Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(13):1822-1833. doi:10.7150/ijbs.27555 This issue Cite
Research Paper
Prior Knowledge Driven Joint NMF Algorithm for ceRNA Co-Module Identification
Jin Deng1, Wei Kong1 ![]() , Shuaiqun Wang1, Xiaoyang Mou2, Weiming Zeng1
, Shuaiqun Wang1, Xiaoyang Mou2, Weiming Zeng1
1. College of Information Engineering, Shanghai Maritime University, 1550 Haigang Ave., Shanghai 201306, P. R. China;
2. Department of Biochemistry, Rowan University and Guava Medicine, Glassboro, New Jersey 08028, USA.
Received 2018-5-30; Accepted 2018-8-30; Published 2018-10-19
Abstract

MRNA and lncRNA serve as a type of endogenous RNA in cell, which can competitively bind to the same miRNA through miRNA response elements (MREs), thereby regulating their respective expression levels, playing an important role in post-transcriptional regulation, and regulating the progress of tumors. The proposed competing endogenous RNA (ceRNA) hypothesis provides novel clues for the occurrence and development of tumors, but the integrative analysis methods of diverse RNA data are significantly limited. In order to find out the relationship among miRNA, mRNA and lncRNA, the previous studies only used individual dataset as seeds to search two other related data in the database to construct ceRNA network, but it was difficult to identify the synchronized effects from multiple regulatory levels. Here, we developed the joint matrix factorization method integrating prior knowledge to map the three types of RNA data of lung cancer to the common coordinate system and construct the ceRNA network corresponding to the common module. The results show that more than 90% of the modules are closely related to cancer, including lung cancer. Furthermore, the resulting ceRNA network not only accurately excavates the known correlation of the three types of RNA molecular, but also further discovers the potential biological associations of them. Our work provides support and foundation for future biological validation how competitive relationships of multiple RNAs affects the development of tumors.
Keywords: miRNA, mRNA, lncRNA, ceRNA, tumor
Introduction
Recent studies have shown that non-coding RNA plays a pivotal role in cell differentiation, metabolism and other life activities [1]. Among them, long non-coding RNA (lncRNA) is characterized by multiple types, multiple modes of action, and numerous quantities, and it is involved in a variety of molecular mechanisms of expression regulation [2-3]. In addition to being directly involved in gene expression regulation, lncRNA can also serve as a competitive RNA that competes with other RNA transcripts for the same miRNA, thereby enabling mutual exchange and regulation [4]. The competing endogenous RNA (CeRNA) hypothesis states that transcripts such as lncRNAs, transcribed pseudogenes, or messenger RNAs (mRNAs) can serve as microRNA response elements(MREs) which competitively bind miRNAs with the same MRE to regulate gene expression levels, thereby affecting cell function [5]. A series of recent studies on lncRNAs and tumors have shown that lncRNAs and miRNAs can also influence tumor formation and progression through mutual regulation [6-7]. However, the function of most lncRNAs is still unknown as well as the interaction between tumors and regulatory mechanisms of three RNA molecules is unclear.
Constructing ceRNA networks helps researchers better understand the interaction mechanisms between the three types of RNA. The emergence of miRNA-mRNA, miRNA-lncRNA and mRNA-lncRNA databases has facilitated the construction of ceRNA networks, such as starBase2.0 [8], miRSponge [9], and CircNet [10]. Most studies, including the above-mentioned databases, are merely based on individual dataset to find other types of data related to it in the database to build regulatory network or competing network. But this method ignores an important issue, that is, the same RNA molecule may have different expression values in different tumors and its function is also different, even the same molecule affects the development of the disease by affecting different molecules. For example, the expression of miR-26 family members involves in multiple types of cancer. On the one hand, miR-26 can mediate the tumor suppressive activity in intestinal epithelium by inhibiting Cyclin D2 (CCND2), Enhancer of zeste homolog 2 (EZH2) and other anti-proliferative target genes [11]. On the other hand, miR-26 can show oncogenic activity by down-regulating tumor suppressor genes in lung cancer and glioma [11]. What's more, miR-221/222 acts as an oncogene in the liver but as a tumor suppressor in other tissues such as tongue squamous cell carcinoma [12-13].
Therefore, it is more advantageous to extract common features from multiple data fusions. Considering that different types of data have different scales and characteristics, it is not possible to simply aggregate multiple data for analysis. Many of the existing algorithms are applied to multi-data fusion [14-16], but these algorithms only perform common module extraction of two types of data, especially the extraction of miRNA-gene common modules. There are few algorithms for extracting common modules of three types of data. Zhang et al. used the joint NMF algorithm to extract common modules (co-modules) of the miRNA-gene-methylation data and found that the module closely related to ovarian cancer can be effectively extracted [17]. However, the correlation between data, including the regulatory relationship of miRNA-gene and the interaction between proteins, has not been taken into account.
In this paper, we propose a computational framework that integrates three kinds of RNA data to extract ceRNA co-modules, that is to transmit multiple dimensions of data to the same space for co-module extraction. Based on the integration of miRNA, mRNA and lncRNA expression data using a non-negative matrix factorization (NMF) framework, three kinds of prior knowledge including miRNA-mRNA, miRNA-lncRNA and protein-protein interaction (PPI) are integrated together in a regular manner. Then an optimization model is built to find the optimal solution through an iterative algorithm. We downloaded three data from the same sample of lung adenocarcinoma (LUAD) from The Cancer Genome Atlas (TCGA) publication and performed differential RNA extraction, then applied it to the algorithm to identify the ceRNA co-module. The results showed that these modules were found to be closely related to LUAD in three dimensions, such as PBK and PVT1 competitively bind miR-372. The PDZ-binding kinase (PBK) gene affects cell proliferation, viability, and prognosis of LUAD, the miR-372 silences in lung cancer cells by histone modification and lncRNA PVT1 may be a new biomarker and potential therapeutic target for lung cancer intervention. The three dimensions of data in the same module are also strongly correlated. Moreover, the ceRNA network is constructed through modules to discover potential lung cancer-related molecules including several genes the expression values of which are closely related to the patient's survival time or the patient's smoking habits.
Methods
In this section, we describe the framework for the simultaneous integration of three RNA data to identify ceRNA co-modules as shown in Figure 1. Subsequently, we introduce the algorithm proposed in this paper in detail, including the definition of the objective function that joins the prior knowledge and the derivation of the multiplication iterative update rule.
Flowchart showing the identification of ceRNA co-modules.
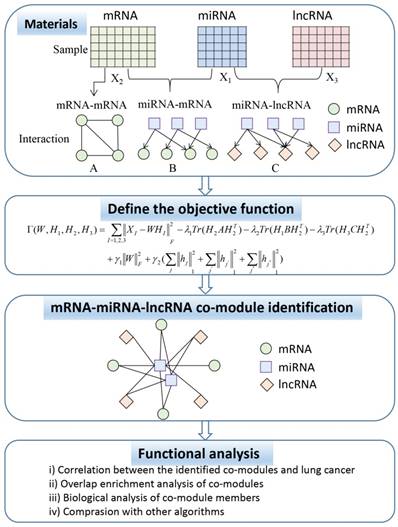
In Figure 1, the ceRNA co-module is defined as a combination of three RNAs, including mRNA, miRNA, and lncRNA. The input includes two kinds of data, one is the expression profile of the three RNAs measured on the same set of samples (represented by the matrices X1, X2, and X3), and the other is the prior knowledge of the relationship between the three sets of data, including mRNA-mRNA (represented by matrix A), miRNA-mRNA (represented by matrix B), and miRNA-lncRNA (represented by matrix C), which are measured on the same set of samples. Then, the three RNA expression matrices are decomposed into a common basis W and three coefficient matrices H1, H2, and H3. At the same time, prior knowledge is incorporated into the framework through network regularization constraints and sparse constraints is applied to this framework so as to obtain easily interpretable solutions. The decomposed matrix component provides information on the mRNA-miRNA-lncRNA module. Then, through several functional analysis of the elements in the module, the performance of the proposed algorithm is verified.
Joint NMF Analysis Algorithm
Assume that X1, X2 and X3 represent three types of RNA profiles of the same samples. To extract the multidimensional module in three data matrices, we use the joint decomposition framework to decompose the original matrices into a common base matrix W and different coefficient matrices HI (I=1,2,3):
XI ≈ WHI, W ≥ 0, HI ≥ 0, I = 1,2,3 (1)
then the objective function of Joint NMF is defined as,
(2)
Lee and Seung designed a multiplication algorithm to minimize the Euclidean error function [18]. The matrices W and HI are first initialized randomly and then iteratively updates W and HI to minimize the Euclidean distance function. And W and H are updated at each step by a generalized multiplication update rule:
,
(3)
According to the above update rules, we can find the matrix decomposition of the optimal value W and H to be the input of further analysis.
Joint Sparse Network-Regularization Multiple NMF (Joint SNMNMF)
The use of the traditional NMF algorithm for the extraction of the common module more reflects the independence of the data, that is, although sharing the same base matrix W, the independence is still strong, and our goal is to find co-modules that are more related to each other. With this limitation, the addition of prior knowledge into the objective function will make it easier to adapt to the biological solution. In addition to improving the biological relevance of the results, these limitations can also improve the efficiency of the co-modules by reducing the large search space.
Prior knowledge includes known or predicted miRNA-mRNA interactions, miRNA-lncRNA and mRNA-mRNA interactions. Assume A is the mRNA-mRNA interaction adjacency matrix, B is the miRNA-mRNA interaction adjacency matrix, and C is the miRNA-lncRNA interaction adjacency matrix. These interactions can be encoded by the following objective functions:
(4)
Where a, b, and c are elements of adjacency matrices, 1 means related, including regulation or protein interaction, 0 means no relationship. hi represents the i-th row of H and hj represents the j-th column of H.
Then, we define the objective function in the same optimization function:
(5)
The parameter λ is the weight for the relationship.
An important feature of the NMF algorithm is to sparse the data to locally discovery certain data features. However, NMF algorithm is sensitive to data quality and algorithms chosen by the researcher. For the NMNMF algorithm, we adopt a method proposed by Kim and Park to sparse the matrix HI while controlling the sparsity of W and H [19]. This method is formulated as follows:
(6)
Where γ1>0 is used to constrain ||W||F, γ2>0 is used to balance the trade-off between the accuracy of the approximation and the sparsity of H.
The SNMNMF algorithm was proposed by Zhang et al. to solve the extraction of miRNA-gene modules [20]. Based on this, we extended the co-module extraction including three types of data. Therefore, the objective function Γ can be redefined as follows:
(7)
Let 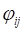 and
and 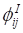 be constrained Lagrange multipliers of
be constrained Lagrange multipliers of 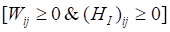 :
:
(8)
where 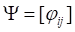 ,
, 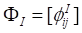
The partial derivatives of L with respect to W and HI are:
(9)
Based on Karush-Kuhn-Tucher (KTT) condition, 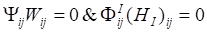 . We can obtain the equation of Wij and (HI)ij:
. We can obtain the equation of Wij and (HI)ij:
(10)
Then, the update rules of W and H are as follows,
(11)
Therefore, according to continuously updating W and H, let it satisfy the convergence rule, including the relative error reaching the set value or the number of iterations reaching the set number of times.
An important step in the SNMNMF algorithm is the selection of parameters, including k, λ, γ. The selection of k is determined by the number of mRNA enrichment pathways, k=41 in this study. Considering there is a certain degree of similarity of mRNA and lncRNA, that is, both are regulated by miRNAs. Therefore, the selection rules of the original SNMNMF algorithm are applied to select the constraint parameters λ and γ are λ1=0.0001, λ2=λ3=0.01, and γ1=γ2=10.
Selection of module elements
Through the above algorithms, we get the final W and H. The original three data matrices share a basic matrix, W. Acting different coefficient matrices on the RNA members in the modules. Considering the interrelationships between RNAs, some RNAs may not belong to any module or may belong to multiple modules. Therefore, we use the Z-score as a measure, Zij=(Xij-μi)/σi, where Xij is element in H, μ refers to the average of feature j in H, and σ refers to standard deviation. For each element, if its z-score is greater than the set threshold T, it is deemed to have the qualification to be assigned to the module. For the selection of T value, according to compare the result of this algorithm with the result of random assignment of 100 elements to the module. The criteria of the selected value consist of two parts. One is that the number of modules that can enrich pathways or biological processes is as many as possible. The other is that the result of the algorithm is more non-random. In this paper, the value of T is chosen as 3.
Results and Discussion
Data Sources and Preprocessing
We downloaded LUAD transcript data and miRNA sequencing data from the TCGA database (https://cancergenome.nih.gov/) and then isolated lncRNA and mRNA data from the transcript data. Considering the NMF algorithm requires that the three data have the same dimensionality, that is, the number of samples corresponding to the three types of data is the same, we retained 512 tumor tissue samples and 20 control samples containing three kinds of RNA data. Moreover, the regulatory data of miRNA-lncRNA was downloaded from miRcode [21] and PPI data was downloaded from Human Protein Reference Data database [22]. In order to find the target mRNAs of miRNA in the network, we use the starbase database [8] to perform miRNA 3p and 5p annotation. For labeled miRNA, target mRNAs were searched from miRDB [23], miRTarBase [24] and TargetScan databases [25], and then their intersections were taken.
To better understand those RNAs that have undergone significant changes in tumor and control tissues, the raw counts of RNA were applied to screen RNAs with significantly differential expression (DE) using the DESeq2 R package developed by Michael I Love, Wolfgang Huber, and Simon Anders [26]. This method uses shrinkage estimation to spread and fold change (FC) to improve the stability and interpretability of the DESeq-based estimation. Those RNAs were removed when log2FC ranged from -2 to 2 and P-value was higher than 0.05, 1483 DE lncRNA, 133 DE miRNA and 2151 DE mRNA were obtained.
Statistical significance of vertical correlation in co-module
In order to better understand the relationship between RNAs in tumors, three DE RNA raw count data with normalization from tumor samples were as the input data of the matrix decomposition. Afterwards, we obtained 41 modules. Since one of the modules could not contain any miRNA, 40 modules, each of which containing an average of 3.5 miRNA, 49.7 mRNA, and 34.3 lncRNA were retained. In Figure 2, histogram of sample-wise correlations of original and reconstructed miRNA, mRNA and lncRNA profiles across 512 samples were constructed to prove that our algorithm has a certain degree of robustness.
A) Histogram of sample-wise correlations of original and reconstructed miRNA, mRNA and lncRNA profiles across 512 samples, the red line represents the standard deviation; B) Original data are plotted against the reconstructed miRNA, mRNA and lncRNA profiles for three samples.
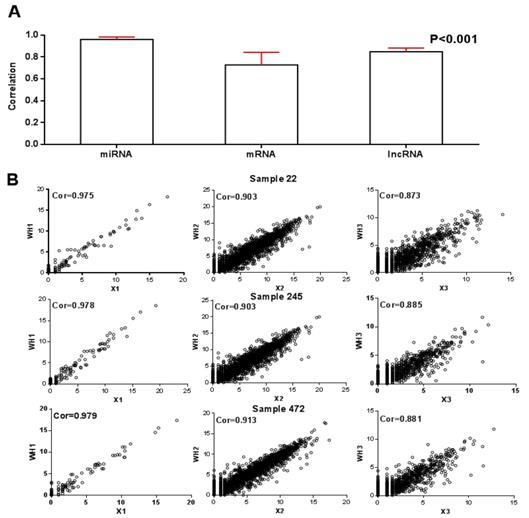
For these 40 modules, we first calculated the correlation between the product of WHI after decomposition and the original RNA data matrix XI. The average correlations of miRNA, mRNA and lncRNA were 0.96, 0.73 and 0.85 respectively, as shown in Figure 2A. On this basis, after randomly selecting three RNA data from three samples, we plotted the correlation between the reconstructed matrix and the original matrix in Figure 2B. It is obvious that the difference between the reconstruction matrix and the original matrix was small. It proved that this algorithm was robust.
Correlation between the identified co-modules and Lung cancer
On this basis, in order to verify the correlation between the modules and lung cancer, we analyzed whether the various data in the obtained co-module are related to cancer alone from the three dimensions of RNA. For miRNA, since no more than five miRNAs are present in the module, if one miRNA is shown to be cancer-related, the module is associated with cancer in the miRNA dimension. For mRNA, if more than 80% of mRNAs are associated with cancer, the module is considered to be associated with cancer, and if more than 50% of mRNAs are associated with lung cancer, the module is considered to be associated with lung cancer. For lncRNAs, considering the limited function currently searched of lncRNA, we used lncRNAdisease [27] web service to search for cancer-related lncRNAs. If there is an lncRNA associated with cancer, this module is considered to be related to this cancer. Figure 3 plots the number of modules associated with cancers including lung cancer in different dimensions. From different dimensions, for miRNA, 36 of 40 modules can be found cancer-associated members, 28 of which are associated with lung cancer, account for 70% of the total number of modules. It could be found that more than 80% of mRNAs in all modules are related to cancer, 25 of which are associated with lung cancer. For lncRNA, we found that 26 modules are related to cancer. Among them, 12 modules are directly related to lung cancer, accounting for 30% of the total module.
A) The number of cancer-related modules from three dimensions B) The number of lung cancer-related modules from three dimensions.
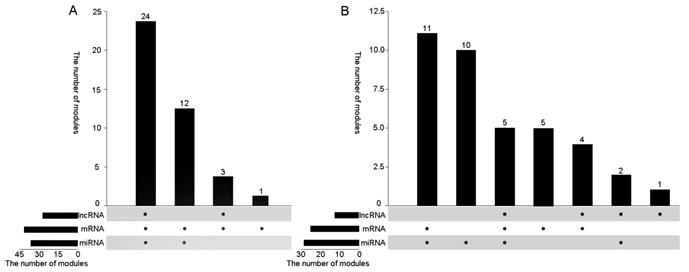
Figure 3A shows to verify whether the module is associated with cancer from the three RNA dimensions, there are 39 modules that can be validated for cancer at least from two dimensions, 23 of which can be proved from three dimensions, exceeding 50%. In addition, in the 25th module that failed to be assigned to miRNAs but most of its mRNAs were also found to be associated with cancer. Figure 3B shows to verify whether the 40 modules are associated with lung cancer from three dimensions. More than half of the modules are proved to be related to lung cancer from two dimensions at least. Among them, three modules were found to be associated with lung cancer in all three dimensions. Only two modules were not directly related to lung cancer from the data point of view. Therefore, using our algorithm can effectively find diseased-related co-modules.
Overlap enrichment analysis of co-modules
The enrichment analysis in a single dimension can only show that the extracted modules can be found in each dimension to be related to the pathogenesis of lung cancer, and all show a performance uniformity. To better explore the performance of co-modules, we explored the relevance of the three dimensions through the enrichment analysis of the three types of data in the module. DIANA-miRPath [28] was applied to find the target mRNAs to obtain enrichment analysis that were thought to be miRNA enrichment pathways or biological processes. WGCNA R package [29] was applied to calculate the correlation of lncRNA and mRNA in all co-modules, then the enrichment analysis with the most relevant mRNA for each lncRNA was thought to be lncRNA enrichment analysis. Therefore, each type of data is based on mRNA data for functional enrichment analysis. At the same time, as shown in Table 1, co-modules with overlapping mRNAs for any two types of data were selected to show functional enrichment overlaps, overlapped mRNA enrichment pathways and biological processes.
Overlapping gene enrichment or enrichment overlap of co-modules
| Module | mRNA | miRNA (mRNA) | lncRNA (mRNA) | Overlapping gene enrichment or enrichment overlap |
|---|---|---|---|---|
| 1 | 49 | 4(1390) | 31(29) | Cell-cell signaling; leukocyte migration; metabolic process |
| 4 | 4 | 3(576) | 38(37) | Cytokine-mediated signaling pathway; PI3K-Akt signaling pathway; regulation of immune response |
| 7 | 60 | 4(714) | 37(35) | Cellular protein metabolic process; platelet degranulation; platelet activation; transcription from RNA polymerase II promoter |
| 11 | 56 | 5(432) | 41(40) | Metabolic pathway; Central carbon metabolism in cancer; blood coagulation; platelet activation |
| 12 | 43 | 6(2037) | 40(37) | Viral carcinogenesis; RNA transport; Transcriptional misregulation in cancer; DNA-templated transcription, initiation |
| 17 | 54 | 2(272) | 36(35) | Neuroactive ligand-receptor interaction; transport |
| 26 | 53 | 4(1291) | 33(30) | Positive regulation of transcription from RNA polymerase II promoter |
| 32 | 39 | 5(848) | 40(38) | Innate immune response |
It can be seen from Table 1 that the overlapped enrichment is mainly for cell metabolism, cancer reactions, immune-related pathways and biological processes. These pathways or biological processes are closely related to the pathogenesis of lung cancer [30]. In addition, the PI3K/AKT pathway is involved in the regulation of many biological processes. By affecting the activation state of various downstream effector molecules, it plays a key role in inhibiting apoptosis and promoting proliferation in cells [31]. It shows that it is closely related to the occurrence and development of lung cancer [32]. Therefore, it is further illustrated that our algorithm can effectively extract co-modules directly related to lung cancer from three dimensions.
CeRNA network construction and analysis
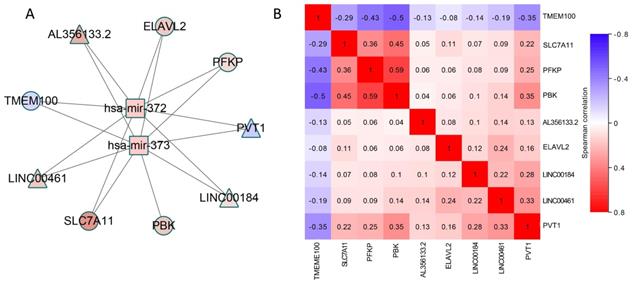
For the module obtained by the algorithm, in order to further dig the mutual relationship of the three types of data in the common module. We have drawn two types of ceRNA networks, with modules 9 and 11 being two typical types. On the one hand, as shown in Figure 4, this ceRNA network includes 2 miRNAs, 4 mRNAs and 4 lncRNAs, all of which show significant changes in expression level.
A) CeRNA network constructed by using module 11, the color of nodes represents the expression change of RNA compared to control samples, and Red (blue) indicates a significant increase (decrease). Square node represents miRNA, circular node denotes mRNA and diamond node represents lncRNA. The edge denotes the regulatory relationship between the two types of RNA. B) The Spearman correlation of lncRNAs and mRNAs in ceRNA network. Red (blue) indicates positive (negative) correlation.
According to review the literature, we found that PDZ-binding kinase (PBK) is highly expressed in many types of tumors, in which pbk is positively correlated with mutant p53 and influences cell proliferation, viability, and prognosis of LUAD [33]. The anti-Hu antibody found in newly diagnosed small cell lung cancer (SCLC) is often associated with low titer of paraneoplastic encephalomyelitis/sensory neuropathy (PEM/PSN) and anti-Hu antibodies. D'Alessandro V et al. used real-time PCR to study peripheral blood samples from untreated lung cancer patients and healthy individuals. HuB (ELAV2) mRNA levels were measured using an absolute quantitative method and it was found that 60% of lung cancer patients showed increased levels of HuB transcripts [34]. Frullanti E et al. compared the transcriptomes of 60 ADCA smokers and 60 I> I patients who were not involved in the lung samples from the Phase I clinical trial, then found that TMEM100 inhibited colony formation of lung cancer cell lines transfected to overexpress the genes, indicating its potential for tumor suppressive activity [35]. By altering the glycolytic flux as a marker of cancer, the PFKP somatic mutation blueprint and catalytic site can guide the therapeutic targeting of PFK1 activity to control abnormal glucose regulation in the disease [36]. MiR-372 and 373 are the same miRNA family with the same precursors. Both are transcribed from chromatin 19q13.42 [37]. There is an evidence that miR-372 transcription down-regulates large tumor suppressor homolog 2 (Lats2), leading to tumorigenesis and proliferation [38]. MiR-372 was bound directly to its 3' untranslated region (3'UTR) and as an upstream target inhibited the expression of ATAD2, which is highly expressed in HCC and exerts a proto-oncogene effect in hepatocarcinogenesis [39]. It was found that miR-373 is silenced in lung cancer cells by histone modifications, and its function as a tumor suppressor and a negative regulator of the mesenchymal phenotype was identified by the downstream IRAK2 and LAMP1 target genes [40]. For lncRNAs, LINC00461 may be involved in tumorigenesis because siRNA depletion inhibits glioma cell division. Transcripts can also bind to and regulate the activity of miR-411-5p and argonaut 2, thereby altering the expression of genes involved in tumor growth. Many current studies have shown that PVT1 is associated with lung cancer, especially non-small cell lung cancer. lncRNA PVT1 is significantly up-regulated in NSCLC tissues and may be a novel biomarker and potential therapeutic target for NSCLC intervention [41].
What's more, most of these elements have been verified to be associated with lung cancer. Therefore, more biological verification is needed in the future to prove their competitive relationship. It also could be seen from Figure 4B that a correlation analysis of the mRNA-lncRNA pairs with potentially competing relationships was performed. We found a significant correlation between multiple pairs of RNA expression values, which also demonstrated the rationality of the ceRNA hypothesis. In particular, for PVT1, the four mRNAs in the network have a strong correlation with it. This suggests that PVT1 may affect the expression of PBK, SLC7A11, ELAVL2 and PFKP through competitive binding of has-mir-372 and has-mir-373.
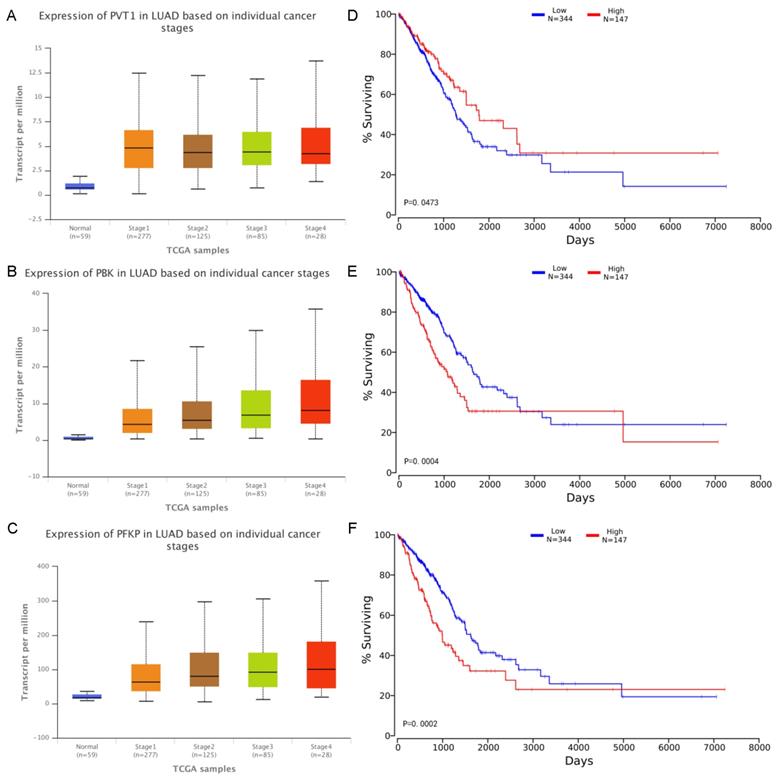
From the above analysis, most of RNA molecules can be found in the literature were closely related to lung cancer, in order to further analyze the correlation between several molecules in the network and LUAD, we have considered the expression values of these molecules in each pathological stage and effects of RNA expression level on LUAD patient survival. The expression values and corresponding survival curves for two mRNA and one lncRNA at different stages was constructed using UALCAN [42] and OncoLnc [43] in Figure 5. There was a significant correlation between the expression values of the three and the patient's survival time. Patients with high expression of PVT1 and low expression of mRNA have a better survival rate during 9 years. In particular, for PBK and PFKP, we found that the higher the degree of deterioration of the LUAD patients, the higher the expression level of the mRNA, consistent with the survival curve results. Therefore, the module can effectively excavate RNA closely related to lung cancer.
A-C Expression of RNA on individual cancer stages. The x-axis indicates different pathological stages, and the five colors indicate normal, stage 1, stage 2, stage 3 and stage 4 respectively. The y-axis represents transcripts per million (TPM) of each RNA molecule. D-F Effects of RNA expression level on LUAD patient survival. The x-axis shows the patient's survival time, and the y-axis shows the Kaplan-Meier (KM) survival probability.
On the other hand, some co-modules may not be able to find a direct link between the RNA molecules or the direct association only contains two kinds of data. As shown in Figure 6, co-module 9 can find miRNA-regulated lncRNAs, but mRNAs directly controlled by miRNAs may be difficult to find. However, the miRNA-regulated target mRNAs have a certain degree of association with the mRNA in the co-module, including activation, inhibition, and co-expression. Since the regulatory information of lncRNAs by miRNAs is currently not comprehensive enough, we can find some lncRNAs that are closely related to lung cancer and are controlled by differential miRNAs but some mRNAs are indirectly controlled by miRNAs, and then some potential biological relationships are discovered.
CeRNA network constructed using module 9, the color of nodes represents the expression change of RNA compared to control samples, and Red (blue) indicates a significant increase (decrease). White node denotes these RNA is not in the co-module. Square node represents miRNA, circular node denotes mRNA and diamond node represents lncRNA. The edge denotes the relationship including regulation, co-expression, activation and inhibition between the two types of RNA.
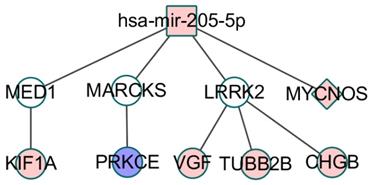
Zarogoulidis P et al. studied the effect of two different miRNA members miR-205 and tumor suppressor miR-218 on lung cancer cell proliferation, invasion and apoptosis after carboplatin treatment. The results showed that ectopic miR-218 overexpression reduced cell proliferation, Invasion and migration, while miR-205 rescues the inhibitory effect of miR-218 by altering the expression levels of the pro-apoptotic proteins PARP, Caspase 3, Bax and upregulating the anti-apoptosis markers Mcl-1 and Survivin [44]. MiR-205 targeting mediator 1 (MED1, also known as TRAP220 or PPARBP) resulted in a decrease in MED1 mRNA levels, as well as total and active phosphorylated MED1 protein [45]. Knockdown of MED1 in lung cancer cell lines leads to increased cell migration and invasion. MED1-depleted cells showed increased metastasis in xenograft tumor models and in vivo metastatic assays [46]. In addition, MED1 and KIFIA have been verified to have significant interactions [47], and compared with benign tumors, the frequency and level of methylation of KIF1A in cancer samples are significantly increased [48]. In the immune-related pathway FcγR-mediated phagocytosis, PRKCE inhibits MARCKS expression through phosphorylation and MARCKS is also regulated by mir-205. Chen CH et al. demonstrated that higher levels of phosphorylated MARCKS are associated with shorter overall survival in lung cancer patients [49]. Cho HJ et al. found that miRNA-205 inhibits the expression of LRRK2 protein through the conserved binding site of the 3' untranslated region (UTR) of LRRK2 gene [50]. LRRK2 has co-expression relationship with three genes in the ceRNA. Three genes are involved in the development of tumors, and the expression of CHGB is associated with malignant tumors and metastases [51].
Although VGF and PRKCE have not been found to be directly related to lung cancer, we found that the expression level is closely related to the patient's survival and smoking habits. The relationship between the expression of two mRNAs and smoking habits of patients was plotted in the Figure 7, and the survival analysis was corresponding to the right side.
A-B Expression of RNA in the samples with different smoking habits. The x-axis indicates different smoking habits, and the five colors indicate normal, smoker, Non-smoker, reformed smoker (< 15 years) and reformed smoker (> 15 years) respectively. The y-axis represents transcripts per million (TPM) of each RNA molecule. C-D Effects of RNA expression level on LUAD patient survival. The x-axis shows the patient's survival time, and the y-axis shows the Kaplan-Meier (KM) survival probability.
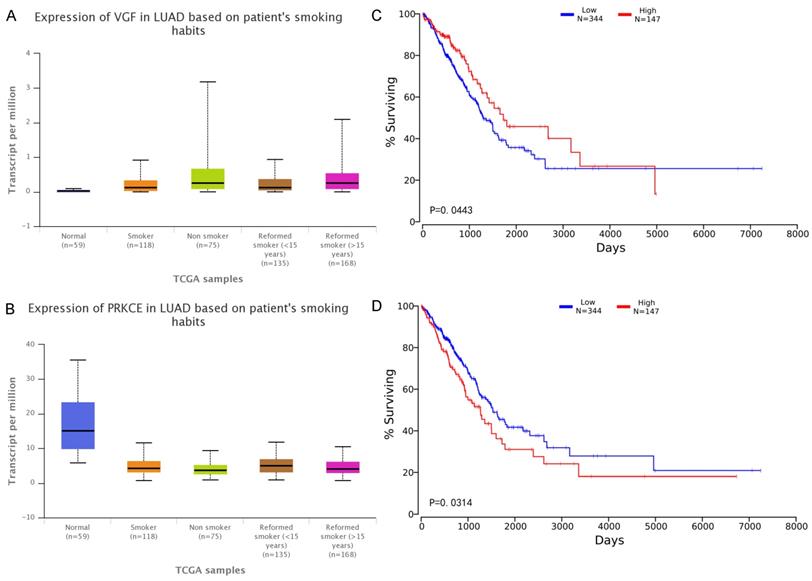
VGF has a higher expression value (significant p-value is 0.035) in the tumor tissues of smokers, but the prognosis of high expression is better than the prognosis of low expression. It is worth mentioning that the expression of PRKCE in control tissues is significantly higher than that in tumor tissues (whether or not the patient is smoking). The expression of PRKCE in tumor tissue of patients without smoking preference was significantly higher than that of smokers (significant p-value is 0.019). The PRKCE expression was highest in the tumor tissue of patients with a smoking history of less than 15 years, which was significantly higher than the smoking history of more than 15 years (significant p-value is 0.036). These results confirm that most of the elements of the co-modules identified in this study can be found to play a role in cancer, especially in lung cancer. Furthermore, the experimental results show that the proposed method not only can find RNA molecules that have been proved to be closely related to LUAD, but also can effectively identify RNA molecules that are related to LUAD but have not been confirmed yet, and provide better clues for subsequent researchers.
Comparison with other algorithms
In order to prove the effectiveness of our algorithm, the KEGG pathway and biological process enrichment rates under different thresholds T are applied to compare our algorithm with joint NMF algorithm due to there are fewer algorithms for three data fusion. From Figure 8, it could be found that each of the results is better than the joint NMF algorithm.
(A) The mRNA modules with respect to GO biological process enrichment ratio with joint NMF comparing with joint NMF. (B) The mRNA modules with respect to KEGG pathway enrichment ratio comparing with joint NMF. The x-axis represents the threshold for determining module membership. The y-axis represents the enrichment rate of the module. We can see that the proposed model is superior to the joint NMF algorithm in each cutoff T.
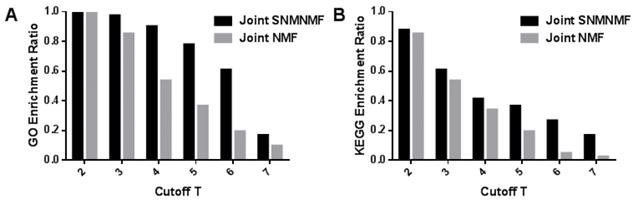
Therefore, the NMF algorithm added with prior knowledge is obviously superior to the traditional NMF algorithm in extracting the performance of the common module. The results of joint NMF algorithm have been compared with random results, which proves that joint NMF algorithm has certain non-randomness [17]. Not surprisingly, our algorithm can effectively extract biologically relevant information.
Conclusion
Disorders of ceRNA networks can lead to human diseases including cancer. As a major member of the intra-competitive network, lncRNA often binds miRNA-related sites in the cell to act as miRNA sponges, thereby eliminating miRNAs' inhibitory effects on their target genes and increasing the target gene expression levels. Constructing ceRNA networks helps us better understand the relationship between molecules in the progression of tumors. It is found that the systematic analysis of multidimensional data of the biological-related combinatorial model can unearth more complex biological associations. However, most of the past researches are based on individual or two-dimensional data, and the rare research on three-dimensional data merely considers the features of the data independence and neglects the correlation between three types of data, which makes it difficult to verify the discovered complex relationships. In this paper, three kinds of LUAD RNA data were combined to extract co-modules using joint SNMNMF algorithm and construct ceRNA networks. In addition, the association between the three types of data is incorporated into the algorithm framework, which is more suitable for not only problems involving multidimensional genomic data, that is, to analyze multiple variables on the same set of samples, but also identification of known relationships between variables including miRNA-mRNA, miRNA-lncRNA and mRNA-mRNA.
Although the results show that this algorithm can effectively obtain the miRNA-lncRNA-mRNA co-module and extract the core ceRNA network from co-modules. Furthermore, it was found that members of the network are closely related to lung cancer. However, the competitive relationship between RNA molecules is not limited to these three RNAs, such as circRNA can also competitively inhibit the transcriptional regulation of miRNA. CiRS-7 contains a series of miR-7 binding sites, it can serve as endogenous miRNA sponges to inhibit miR-7 activity, and miR-7 is an important regulator of various cancer-related pathways. In future studies, it would make more sense to apply the proposed method to more data sources simultaneously in order to obtain a more comprehensive competing endogenous RNA network and to discover more complex biological molecular associations.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 61271446 and 31170952) and Natural Science Foundation of Shanghai (No. 18ZR1417200).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. [J]. Nature Reviews Genetics. 2014;15(1):7-21
2. Quinn J J, Chang H Y. Unique features of long non-coding RNA biogenesis and function. [J]. Nature Reviews Genetics. 2015;17(1):47
3. Batista P J, Chang H Y. Long noncoding RNAs: cellular address codes in development and disease[J]. Cell. 2013;152(6):1298-307
4. Thomson D W, Dinger M E. Endogenous microRNA sponges: evidence and controversy[J]. Nature Reviews Genetics. 2016;17(5):272
5. Leonardo Salmena, Laura Poliseno, Yvonne Tay, Lev Kats, Pier Paolo Pandolfi. ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? [J]. Cell. 2011;146(3):353-8
6. Huang X, Xiao R, Shan P. et al. Uncovering the roles of long non-coding RNAs in cancer stem cells[J]. Journal of Hematology & Oncology. 2017;10(1):62
7. Liu C, Tang D G. MicroRNA regulation of cancer stem cells. [J]. Cancer Research. 2011;71(18):5950
8. Li J H, Liu S, Zhou H. et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data[J]. Nucleic Acids Research. 2014;42(Database issue):D92
9. Wang P, Zhi H, Zhang Y. et al. miRSponge: a manually curated database for experimentally supported miRNA sponges and ceRNAs[J]. Database the Journal of Biological Databases & Curation. 2015;2015:bav098
10. Liu Y C, Li J R, Sun C H. et al. CircNet: a database of circular RNAs derived from transcriptome sequencing data[J]. Nucleic Acids Research. 2016;44(D1):D209
11. Zeitels L R, Acharya A, Shi G. et al. Tumor suppression by miR-26 overrides potential oncogenic activity in intestinal tumorigenesis. [J]. Genes Dev. 2014;28(23):2585-2590
12. Rong M, Chen G, Dang Y. Increased MiR-221 expression in hepatocellular carcinoma tissues and its role in enhancing cell growth and inhibiting apoptosis in vitro[J]. Bmc Cancer. 2013;13(1):21
13. Liu X, Yu J, Jiang L. et al. MicroRNA-222 Regulates Cell Invasion by Targeting Matrix Metalloproteinase 1 (MMP1) and Manganese Superoxide Dismutase 2 (SOD2) in Tongue Squamous Cell Carcinoma Cell Lines[J]. Cancer Genomics & Proteomics. 2009;6(3):131-139
14. Ernst J, Beg Q K, Kay K A. et al. A Semi-Supervised Method for Predicting Transcription Factor-Gene Interactions in Escherichia coli[J]. Plos Computational Biology. 2008;4(3):e1000044
15. Glass K, Huttenhower C, Quackenbush J. et al. Passing Messages between Biological Networks to Refine Predicted Interactions[J]. Plos One. 2013;8(5):59-59
16. Zhang S, Li Q, Liu J. et al. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules[J]. Bioinformatics. 2011;27(13):i401-i409
17. Zhang S, Liu C C, Li W. et al. Discovery of multi-dimensional modules by integrative analysis of cancer genomic data[J]. Nucleic Acids Research. 2012;40(19):9379-91
18. Paatero P, Tapper U. Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values[J]. Environmetrics. 1994;5(2):111-126
19. Kim H, Park H. Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis. [J]. Bioinformatics. 2007;23(12):1495-1502
20. Zhang S, Li Q, Liu J. et al. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules. [J]. Bioinformatics. 2011;27(13):i401-i409
21. Jeggari A, Marks D S, Larsson E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome[J]. Bioinformatics. 2012;28(15):2062
22. Keshava Prasad T S, Goel R, Kandasamy K. et al. Human Protein Reference Database-2009 update. [J]. Nucleic Acids Research. 2009;37(Database issue):767-72
23. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. [J]. Nucleic Acids Research. 2015;43(Database issue):D146
24. Chou C H, Chang N W, Shrestha S. et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database[J]. Nucleic Acids Research. 2016;44(Database issue):D239
25. Garcia D M, Baek D, Shin C. et al. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. [J]. Nature Structural & Molecular Biology. 2011;18(10):1139-1146
26. Love M I, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2[J]. Genome Biology. 2014;15(12):550
27. Chen G, Wang Z, Wang D. et al. LncRNADisease: a database for long-non-coding RNA-associated diseases[J]. Nucleic Acids Research. 2013;41(Database issue):983-6
28. Vlachos I S, Zagganas K, Paraskevopoulou M D. et al. DIANA-miRPath v3.0: deciphering microRNA function with experimental support[J]. Nucleic Acids Research. 2015;43(1):460-6
29. Langfelder P, Horvath S. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform 9: 559[J]. Bmc Bioinformatics. 2008;9(1):559
30. Hampton T. Lung Cancer Pathway[J]. Jama the Journal of the American Medical Association. 2010;303(21):2129-2129
31. Luo J, Manning B D, Cantley L C. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. [J]. Cancer Cell. 2003;4(4):257-262
32. Xie M, He J, He C. et al. γ Secretase Inhibitor BMS-708163 Reverses Resistance to EGFR Inhibitor via the PI3K/Akt Pathway in Lung Cancer[J]. Journal of Cellular Biochemistry. 2015;116(6):1019-27
33. Lei B, Qi W, Zhao Y. et al. PBK/TOPK expression correlates with mutant p53 and affects patients' prognosis and cell proliferation and viability in lung adenocarcinoma[J]. Human Pathology. 2015;46(2):217-224
34. Vito D, Anna M L, Massimiliano C. et al. Molecular Detection of Neuron-Specific ELAV-Like-Positive Cells in the Peripheral Blood of Patients with Small-Cell Lung Cancer[J]. Cellular Oncology. 2008;30(4):291-297
35. Frullanti E, Colombo F, Falvella F S. et al. Association of lung adenocarcinoma clinical stage with gene expression pattern in noninvolved lung tissue[J]. International Journal of Cancer. 2012;131(5):E643-E648
36. Webb B A, Forouhar F, Szu F E. et al. Structures of human phosphofructokinase-1 and atomic basis of cancer-associated mutations[J]. Nature. 2015;523(7558):111-114
37. Lai J H, She T F, Juang Y M. et al. Comparative proteomic profiling of human lung adenocarcinoma cells (CL 1-0) expressing miR-372[J]. Electrophoresis. 2012;33(4):675-688
38. Wu G, Liu H, He H. et al. miR-372 down-regulates the oncogene ATAD2 to influence hepatocellular carcinoma proliferation and metastasis[J]. Bmc Cancer. 2014;14(1):1-11
39. Seol H S, Akiyama Y, Shimada S. et al. Epigenetic silencing of microRNA-373 to epithelial-mesenchymal transition in non-small cell lung cancer through IRAK2 and LAMP1 axes[J]. Cancer Letters. 2014;353(2):232-241
40. Kwak M S, Lee D H, Cho Y. et al. Association of polymorphism in pri-microRNAs-371-372-373 with the occurrence of hepatocellular carcinoma in hepatitis B virus infected patients. [J]. Plos One. 2012;7(7):e41983
41. Yang Y R, Zang S Z, Zhong C L. et al. Increased expression of the lncRNA PVT1 promotes tumorigenesis in non-small cell lung cancer[J]. International Journal of Clinical & Experimental Pathology. 2014;7(10):6929
42. Chandrashekar D S, Bashel B, Sah B. et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. [J]. Neoplasia. 2017;19(8):649-658
43. Anaya J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs[J]. Peerj Computer Science. 2016;2(2):e67
44. Zarogoulidis P, Petanidis S, Kioseoglou E. et al. MiR-205 and miR-218 expression is associated with carboplatin chemoresistance and regulation of apoptosis via Mcl-1 and Survivin in lung cancer cells. [J]. Cellular Signalling. 2015;27(8):1576-1588
45. Hulf T, Sibbritt T, Wiklund E D. et al. Epigenetic-induced repression of microRNA-205 is associated with MED1 activation and a poorer prognosis in localized prostate cancer[J]. Oncogene. 2013;32(23):2891
46. Kim H J, Roh M S, Son C H. et al. Loss of Med1/TRAP220 promotes the invasion and metastasis of human non-small-cell lung cancer cells by modulating the expression of metastasis-related genes. [J]. Cancer Letters. 2012;321(2):195-202
47. Albers M, Kranz H, Kober I. et al. Automated yeast two-hybrid screening for nuclear receptor-interacting proteins. [J]. Molecular & Cellular Proteomics. 2005;4(2):205-213
48. Brait M, Loyo M, Rosenbaum E. et al. Correlation between BRAF mutation and promoter methylation of TIMP3, RARβ2 and RASSF1A in thyroid cancer[J]. Epigenetics. 2012;7(7):710-719
49. Chen C H, Statt S, Chiu C L. et al. Targeting myristoylated alanine-rich C kinase substrate phosphorylation site domain in lung cancer. Mechanisms and therapeutic implications[J]. Am J Respir Crit Care Med. 2014;190(10):1127-1138
50. Cho H J, Liu G, Jin S M. et al. MicroRNA-205 regulates the expression of Parkinson's disease-related leucine-rich repeat kinase 2 protein[J]. Human Molecular Genetics. 2013;22(3):608
51. Weisbrod A B, Zhang L, Jain M. et al. Altered PTEN, ATRX, CHGA, CHGB & TP53 Expression are Associated with Aggressive VHL-Associated Pancreatic Neuroendocrine Tumors[J]. Hormones & Cancer. 2013;4(3):165-175
Author contact
![]() Corresponding author: Wei Kong: weikongedu.cn
Corresponding author: Wei Kong: weikongedu.cn