martinez R, Covarrubias L. Interdigital cell death function and regulation: new insights on an old programmed cell death model. Dev Growth Differ. 2011;53(2):245-258
161. Leslie KE. The events of normal and abnormal postpartum reproductive endocrinology and uterine involution in dairy cows: a review. Can Vet J. 1983;24(3):67-71
162. Morrow DA, Roberts SJ, McEntee K. A review of postpartum ovarian activity and involution of the uterus and cervix in cattle. Cornell Vet. 1969;59(1):134-154
163. Gier HT, Marion GB. Uterus of the cow after parturition: involutional changes. Am J Vet Res. 1968;29(1):83-96
164. Morrow DA, Roberts SJ, McEntee K. Pospartum ovarian activity and involution of the uterus and cervix in dairy cattle. II. Involution of uterus and cervix. Cornell Vet. 1969;59(2):190-198
165. O'Brien J, Martinson H, Durand-Rougely C, Schedin P. Macrophages are crucial for epithelial cell death and adipocyte repopulation during mammary gland involution. Development. 2012;139(2):269-275
166. Luke CJ, Silverman GA. Necrotic cell death: harnessing the Dark side of the Force in mammary gland involution. Nat Cell Biol. 2011;13(3):197-199
167. Watson CJ. Post-lactational mammary gland regression: molecular basis and implications for breast cancer. Expert Rev Mol Med. 2006;8(32):1-15
168. Strange R, Li F, Saurer S, Burkhardt A, Friis RR. Apoptotic cell death and tissue remodelling during mouse mammary gland involution. Development. 1992;115(1):49-58
169. de la Grandmaison GL, Marchaut J, Watier L, Mediouni Z, Charlier P. Frequency and nature of testicular and paratesticular lesions in forensic autopsies. Med Sci Law. 2013;53(4):208-212
170. Pop OT, Cotoi CG, Plesea IE, Enache SD, Popescu FC, Enache MA. et al. Correlations between intralobular interstitial morphological changes and epithelial changes in ageing testis. Rom J Morphol Embryol. 2011;52(1 Suppl):339-347
171. Harbitz TB. Testis weight and the histology of the prostate in elderly men. An analysis in an autopsy series. Acta Pathol Microbiol Scand A. 1973;81(2):148-158
172. Ishii T, Sternby NH. Pathology of centenarians. II. Urogenital and digestive systems. J Am Geriatr Soc. 1978;26(9):391-396
173. Baker HW, Hudson B. Changes in the pituitary-testicular axis with age. Monogr Endocrinol. 1983;25:71-83
174. Motta PM, Heyn R, Makabe S. Three-dimensional microanatomical dynamics of the ovary in postreproductive aged women. Fertil Steril. 2002;78(2):360-370
175. Li X, Shu R, Filippatos G, Uhal BD. Apoptosis in lung injury and remodeling. J Appl Physiol (1985 ). 2004;97(4):1535-1542
176. Savill J. Apoptosis and the kidney. J Am Soc Nephrol. 1994;5(1):12-21
177. Savill J, Gregory C, Haslett C. Cell biology. Eat me or die. Science. 2003;302(5650):1516-1517
178. Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407(6805):784-788
179. Ren Y, Savill J. Apoptosis: the importance of being eaten. Cell Death Differ. 1998;5(7):563-568
180. Savill J. Apoptosis. Phagocytic docking without shocking. Nature. 1998;392(6675):442-443
181. Liao DJ, Dickson RB. Cell death in MMTV-c-myc transgenic mouse mammary tumors may not be typical apoptosis. Lab Invest. 2003;83(10):1437-1449
182. Wyllie AH. Apoptosis: an overview. Br Med Bull. 1997;53(3):451-465
183. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239-257
184. Rous P, Kidd JG. CONDITIONAL NEOPLASMS AND SUBTHRESHOLD NEOPLASTIC STATES: A STUDY OF THE TAR TUMORS OF RABBITS. J Exp Med. 1941;73(3):365-390
185. Duesberg P, Mandrioli D, McCormack A, Nicholson JM. Is carcinogenesis a form of speciation? Cell Cycle. 2011;10(13):2100-2114
186. Huuhtanen RL, Blomqvist CP, Bohling TO, Wiklund TA, Tukiainen EJ, Virolainen M. et al. Expression of cyclin A in soft tissue sarcomas correlates with tumor aggressiveness. Cancer Res. 1999;59(12):2885-2890
187. Knauss S, Klein A. From aneuploidy to cancer: the evolution of a new species? J Biosci. 2012;37(2):211-220
188. Vincent MD. Cancer: beyond speciation. Adv Cancer Res. 2011;112:283-350
189. Mohamed MS, Bishr MK, Almutairi FM, Ali AG. Inhibitors of apoptosis: clinical implications in cancer. Apoptosis. 2017;22(12):1487-1509
190. Cassier PA, Castets M, Belhabri A, Vey N. Targeting apoptosis in acute myeloid leukaemia. Br J Cancer. 2017;117(8):1089-1098
191. Nonnenmacher L, Hasslacher S, Zimmermann J, Karpel-Massler G, La Ferla-Bruhl K, Barry SE. et al. Cell Death Induction in Cancer Therapy - Past, Present, and Future. Crit Rev Oncog. 2016;21(3-4):253-267
192. Westhoff MA, Marschall N, Debatin KM. Novel Approaches to Apoptosis-Inducing Therapies. Adv Exp Med Biol. 2016;930:173-204
193. Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M. et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642-doi 10.1038/436642a
194. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130(2):223-233
195. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10(1):51-57
196. Ruhland MK, Coussens LM, Stewart SA. Senescence and cancer: An evolving inflammatory paradox. Biochim Biophys Acta. 2016;1865(1):14-22
197. Sieben CJ, Sturmlechner I, van de Sluis B, van Deursen JM. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018 -pii: S0962-8924(18)30070-9. doi: 10.1016/j.tcb.2018.04.006
198. Zellmer L, Han YP, Chen LC, Xu NZ, Liao DJ. Does the cytochrome c-caspase pathway of cell death occur physiologically in animals? Journal Tumor Med Prev. 2017;1(2):JTMP.MS.ID.555557.pdf
199. Wietecha MS, Cerny WL, DiPietro LA. Mechanisms of vessel regression: toward an understanding of the resolution of angiogenesis. Curr Top Microbiol Immunol. 2013;367:3-32
200. DiPietro LA. Angiogenesis and wound repair: when enough is enough. J Leukoc Biol. 2016;100(5):979-984
201. Forrest L. Current concepts in soft connective tissue wound healing. Br J Surg. 1983;70(3):133-140
202. Shoshan S. Wound healing. Int Rev Connect Tissue Res. 1981;9:1-26
203. Galluzzi L, Bravo-San Pedro JM, Kepp O, Kroemer G. Regulated cell death and adaptive stress responses. Cell Mol Life Sci. 2016;73(11-12):2405-2410
204. Tait SW, Green DR. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol. 2013:5 (9):-doi: 10.1101/cshperspect.a008706
205. Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87(7):1157-1180
206. Logue SE, Cleary P, Saveljeva S, Samali A. New directions in ER stress-induced cell death. Apoptosis. 2013;18(5):537-546
207. Fulda S. Exploiting mitochondrial apoptosis for the treatment of cancer. Mitochondrion. 2010;10(6):598-603
208. Ravanan P, Srikumar IF, Talwar P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017;188:53-67
209. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486-541
210. Tsapras P, Nezis IP. Caspase involvement in autophagy. Cell Death Differ. 2017;24(8):1369-1379
211. Tait SW, Green DR. Caspase-independent cell death: leaving the set without the final cut. Oncogene. 2008;27(50):6452-6461
212. Chang YH, Lin HH, Wang YK, Chiu WT, Su HW, Tang MJ. Activation of caspase-8 and Erk-1/2 in domes regulates cell death induced by confluence in MDCK cells. J Cell Physiol. 2007;211(1):174-182
213. Chow M, Rubin H. Relation of the slow growth phenotype to neoplastic transformation: possible significance for human cancer. In Vitro Cell Dev Biol Anim. 1999;35(8):449-458
214. Garrido C, Ottavi P, Fromentin A, Hammann A, Arrigo AP, Chauffert B. et al. HSP27 as a mediator of confluence-dependent resistance to cell death induced by anticancer drugs. Cancer Res. 1997;57(13):2661-2667
215. Hosick HL. Spontaneous cell loss during growth of postconfluent primary cultures from mammary adenocarcinomas. Cancer Res. 1976;36(9 pt.1):3126-3130
216. Padron JM, van der Wilt CL, Smid K, Smitskamp-Wilms E, Backus HH, Pizao PE. et al. The multilayered postconfluent cell culture as a model for drug screening. Crit Rev Oncol Hematol. 2000;36(2-3):141-157
217. Rubin H. The role of selection in progressive neoplastic transformation. Adv Cancer Res. 2001;83:159-207
218. Rubin H. Cell damage, aging and transformation: a multilevel analysis of carcinogenesis. Anticancer Res. 1999;19(6A):4877-4886
219. Aits S, Jaattela M. Lysosomal cell death at a glance. J Cell Sci. 2013;126(Pt 9):1905-1912
220. Kirkegaard T, Jaattela M. Lysosomal involvement in cell death and cancer. Biochim Biophys Acta. 2009;1793(4):746-754
221. Morris G, Walker AJ, Berk M, Maes M, Puri BK. Cell Death Pathways: a Novel Therapeutic Approach for Neuroscientists. Mol Neurobiol. 2017 -doi: 10.1007/s12035-017-0793-y
222. Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta. 2012;1824(1):22-33
223. Serrano-Puebla A, Boya P. Lysosomal membrane permeabilization as a cell death mechanism in cancer cells. Biochem Soc Trans. 2018;46(2):207-215
224. Ashkenazi A, Salvesen G. Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol. 2014;30:337-356
225. Driscoll PC. Structural studies of death receptors. Methods Enzymol. 2014;545:201-242
226. Holland PM. Death receptor agonist therapies for cancer, which is the right TRAIL? Cytokine Growth Factor Rev. 2014;25(2):185-193
227. Lavrik IN. Systems biology of death receptor networks: live and let die. Cell Death Dis. 2014;5:e1259-doi 10.1038/cddis.2014.160
228. Micheau O, Shirley S, Dufour F. Death receptors as targets in cancer. Br J Pharmacol. 2013;169(8):1723-1744
229. Sobrido-Camean D, Barreiro-Iglesias A. Role of Caspase-8 and Fas in Cell Death After Spinal Cord Injury. Front Mol Neurosci. 2018;11:101-doi 10.3389/fnmol.2018.00101
230. Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev. 2017;277(1):76-89
231. Ukrainskaya VM, Stepanov AV, Glagoleva IS, Knorre VD, Belogurov AAJ, Gabibov AG. Death Receptors: New Opportunities in Cancer Therapy. Acta Naturae. 2017;9(3):55-63
232. Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11(9):3155-3162
233. Pfeffer CM, Singh ATK. Apoptosis: A Target for Anticancer Therapy. Int J Mol Sci. 2018:19 (2):-pii: E448. doi: 10.3390/ijms19020448
234. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460-3470
235. Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR. et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62-72
236. Martinvalet D. The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death Dis. 2018;9(3):336-doi 10.1038/s41419-017-0237-7
237. Patel S, Sharma D, Kalia K, Tiwari V. Crosstalk between endoplasmic reticulum stress and oxidative stress in schizophrenia: The dawn of new therapeutic approaches. Neurosci Biobehav Rev. 2017;83:589-603
238. Wu H, Ng BS, Thibault G. Endoplasmic reticulum stress response in yeast and humans. Biosci Rep. 2014:34 (4):-pii: e00118. doi: 10.1042/BSR20140058
239. Lou X, Zhang J, Liu S, Xu N, Liao DJ. The other side of the coin: The tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13(11):1677-1693
240. Wang Z, Figueiredo-Pereira C, Oudot C, Vieira HL, Brenner C. Mitochondrion: A Common Organelle for Distinct Cell Deaths? Int Rev Cell Mol Biol. 2017;331:245-287
241. Welchen E, Gonzalez DH. Cytochrome c, a hub linking energy, redox, stress and signaling pathways in mitochondria and other cell compartments. Physiol Plant. 2016;157(3):310-321
242. Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25(1):104-113
243. Beckta JM, Ahmad SF, Yang H, Valerie K. Revisiting p53 for cancer-specific chemo- and radiotherapy: ten years after. Cell Cycle. 2014;13(5):710-713
244. Ozaki T, Nakagawara A. p53: the attractive tumor suppressor in the cancer research field. J Biomed Biotechnol. 2011;2011:603925
245. Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. 2016:6 (5)
246. Yang M, Wu J, Wu SH, Bi AD, Liao DJ. Splicing of mouse p53 pre-mRNA does not always follow the "first come, first served" principle and may be influenced by cisplatin treatment and serum starvation. Mol Biol Rep. 2012;39(9):9247-9256
Author contact
![]() Corresponding authors: Xixi Dou, Key Laboratory of Biopharmaceuticals, Shandong Academy of Pharmaceutical Science, Jinan 250101, Shandong Province, P.R. China. Email: douxxcom or Lichan Chen, College of Chemical Engineering, Huaqiao University, Xiamen 361021, Fujian Province, P.R. China. Email: lchenedu.cn or Joshua Liao, Department of Pathology, Guizhou Medical University Hospital, Guiyang 550004, Guizhou Province, P.R. China. Email: djliaoedu.cn
Corresponding authors: Xixi Dou, Key Laboratory of Biopharmaceuticals, Shandong Academy of Pharmaceutical Science, Jinan 250101, Shandong Province, P.R. China. Email: douxxcom or Lichan Chen, College of Chemical Engineering, Huaqiao University, Xiamen 361021, Fujian Province, P.R. China. Email: lchenedu.cn or Joshua Liao, Department of Pathology, Guizhou Medical University Hospital, Guiyang 550004, Guizhou Province, P.R. China. Email: djliaoedu.cn