Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(13):1845-1858. doi:10.7150/ijbs.27661 This issue Cite
Research Paper
The HDAC Inhibitor Quisinostat (JNJ-26481585) Supresses Hepatocellular Carcinoma alone and Synergistically in Combination with Sorafenib by G0/G1 phase arrest and Apoptosis induction
Bin He1,2,3,4,6*, Longfei Dai1,2,3,4,6*, Xiaoqian Zhang5,6*, Diyu Chen1,2,3,4,6, Jingbang Wu1,2,3,4,6, Xiaode Feng1,2,3,4,6, Yanpeng Zhang1,2,3,4,6, Haiyang Xie1,2,3,4,6, Lin Zhou1,2,3,4,6, Jian Wu1,2,3,4,6 ![]() , Shusen Zheng1,2,3,4,6
, Shusen Zheng1,2,3,4,6 ![]()
1. Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University
2. NHFPC Key Laboratory of Combined Multi-organ Transplantation
3. Key Laboratory of the diagnosis and treatment of organ Transplantation, CAMS
4. Key Laboratory of Organ Transplantation, Zhejiang Province
5. State Key Laboratory for Diagnosis and Treatment of Infectious Diseases,
6. Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310003, China
*Bin He, Longfei Dai and Xiaoqian Zhang contributed equally to this study.
Received 2018-6-5; Accepted 2018-8-30; Published 2018-10-20
Abstract

The high activity of Histone deacetylases (HDACs) in hepatocellular carcinoma (HCC) usually positively correlates with poor prognosis of patients. Accordingly histone deacetylases inhibitors (HDACis) are considered to be potential agents treating patients with HCC. In our study, we evaluated effect of quisinostat alone and in combination with sorafenib in HCC cells via inducing G0/G1 phase arrest through PI3K/AKT/p21 pathway and apoptosis by JNK/c-Jun/caspase3 pathway in vitro and in vivo. The proliferation assay and flow cytometry were used to measure the viability, cell cycle and apoptosis. And Western blot assay was carried out to determine expression alternations of related proteins. Moreover HCCLM3 xenograft was further performed to detect antitumor effect of quisinostat in vivo. Here, we found that quisinostat impeded cell proliferation, and remarkably induced G0/G1 phase arrest and apoptosis in HCC cells in a dose-dependent manner. G0/G1 phase arrest was observed by alterations in PI3K/AKT/p21 proteins. Meanwhile the JNK, c-jun and caspase-3 were activated by quisinostat in a dose-dependent manner. Correspondingly quisinostat facilitated G0/G1 cycle arrest and apoptosis in HCC cells through PI3K/AKT/p21 pathways and JNK/c- jun/caspase3 pathways. Moreover, the potent tumor-suppressive effects facilitated by quisinostat, was significantly potentiated by combination with sorafenib in vitro and vivo. The combination treatment of quisinostat and sorafenib markedly suppressed cell proliferation and induced apoptosis in a synergistic manner. Moreover the therapy of quisinostat combined with sorafenib could apparently decrease tumor volume of a HCCLM3 xenograft model. Our study indicated that quisinostat, as a novel chemotherapy for HCC, exhibited excellent antitumor activity in vitro and vivo, which was even enhanced by the addition of sorafenib, implying combination of quisinostat with sorafenib a promising and alternative therapy for patients with advanced hepatocellular carcinoma.
Keywords: Histone deacetylases (HDACs), Histone deacetylases inhibitors (HDACis), Hepatocellular Carcinoma cells (HCC cells), quisinostat (JNJ-26481585), sorafenib, synergistic effect
Introduction
Hepatocellular carcinoma (HCC) is the second deadly cancer worldwide, which accounts for 90% of primary liver cancers[1-3]. The curative therapies for early-stage HCC are resection and liver transplantation, suitable only for part of patients, owing to high recurrence rates of HCC[4]. Consequently it is urgent to carry out innovative systemic therapies for patients at an advanced disease stage.
Recently histone deacetylases inhibitors (HDACis) are recognized to be effective antitumor agents in therapeutic strategy, for several studies of malignant hematological cancers and solid tumors have verified their excellent anticancer abilities and efficient chemotherapeutic potentials in early-phase clinical trials[5]. HDACis executed their antitumor functions by catalyzing a serial of process in which acetyl groups in histones were removed from lysine residues, resulting in chromatin condensation as well as transcriptional inhibition of target genes, especially tumor suppressor genes[6]. It was verified that HDAC expression positively correlated with higher incidences of HCC, cell invasion and metastasis and poor prognosis after liver resection[7]. Studies showed that expressions of HDAC1-3 were predominantly high in HCC samples of patients and that repression of HDAC with trichostatin A, a HDAC inhibitor, or transfection with miR-449 significantly increased apoptosis and reduced proliferation in HCC cells[8]. Meanwhile it was also reported expression levels of homeobox protein PROX1 and HDAC1 synergistically exhibited prognostic value by predicting survival after operation and early recurrence of HCC[9]. In addition studies showed miR-200a could impede proliferation and migration of HCC cells by targeting HDAC4/Sp1/miR-200a network to suppress expression of HDAC4[10]. Quisinostat (JNJ-26481585), as a more efficient second-generation HDACi, has been explored to boost pharmacodynamic response and to enhance efficacy at nanomolar concentrations in tumor models[11-13].
Currently, sorafenib is used as a first-line treatment to patients with advanced hepatocellular carcinoma approved by the U.S. Food and Drug Administration (FDA) [14, 15]. However, it remains inefficient to a number of patients that seem to react to sorafenib and then response insensitive, contributing to tumor metastasis and progression[15].
Recent evidences suggested that epigenetic mechanisms, such as histone deacetylations, frequently involved in resistance and insensitivity against antitumor drugs[16]. In order to deal with drug resistance, HDACis were employed to combine with alternative chemotherapeutic drugs as a promising strategy, since combination therapy could lower the dose of both anti-cancer agents[17]. Moreover reports have been illustrated combination of sorafenib with HDACis performed synergistic or additive effects, and studies have also showed patients with advanced HCC could benefit from the combination therapy.[18-20].
Our research herein showed several HDAC family members, HDAC1, HDAC2 and HDAC4, were aberrantly expressed in HCC tissues with significant correlation to poor prognosis. Furthermore we provided evidences that quisinostat had consistent antitumor efficacy in HCC cells through facilitating G0/G1 phase arrest via PI3K/AKT/p21 pathway and apoptosis via JNK/c-Jun/caspase3/PARP pathway, which was further potentiated when combined with sorafenib. In addition the combination treatment inhibited tumor growth of HCCLM3 xenografts more effectively than each agent alone did. In summary, these findings indicated that our combination therapy significantly potentiated development of promising therapeutic applications against HCC and established a rationale for clinical studies.
Materials and methods
Patients and specimens
This project was approved by the First Affiliated Hospital of Zhejiang University Ethics Committee, and informed consents were achieved from each patient according to institutional review board protocols. All cancer tissue samples and peritumoral tissue samples were obtained from 111 patients who underwent hepatocellular curative resection during January 2009 to December 2014.
Reagents
Sorafenib, quisinostat (JNJ-26481585), JNK inhibitor SP600125, AKT inhibitor MK2206 2HCL and Caspase inhibitor Z-VAD-FMK were from Selleckchem (Houston, Texas, USA). Dimethylsulfoxide was purchased from Sigma-Aldrich (St. Louis, MO, USA). Minimum Essential Medium Eagle (MEM), fetal bovine serum (FBS) were from Gibco Life Technologies (Grand Island, NY, USA). The Cell Counting Kit-8 was purchased from Dojindo (Kumamoto, Japan). The cell cycle staining kit was from MultiSciences (Hangzhou, China). The Annexin V-FITC apoptosis detection kit was obtained from KeyGen Biotech (Nanjing, China). The EdU Apollo567 In Vitro Imaging Kit was from Ribobio (Guangzhou, China). The BCA protein assay kit was from Thermo Fisher Scientific Inc (Waltham, Massachusetts, USA). The following antibodies were used: HDAC1, HDAC2, HDAC4 (Proteintech), p21Cip1, CyclinD1, CyclinE1, CyclinA2, cdk2, cdk4, cdk6, Cleaved-caspase-3, Cleaved-caspase-9, Caspase-3, Caspase-9, Cleaved-PARP, PARP, phospho-JNK, JNK, PI3K-p110, PI3K- p85, phospho-AKT473, phospho-c-jun, Bcl-xl, Bcl2, Bax, Survivin, Ki67, GAPDH (Cell Signaling Technology).
Tissue microarrays
HDAC1, HDAC2 and HDAC4 expressions in 111 cases of paired HCC that underwent surgical operation were evaluated by tissue microarray chips. Follow-up information was obtained by reviewing patients' medical records up to termination date (August 16, 2016) or death.
Cell culture
Human HCC cell lines (HCCLM3, SK-hep-1, Hep-3B, Huh7 and SMMC-7721) were obtained from the Type Culture Collection at the Chinese Academic of Sciences (Shanghai, China). All cell lines were cultured in MEM supplemented with 10% FBS as well as 1% penicillin along with 1% streptomycin at 37°C in a humidified atmosphere 5% CO2 incubator.
Cell proliferation assay
Cells were seeded in 96-well plates with 200 µl of medium at a density of 4000 cells each well. Then cells were treated with various concentrations of quisinostat for 48 h. Cell proliferation was measured by the Cell Counting Kit-8 (CCK-8). After the treatment, each well was supplemented with 10 µl CCK-8 solution and incubated for 2h. The absorbance was detected using a microplate reader at 450 nm (BioTek Instruments, Inc. Winooski, VT, USA). In addition, EdU Apollo567 was used to assess effect of quisinostat on cell viability. For both HCCLM3 and SMMC-7721, 5x104 cells were cultured in 6-well plates respectively and treated with various concentrations of quisinostat (12.5 nM, 25 nM and 50 nM) or DMSO. After incubation with quisinostat for 48h the immunoassay was performed.
Determination of synergism
The combination index (CI) was introduced here to assess the drug interactions according to Chou and Talalay's method [21]. It is concluded that when a CI is less than 0.90 there is usually a synergism effect between related drugs, a CI more than 1.10 implies antagonism and between 0.90 and 1.10 manifests an additive effect. And Calcusyn software (Biosoft, Oxford, UK) was employed to analyse the obtained data.
Colony-forming assay
HCCLM3 and SMMC-7721 cells were seeded in 6-well plate with 1000 cells each well. After incubation, cells were treated with different concentrations of quisinostat for 10 days. After washed by phosphate buffered saline (PBS), cells were fixed with methanol for 20 min and stained with crystal violet for 15 min. The number of colonies (>50 cells/colony) were counted.
Cell cycle and apoptosis evaluation
Briefly, cells were seeded into 6-well plates, and treated with different concentrations of quisinostat for 48 h. For cell cycle analysis, cells then were fixed by 70% ice-cold ethanol, followed by incubating in 4°C overnight. On the next day, washed with PBS and cultured for 30 min with 100 μL RNase, cells were resuspended by 400 μL PI at room temperation in the dark for 30 min. At last BD LSR II flow cytometer was introduced to analyze cell cycle distribution. For apoptosis analysis, cells were then trypsinized and pelleted by centrifugation and washed with PBS twice. Afterward, cells were stained using 5 μL of Annexin V-FITC and 5 μL of PI solution at room temperation for 15min in the dark, followed by adding 400 μL of Annexin V Binding Buffer. Similarly, BD LSR II flow cytometer (BD Biosciences, San Jose, CA, USA) was used to analyze apoptosis.
Western blot analysis
The RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) was applied to lyse cells for 20 min on ice. Then lysed cells were centrifugated at 16 000 g at 4°C for 15 minutes. Once supernatant obtained, BCA assay kit was used to measure protein concentration. And 30 μg protein of cell extracts were degenerated by LDS sample buffer (Invitrogen, Carlsbad, CA, USA) at 90°C for 10 minutes. The same total quantity of proteins was loaded onto a SDS-PAGE in 10% Bis-Tris gel (Invitrogen). Then, lysates were transferred to PVDF membranes for 1 h. After blocking for 1 h, they were incubated with primary antibody (1:1000) at 4°C overnight. Subsequently secondary antibody (1:3000) was employed for incubating with membranes at 25˚C for 1 h. Immunoreactive bands were detected by Enhanced chemiluminescence (ECL) (Guge Biotechnology, China) and photographed with an image acquisition system.
Xenograft model analysis
All animal experimental protocols were approved by the Animal Experimental Ethics Committee of the First Affiliated Hospital of Zhejiang University School of Medicine. The experimental were carried out according to guidelines of the National Institutes of Health. 4 weeks old Male nude athymic mice were from Shanghai Experimental Animal Centre, Chinese Academy of Science. Subcutaneous HCCLM3 tumor tissues were cut into about 1 mm3 pieces. 2-3 pieces of tumor tissues were injected subcutaneously into the immunodeficient mice. After 7 days, the mice were randomly divided into 4 groups that received the following agents by intraperitoneal injection: (i) vehicle (5% [v/v] DMSO), (ii) sorafenib (10 mg/kg), (iii) quisinostat (10 mg/kg), and (iv) quisinostat (10 mg/kg) plus sorafenib (10 mg/kg). The treatments were performed five times per week for 4 weeks. The weight of mice was monitored every two days and tumor volume was detected every three days. All mice were sacrificed after 4 weeks to harvest the tumors. Tumor/volume (mm3) = (l × w2)/2.
Tunel assay
The in situ cell death detection kit (Roche, Germany) was designed as a precise and simple technique for detecting and quantifying apoptotic cell death in tumors isolated from the mice. The isolated tumors were embedded with paraffin and cut into 10-μm thick sections. Tunel assay was carried out referring to manufacturer's instructions.
Immunohistochemical staining and evaluation
At terminal sacrifice, isolated tumors were embedded with paraffin for immunohistochemical staining according to the study described before[22]. Tissue sections were incubated with primary antibodies (p-JNK, cle-Caspase3, cle-PARP, Ki67), followed incubating with a biotinylated secondary antibody. A light microscope was applied to capture the images, which were further analyzed by Image-Pro Plus 4.5 Software.
Statistical analysis
SPSS 19.0 statistical software and GraphPad Prism 6 software were used for statistical analysis. Data are presented as mean ± SD. Differences between two groups were examined using a 2-tailed paired Student's t-test. Survival data were used to establish Kaplan-Meier curves. All experiments were performed in triplicate. And P values <0.05 were considered statistically significant.
Results
HDACs were overexpressed in HCC tissues and correlated with poor prognosis of HCC patients
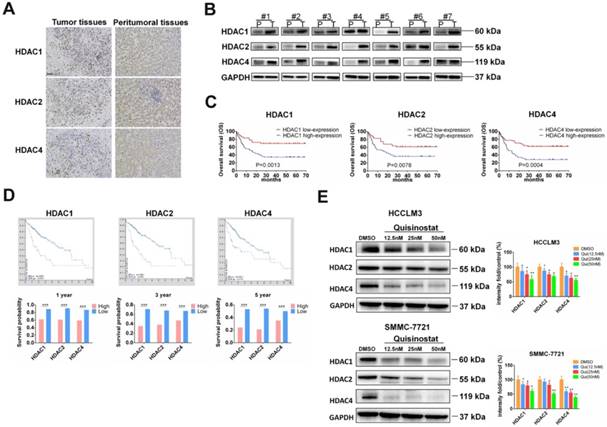
To investigate function of HDACs in progression of HCC, we used Immunohistochemistry and Western blot assay to detect HDACs in paired tumor tissues and peritumoral tissues. We found that HDAC1, HDAC2 and HDAC4 were upregulated in tumor tissues. Both IHC and Western blotting revealed that expressions of HDAC1, HDAC2 and HDAC4 were higher than that of peritumoral tissues (Fig. 1A-B). Next we followed-up these 111 cases and analyzed relationship between HDACs level and prognosis of patients. As Figure 1C showed that those HCC patients who had high expressions of HDAC1, HDAC2 and HDAC4 mainly led to poor overall survival (P=0.0013, 0.0078, 0.0004, respectively). To further analyze the association between HDACs and overall survival, we searched database of human protein atlas (www.proteinatlas.org) and found that patients with high HDACs had poor prognosis, especially in 1-year, 3-year and 5-year survival (Fig. 1D). Also we detected effects of quisinostat on expression levels of HDACs in HCCLM3 and SMMC-7721cells, finding HDAC1, HDAC2 and HDAC4 were decreased by quisinostat in a dose-dependent manner in HCCLM3 and SMMC-7721 cell lines (Fig.1E). Thus we concluded that HDACs could contribute to progression of HCC.
The overexpressions of HDACs in HCC tissues were correlated with poor prognosis of HCC patients. (A)The expressions of HDACs in paired HCC tissues and peritumoral tissues were detected by Immunohistochemistry and (B) Western blot assay. (C) The relationship between HDACs level and prognosis of 111 paired cases were analyzed. (D) The relationship between HDACs and overall survival from database of human protein atlas (www.proteinatlas.org). (E) The effects of quisinostat on the expressions of HDACs in HCC cells. The expression levels of HDAC1, HDAC2 and HDAC4 were suppressed in both HCCLM3 and SMMC-7721 cell lines. Images were photographed with confocal microscope under ×200 magnification. Scale bar, 100 μm. Data were shown as mean ± SD. n = 3; * P < 0.05, ** P < 0.01 and *** P < 0.001 compared with DMSO group.
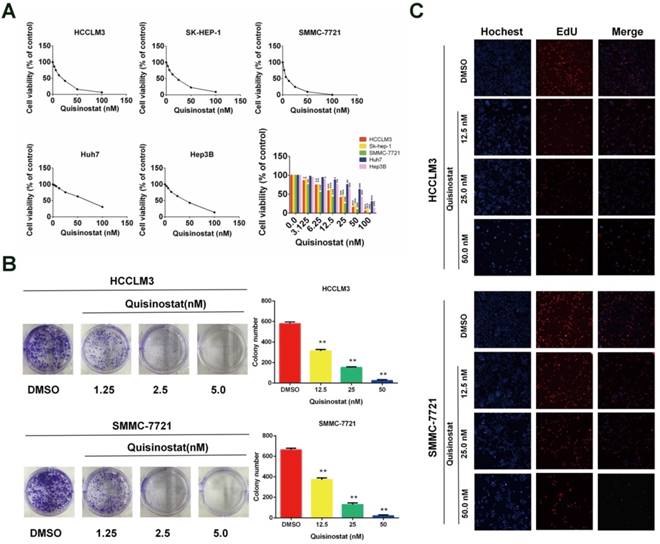
Quisinostat inhibited proliferation of hepatocellular carcinoma cells
We used CCK8 assay to identify influences of quisinostat on proliferation in five human HCC cell lines (HCCLM3, SK-hep-1, Hep-3B, Huh7 and SMMC-7721) respectively (Fig. 2A). It was observed that quisinostat substantially inhibited proliferation of HCC cells in a dose-dependent manner. Thses five HCC cells showed various sensitivities to cytotoxic effects of quisinostat, among which HCCLM3 and SMMC-7721 cells were more sensitive to quisinostat. Therefore HCCLM3 and SMMC-7721cells were used in the following experiments. In addition, as shown in Figure 2B, cells treated with quisinostat exhibited smaller and fewer colonies than DMSO group did. Meanwhile EDU assay was also introduced to measure the proliferation rates of HCC cells and verify that quisinostat repressed proliferation in HCC cells (Fig. 2C). According to results obtained above, it could therefore be concluded that quisinostat did impede proliferation of hepatocellular carcinoma cells.
Effects of Quisinostat on proliferation in HCC cells. (A) Quisinostat inhibited cell proliferation in HCCLM3, Sk-hep-1, Hep-3B, Huh7 and SMMC-7721 cells as a concentration-dependent manner verified by CCK8 assay. (B) Colony formation of HCCLM3 and SMMC-7721 cells in present or absent of quisinostat treatment. (C) EdU assays of incubation with various concentrations of quisinostat (12.5 nM, 25.0 nM, 50.0 nM) for 48 h in HCCLM3 and SMMC-7721 cells, and DMSO as control. Data were shown as mean ± SD. n = 3; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with DMSO group.
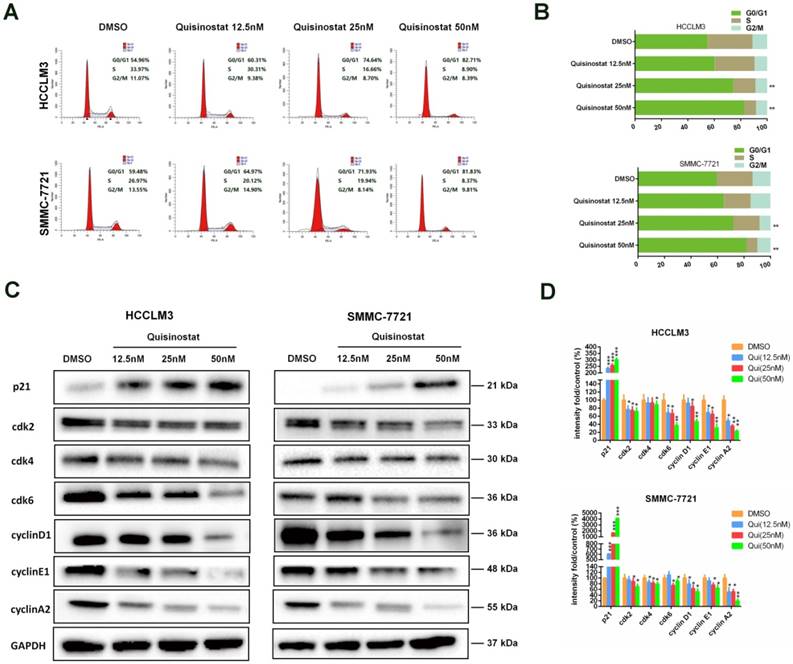
Quisinostat triggered G0/G1 phase arrest in hepatocellular carcinoma cells
Flow cytometry analysis was performed to explore mechanism of cell cycle arrest induced by quisinostat. As shown in Figure 3A-B, in contrast to DMSO group, it was observed that quisinostat substantially induced G0/G1 phase arrest both in HCCLM3 and SMMC-7721 cells. Moreover, in order to verify the influence of quisinostat on cell cycle arrest, expressions of p21, cdk2, cdk4, cdk6, cyclinD1, cyclinE1 and cyclinA2 were detected by Western blotting (Fig. 3C-D). Consequently our results supported that quisinostat did play a role in G0/G1 cell cycle arrest by upregulating expression of p21 as well as downregulating levels of cdk2/cdk4/cdk6/ cyclinD1/cyclinE1/cyclinA2 proteins.
Cell cycle analysis of Quisinostat-treated HCC cells. (A-B) Quisinostat caused a G0/G1 phase arrest. Treatment of HCCLM3 and SMMC-7721 cells with quisinostat (12.5, 25.0 and 50.0nM) for 48h. (C-D) Western blot analysis of p21, cdk2, cdk4, cdk6, cyclinD1, cyclinE1 and cyclinA2 proteins after quisinostat treatment. Data were shown as mean ± SD. n = 3; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with DMSO group.
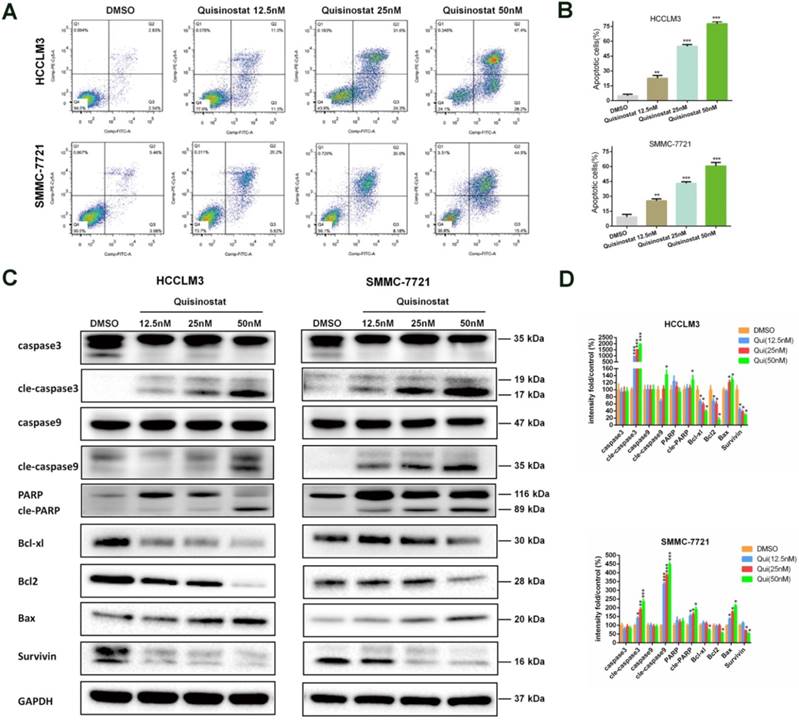
Quisinostat facilitated apoptosis in hepatocellular carcinoma cells
Accordingly apoptosis assay demonstrated that quisinostat could facilitate apoptosis in HCC cells more effectively when compared with DMSO group (Fig. 4A-B). In agreement with the data of apoptosis assay, quisinostat enhanced expression levels of proapoptosis proteins, cleaved-Caspase-3, cleaved-Caspase-9, cleaved-PARP and Bax and decreased levels of antiapoptosis proteins, Bcl-xl, Bcl2 and survivin in HCC cells in contrast to DMSO group (Fig. 4C). Our findings suggested that quisinostat induced apoptotic events both in HCCLM3 and SMMC-7721 cells.
Effects of Quisinostat on apoptosis in HCC cells. (A-B) Treatment of HCCLM3 and SMMC-7721 cells with quisinostat (12.5, 25.0 and 50.0nM) for 48h. FITC and PI staining were applied to evaluate apoptosis facilitated by quisinostat. (C-D) Western blot analysis of Caspase-3, cleaved-Caspase-3, Caspase-9, cleaved-Caspase-9, PARP, cleaved-PARP, Bcl-xl, Bcl2, Bax and survivin protein were carried out after quisinostat treatment. Data were shown as mean ± SD. n = 3; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with DMSO group.
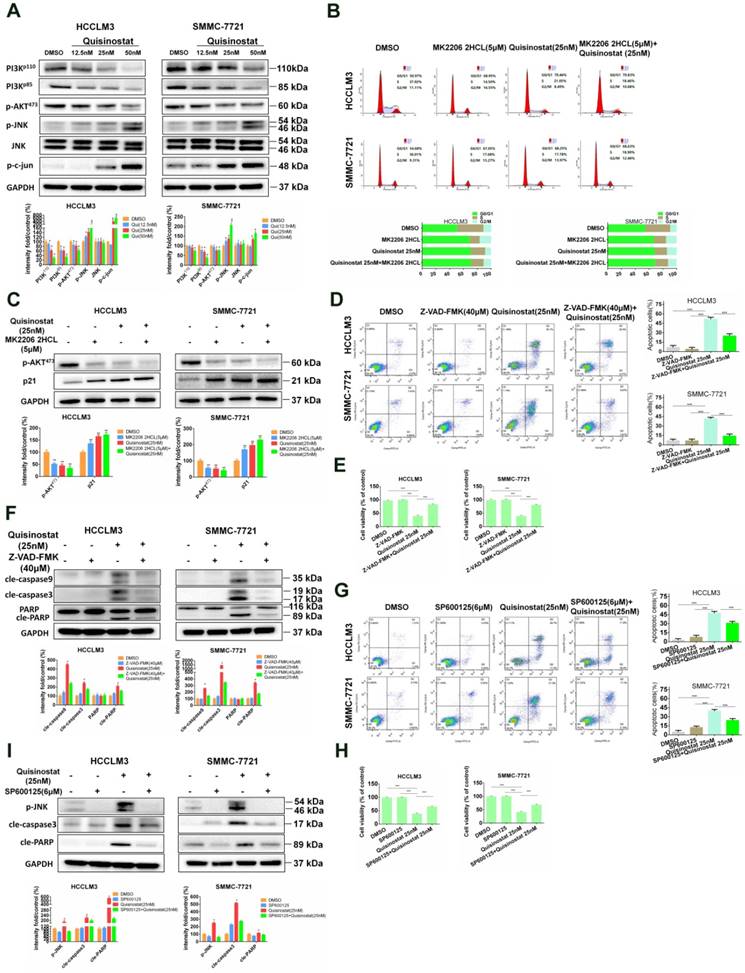
PI3K/AKT and JNK/c-jun in Quisinostat-induced G0/G1 phase arrest and apoptosis
It has been reported that Akt was employed in processes during cell cycle by downregulating p21Cip1[23]. In addition studies also implied that MAPK signaling pathways, especially Erk and JNK, were also engaged in cell proliferation along with apoptosis[24-26]. To analyze the potential molecular mechanism of influences of quisinostat on MAPK signaling pathways, Western blotting was employed to assess expressions of AKT kinase and JNK kinase in HCCLM3 and SMMC-7721 cells. As shown in Figure 5A, PI3K-p110, PI3K-p85 and phosphorylated AKT473 proteins seemed to be downregulated by quisinostat. Meanwhile, quisinostat increased expressions of JNK phosphorylations and c-jun in a dose-dependent manner. To analyse the effect of AKT kinase in cell cycle arrest, we employed MK2206 2HCL, the inhibitor of AKT. We discovered that MK2206 2HCL effectively induced G0/G1 phase arrest in agreement with effect of quisinostat and that combination of quisinostat with MK2206 2HCL did not exhibit a significant statistical difference compared with that of MK2206 2HCL or quisinostat alone (Fig. 5B). In addition MK2206 2HCL could inhibit AKT473 kinase in that expression of phosphorylated AKT473 was remarkably decreased and p21 increased, which was consistent with the effect of quisinostat on AKT kinase (Fig. 5C). These results indicated AKT kinase did play an important role in quisinostat-induced G0/G1 phase arrest. Furthermore, to investigate the role of caspase family in apoptosis, we employed Z-VAD-FMK, the inhibitor of caspase family, and found Z-VAD-FMK restored cell apoptosis and cell proliferation in response to quisinostat, which was supported with Flow cytometric analysis and Western blotting (Fig. 5D-F). Quisinostat remarkably activated cleaved-Caspase3, cleaved-Caspase9 and cleaved-PARP activity that were supressed by Z-VAD-FMK, indicating that Caspase3, Caspase 9, PARP were involved in apoptotic events induced by quisinostat. To explore mechanism of the process, 6 μM SP600125 was introduced to pretreat HCC cells for 5 h, then quisinostat for 48h. We found that SP600125 markedly protected cells from apoptosis stimulated by quisinostat (Fig. 5G-H). Meanwhile it was demonstrated that SP600125 apparently reversed quisinostat-induced activation of JNK phosphorylations, cleaved-Caspase3 and cleaved-PARP, which was evaluated by Western blot analysis (Fig. 5I). Our results suggested that PI3K/AKT and JNK/MAPK pathways could be attributed to the induced G0/G1 cell cycle arrest and apoptosis by quisinostat in hepatocellular carcinoma cells.
PI3K/AKT and JNK/c-Jun pathway facilitated by Quisinostat were required for cell cycle arrest and cell apoptosis. (A) HCCLM3 and SMMC-7721 cells were treated with quisinostat (12.5, 25.0 and 50.0nM) for 48 h. Western blot analysis was used to evaluate expressions of PI3K-p110, PI3K-p85, phosphorylation of AKT473, JNK, phosphorylation of JNK and c-Jun. Data, mean ± SD (n = 3); *, P < 0.05; **, P < 0.01 compared with DMSO group. (B) Cells were preincubated for 5 h with or without MK2206 2HCL(5μM), followed by culturing with 25nM quisinostat for 48 h, then conducting the cell cycle analysis and (C) Expression levels of phosphorylation of AKT473 and p21 proteins were detected by Western Blotting. Data were shown as mean ± SD. n = 3; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with the DMSO group. (D) Cells were pretreated with or without 40μM Z-VAD-FMK for 2 h, followed by culturing with 25nM quisinostat for 48 h to conduct apoptosis analysis, (E) cell proliferation analysis and (F) Expression levels of cle-Caspase9, cle-Caspase3, PARP, and cle-PARP proteins were detected by Western Blotting. Data were shown as mean ± SD. n = 3; *P <0.05; **P < 0.01; ***P < 0.001 compared with Quisinostat group. (G) Cells were pretreated with SP600125 (6μM) for 5 h followed by culturing with 25nM quisinostat for 48 h to conduct apoptosis analysis, (H) cell proliferation analysis and (I) Expression levels of phosphorylation of JNK, cle-Caspase3 and cle-PARP proteins were detected by Western Blotting. Data were shown as mean ± SD. n = 3; *P <0.05; **P < 0.01; ***P < 0.001 compared with Quisinostat group.
Quisinostat and Sorafenib synergistically surpressed proliferation of hepatocellular carcinoma cells via inducing apoptosis
In order to examine antitumor efficiency of combined treatment of sorafenib and quisinostat, a concentration response study was conducted. It was indicated that quisinostat extremely potentiated sorafenib-mediated cytotoxicity in a dose-dependent manner in HCC cells lines (Fig. 6A). Moreover, CompuSyn software was applied to evaluate CI values of each agent, thus to investigate whether quisinostat plus sorafenib act synergistically. Our study demonstrated in HCC cells that the more CI value further inclined to reduce, the higher combined antiproliferative effects obtained, suggesting that there was synergistic effect for combination of quisinostat plus sorafenib (Fig. 6B). The Supplemental Table 1 listed concentrations of quisinostat and sorafenib in each group as well as the combined antiproliferative effect values and Cis, respectively. In summary, we showed that combination therapy of quisinostat plus sorafenib could synergistically reinforce cytotoxicity and antiproliferative efficiency of HCC cells. Next we detected that combination therapy of quisinostat and sorafenib performed an obvious enhancement of apoptotic HCC cells in contrast with single-drug treatment, as shown in Figure 6C. Additionally, in contrast to single-drug alone, the combination strategy increased expressions of JNK phosphorylations, cleaved-PARP, cleaved-Caspase-3 and cleaved-Caspase-9 in HCC cells (Fig. 6D). Consequently it was demonstrated synergistic antiproliferative effects between quisinostat and sorafenib contributed to apoptosis induced by caspase-dependent pathway.
Synergistic interaction between Quisinostat and Sorafenib in HCC cells. (A) A serial of concentrations of sorafenib were employed to treat HCCLM3 and SMMC7721 cells with or without quisinostat for 48 h. Cell proliferation was measured by CCK8 assay. Data were shown as mean ± SD (n = 3); *, P < 0.05 compared with Sorafenib alone. (B) The combination index (CI) values and the fraction affected (Fa) for each dose were applied to create CI plots by CompuSyn software for HCC cells. (C) HCCLM3 and SMMC7721 cells were cultured with indicated agents for 48 h. Apoptosis assay were measured by flow cytometry. Data, mean ± SD (n = 3; Combination vs Quisinostat or combination vs Sorafenib *P<0.05). (D) HCCLM3 and SMMC-7721 cells were cultured with quisinostat and/or sorafenib for 48 h. Cells were analyzed for phosphorylation of JNK, cle-caspase3, cle-caspase9 and cle-PARP by Western blot analysis. Data, mean ± SD (n = 3; Combination vs Quisinostat or combination vs Sorafenib *P<0.05). (E) Cells were preincubated for 5 h with or without SP600125 (6μM), and then treated with quisinostat (12.5 nM) and/or sorafenib(4 μM) for 48 h, followed by apoptosis analysis and (F) Expression levels of apoptosis-related proteins were detected by Western blot analysis. Data, mean ± SD (n = 3; Combination vs Quisinostat or combination vs Sorafenib *P<0.05).
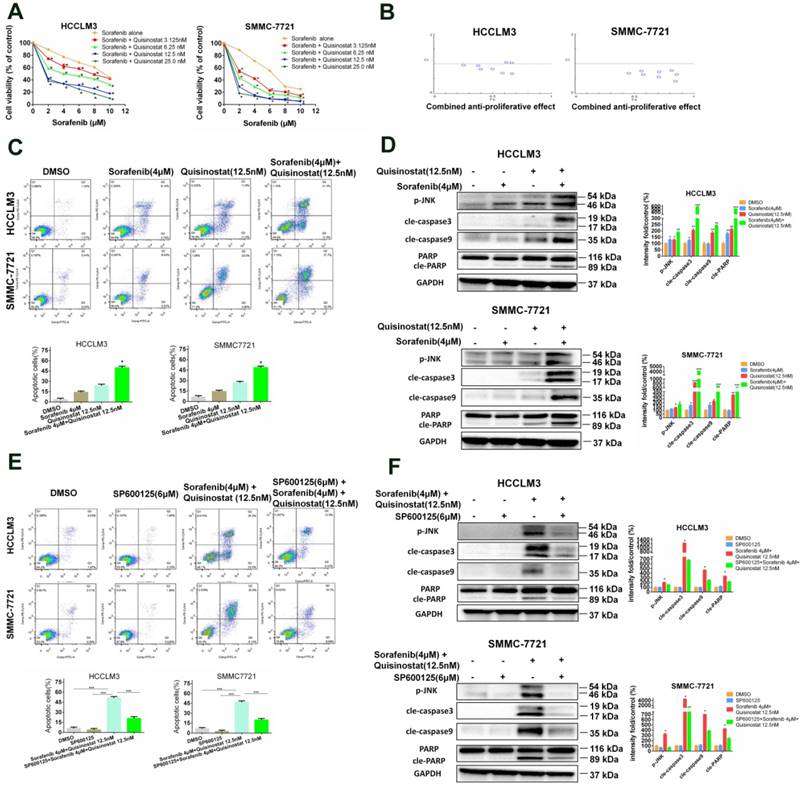
Combination of Quisinostat and Sorafenib potentiated cell apoptosis through JNK pathway
To further investigate potential molecular mechanism, our study devoted to evaluate effects of combination treatment of quisinostat plus sorafenib on JNK kinase activation. We found that SP600125 effectively rescued apoptosis triggered by combination of quisinostat and sorafenib (Fig. 6E). Moreover it was demonstrated that SP600125 remarkably reversed combination of quisinostat and sorafenib induced activation of JNK phosphorylations, cleaved-caspase-3, cleaved-caspase9 and cleaved-PARP evaluated by Western blot analysis (Fig. 6F). Therefore our study confirmed that JNK pathway was involved in combination of quisinostat plus sorafenib to potentiate apoptotic cell death.
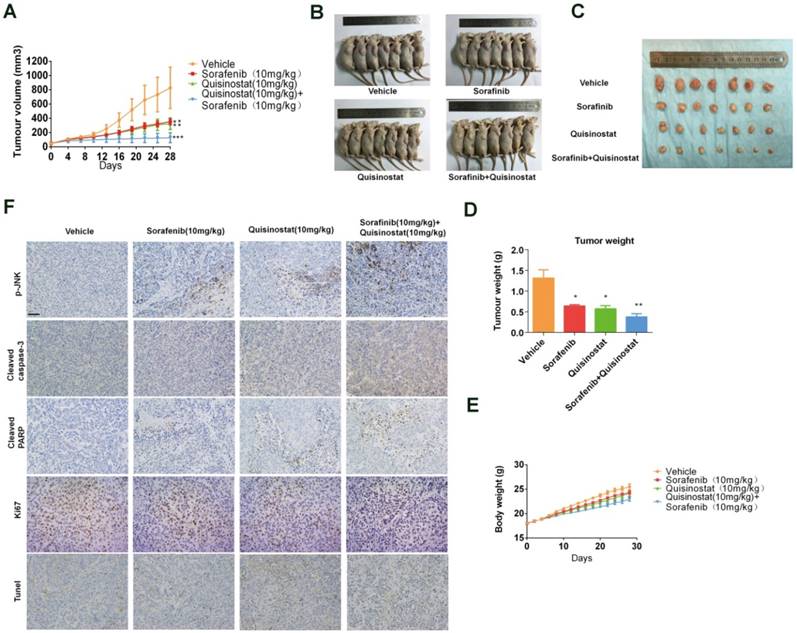
The combination of Quisinostat and Sorafenib inhibited tumor growth in a HCCLM3 xenograft model
We assessed antitumor activity of combination treatment on HCCLM3 xenografts model (Fig. 7). Figure 7A-B indicated that both quisinostat and sorafenib alone markedly repressed tumor growth of HCCLM3 cells in vivo compared with control group. Furthermore combination treatment exhibited enhanced ability to impede tumor growth than each agent alone did. Moreover both of the each drug and combination treatment did not resulted in marked weight loss of the mice, indicating the combination therapy hardly with any toxicity (Fig. 7C). Intratumoral biomarkers were examined by immunohistochemistry assay to explore mechanisms of antitumor effects in vivo. In agreement with the data in vitro, immunohistochemistry of the xenograft tumors suggested that combination treatment significantly induced the upregulation of JNK phosphorylations, cleaved-Caspase3, cleaved-PARP and tunel staining along with the decreased Ki67 expression in tumors, indicating promoted apoptosis in xenograft tumors (Fig. 7D). To sum up, these data demonstrated that combination treatment of quisinostat and sorafenib markedly potentiated antitumor effect on HCCLM3 xenografts.
Antitumor activity of Quisinostat plus Sorafenib in a HCCLM3 xenograft model. Athymic nude mice were subcutaneously implanted with identified pieces of HCCLM3 tumor tissues. Then mice were randomized to four groups (n = 7), intraperitoneally injecting with indicated treatments. (A) Volumes of tumor were detected regularly and calculated. (B) The killed mice bearing the tumors and (C) the excised tumors were shown. (D) Tumor weight were measured regularly and calculated. (E) Body weights were measured once every two days, shown as mean ± SD. (F) IHC analysis of proliferation and apoptosis in HCCLM3 xenograft tumors. Once harvested, tumor tissues were embedded with paraffin and then performed immunostaining for phosphorylation of JNK, cle-caspase3, cle-PARP and Ki67. And apoptotic cells in the livers of mice were detected by the TUNEL assay. Images were photographed with confocal microscope under ×200 magnification. Scale bar, 100 μm. Data were shown as mean ± SD. n = 7. *P <0.05, *P<0.05 compared with control group.
Discussion
HDACs overexpression and/or abnormal hyper-activation have been reported to be involved in mostly malignant characters in HCC cells[27, 28]. Thus HDACis have been poured substantial attention for their considerable potential aiming at epigenetic aberrations employed in the growth and metastasis of cancers [29]. Consequently it was verified that HDACis shared a multitude of antitumor abilities, especially promoting cell cycle arrest, apoptosis, differentiation and senescence along with altered angiogenesis[30, 31]. Cell senescence is recognized as senescent morphology as well as loss of proliferative capability permanently. It has been reported that HDACi sodium butyrate could induce cellular senescence through the p21Waf1 in fibroblast cells[32]. Likewise, HDACis, sodium butyrate (NaB) and panobinostat (LBH589), were also proved to facilitate cell senescence in breast cancer cells owing to upregulation of miR-31 and repression of the polycomb group (PcG) protein BMI1[33]. Moreover cell senescence was evaluated as a natural safeguard against cancer by inhibiting CDK4/6 kinases induced by CDK4/6 inhibitors[34]. Similarly, studies was verified that CDK4/6 inhibitors could function as epigenetic modulators to activate senescence programs in tumor cells[35]. Consistent with these data reported above, in our study, it was observed that quisinostat could significantly reduce the amount of cdk2/cdk4/cdk6 along with elevating p21, indicating inhibition of CDK4/6 kinases and activation of p21 pathway. In summary, we believed that quisinostat could promote cell senescence in which CDK4/6 and p21 pathway may be involved. As an orally available second generation HDACi, quisinostat has implemented the phase 1 clinical evaluation in patients with advanced solid tumors[13], for its specific sensitivity to enzymatic HDAC1 and HDAC2, and broad spectrum activity targeting Class I and Class II isozymes[11].
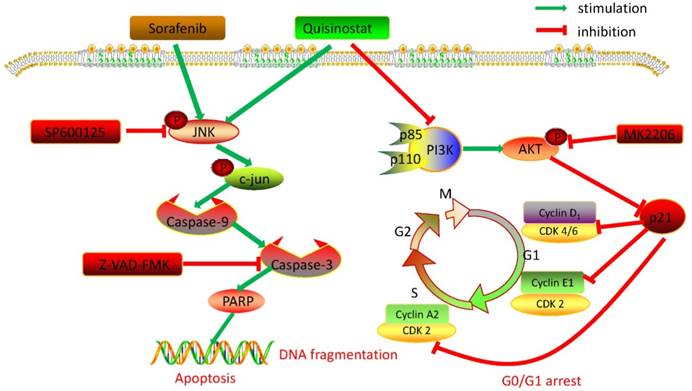
Collectively, we detected expressions of HDACs in 111 paired tumor tissues and peritumor tissues, and found HDAC1, HDAC2 and HDAC4 were overexpressed in HCC tissues. Further statistical analysis manifested that expressions of HDACs were negatively correlated with survival time and overall survival time. Then we demonstrated that quisinostat exhibited remarkable antitumor activity in HCC cells both in vitro and in vivo. Further study showed that quisinostat could regulate cell proliferation, cell cycle arrest and apoptosis via p21 pathway mediated G0/G1 phase arrest and through caspase family induced cell apoptosis. We found that there was a synergistic effect between quisinostat and sorafenib. This combination treatment reduced the dose of both of quisinostat and sorafenib, which would delay the resistance or tolerance in HCC cells thus potentiate antitumor capability in contrast to each single agent. Mechanistically, induction of G0/G1 cell cycle arrest and apoptosis by quisinostat could be attributed to PI3K/AKT/p21 and JNK/c-jun/caspase-3 pathway. Since we found that inhibition of AKT kinase could activate P21 mediated G0/G1 phase arrest and that reperssion of JNK could reverse caspase family regulated apoptosis. Thus, we preliminary proved that quisinostat could impede growth of HCC cells via G0/G1 phase arrest and apoptosis through PI3K/AKT/p21 and JNK/c-jun/caspase-3 signaling pathway (Fig. 8).
Overview of pathways for quisinostat facilitated G0/G1 cell cycle arrest and apoptosis in HCC cells.
It has been previously demonstrated that HDACis exerted their antitumor effects through promoting cell cycle arrest, apoptosis, differentiation and prohibiting tumor vascularization[36]. In our study, cell cycle experiments proved that arrest in G0/G1 phase truly contributed to repression of cell proliferation in a dose-dependent manner, resulting in cell death. Evidences were indicated that during different phases of cell cycle various cyclins could specifically interact with their kinases, CDKs, which, however could be downregulated by p27Kip1 and p21Cip1[37]. In addition studies have demonstrated characterized functions of cdk4/cyclinD1 and cdk2/cyclinE1/cyclinA2 complexes could promote G1 or G1-to-S transition, respectively[38, 39]. Furthermore it has been reported that inhibiting the cdk2/cyclinE1/cyclinA2 complex could lengthen G1 phase since its characterized capability was to control cell cycle progression[40]. Accordingly, in our study we found that quisinostat could significantly reduce the amount of cdk2/cdk4/cdk6 along with elevating expression of p21. In parallel with data reported before we observed that quisinostat significantly reduced expression of cdk2/cyclinE1/cyclinA2 and cdk4/cyclinD1, implying that quisinostat functioned its antitumor effects through cell cycle arrest.
It was proved HDACis played a favorable role in the process relating to cell proliferation and apoptosis in tumor diseases[41, 42]. It has been noted that HDACis could increase sensitivity toward apoptosis induced by TRAIL in cancer cells through intrinsic (mitochondrial) pathway[43]. Studies also showed that HDACis could enhance TRAIL-induced apoptosis by facilitating caspase-9 activation in contrast to trichostatin A and valproic[44]. It was identified that considerable changes were observed in expressions of pro- and antiapoptotic proteins, such as elevated levels of Bax and decreased expression of Bcl-xl[44, 45]. In addition HDACis were reported to inhibit expression levels of antiapoptotic Bcl-2 family proteins, such as Bcl-2 and Bcl-xl, to induce mitochondrial apoptosis[46, 47]. Furthermore, HDACis also could enhance expressions of proapoptotic Bcl-2 family proteins, including Bax, some BH3 proteins and Bak[46, 48]. Accordingly, we suggested that quisinostat facilitated a pleasurable enhancement in apoptosis by a dose-dependent manner through activating caspase family, including PARP, Caspase-3 and Caspase-9. Meanwhile, activation of Bax and suppression of Bcl-xl, Bcl2 and survivin also declared the apoptosis induced by quisinostat.
It is well acknowledged that various environmental singals, such as UV and inflammatory cytokines, could activate JNK substantially and predominantly[49]. Once activated JNK would transfer into the nucleus thus conferring transcription effects on expression of c-Jun[50]. JNK/c- jun pathway was employed in the apoptosis process[51]. Data presented here indicated that JNK/c- jun pathway was attributed to apoptosis induced by quisinostat in HCC cells, which would be reversed by inhibition of JNK/c- jun pathway (Fig. 5), suggesting that JNK pathway was engaged in apoptosis mediated by quisinostat. Furthermore JNK/c- jun pathway was also proved to be activated by sorafenib, its mechanism, however, still remained unclear[52]. Here our data showed that there was synergistic effect for the combination of quisinostat and sorafenib to stimulate JNK/c- jun pathway and apoptosis in HCC cells, which could be reversed by inhibiting JNK/c- jun pathway (Fig. 6). Our study elucidated that JNK/c- jun pathway did play a role in the synergistic apoptotic effect of quisinostat and sorafenib. Nevertheless further studies are needed to determine the importance of JNK/c- jun via overexpression or knockdown experiments by siRNA.
It was reported that PI3K/AKT pathway mediated several cellular functions, especially cell proliferation, cell cycle, apoptosis as well as angiogenesis [53, 54]. Once the signals, such as growth factors, cytokines and antigens, combined with the tyrosine kinase, PI3K was activated thereby in turn facilitating AKT kinase to regulate a serial of cellular functions[55]. In our study we found quisinostat arrested cell cycle at G0/G1 by upregulating p21 and downregulating AKT473 phosphorylation. Meanwhile we discovered inhibition effect of combination of quisinostat and MK2206 2HCL did not exhibit a marked statistical difference in contrast to that of MK2206 2HCL or quisinostat alone, implying inhibition of Akt kinase might contribute to cycle arrest induced by quisinostat. Besides quisinostat inhibited AKT473 phosphorylation, along with decreased expressions of PI3Kp110 and PI3Kp85, revealing that PI3K/AKT did employed in upregulation of p21 and downregulation of cdk2/cyclinE1/cyclinA2 and cdk4/cyclinD1, leading to G0/G1 arrest.
As mentioned above, our observations suggested that quisinostat had consistent antitumor efficacy in HCC cells through G0/G1 arrest and apoptosis, which was further potentiated when combined with sorafenib. Mechanistically, we demonstrated that quisinostat targeted PI3K/AKT/p21 pathway to induce cell cycle arrest, and JNK/c-jun/caspase-3 pathway to promote apoptosis. Additionally combination of quisinostat and sorafenib showed more effective antitumor ability against tumor growth of HCC than each agent alone did in vivo. Thus our data suggested it could be a promising therapy to combine quisinostat with sorafenib for patients with advanced hepatocellular carcinoma. Therefore, our findings provided evidences that quisinostat would be a potential chemotherapeutic drug against HCC and even in combination with sorafenib to enhance its antitumor capability, verifying the possibility for this combination treatment in the future.
Abbreviations
HDACs: Histone deacetylases; HDACi: Histone deacetylases inhibitor; cdk2: Cyclin-dependent kinase-2; cdk4: Cyclin-dependent kinase-4; cdk6: Cyclin-dependent kinase-6; JNK: c-Jun N-terminal kinase; MAPK: Mitogen-activated protein kinase; PI3K: Phosphoinositide 3-kinase.
Supplementary Material
Supplementary table 1.
Acknowledgements
This research was financially supported by Innovative Research Groups of National Natural Science Foundation of China (No.81721091), Major program of National Natural Science Foundation of China (No.91542205), National S&T Major Project (No.2017ZX10203205) and Zhejiang International Science and Technology Cooperation Project (No.2016C04003).
Authors' contributions
HB and DLF carried out the molecular studies, ZXQ drafted the manuscript. CDY and WJB carried out the animal experiments and sample collection. FXD and ZL carried out IHC. ZYP and XHY participated in statistical analysis. WJ and ZSS conceived of the study and participated in its design and coordination. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study has been approved by the authors' institutional review board. All processes involving animals and patients were approved by the ethics committee of the First Affiliated Hospital of Zhejiang University. Animal experiments were performed according to the guidelines of the Experimental Animals Management Committee. Written informed consent was obtained from all subjects.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International journal of cancer. 2010;127:2893-917
2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61:69-90
3. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M. et al. Hepatocellular carcinoma. Nature Reviews Disease Primers. 2016;2:16018
4. Chen CH, Chen MC, Wang JC, Tsai AC, Chen CS, Liou JP. et al. Synergistic interaction between the HDAC inhibitor, MPT0E028, and sorafenib in liver cancer cells in vitro and in vivo. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:1274-87
5. Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:3958-69
6. Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Advances in cancer research. 2004;91:137-68
7. Mann DA. Epigenetics in liver disease. Hepatology (Baltimore, Md). 2014;60:1418-25
8. Buurman R, Gurlevik E, Schaffer V, Eilers M, Sandbothe M, Kreipe H. et al. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology. 2012;143(e15):811-20
9. Liu Y, Zhang JB, Qin Y, Wang W, Wei L, Teng Y. et al. PROX1 promotes hepatocellular carcinoma metastasis by way of up-regulating hypoxia-inducible factor 1alpha expression and protein stability. Hepatology (Baltimore, Md). 2013;58:692-705
10. Yuan JH, Yang F, Chen BF, Lu Z, Huo XS, Zhou WP. et al. The histone deacetylase 4/SP1/microrna-200a regulatory network contributes to aberrant histone acetylation in hepatocellular carcinoma. Hepatology (Baltimore, Md). 2011;54:2025-35
11. Arts J, King P, Mariën A, Floren W, Beliën A, Janssen L. et al. JNJ-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clinical Cancer Research. 2009;15:6841-51
12. Lee J-H, Choy M, Ngo L, Foster S, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proceedings of the National Academy of Sciences. 2010;107:14639-44
13. Venugopal B, Baird R, Kristeleit RS, Plummer R, Cowan R, Stewart A. et al. A phase I study of quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:4262-72
14. Josep M. Llovet MD, Sergio Ricci MD, Vincenzo Mazzaferro MD, Philip Hilgard MD, Edward Gane MD, Jean-Frédéric Blanc MD, et al. Sorafenib in Advanced Hepatocellular Carcinoma. N Engl J Med. 2008;359:378-90
15. Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140:1410-26
16. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69-80
17. Yuan H, Li AJ, Ma SL, Cui LJ, Wu B, Yin L. et al. Inhibition of autophagy signi fi cantly enhances combination therapy with sorafenib and HDAC inhibitors for human hepatoma cells. World journal of gastroenterology. 2014;20:4953-62
18. Cervello M, McCubrey JA, Cusimano A, Lampiasi N, Azzolina A, Montalto G. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget. 2012;3:236-60
19. Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A. et al. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. Journal of hepatology. 2012;56:1343-50
20. Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP. et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clinical cancer research. 2008;14:5385-99
21. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews. 2006;58:621-81
22. Lai CY, Tsai AC, Chen MC, Chang LH, Sun HL, Chang YL. et al. Aciculatin induces p53-dependent apoptosis via MDM2 depletion in human cancer cells in vitro and in vivo. PloS one. 2012;7:e42192
23. Jain MV, Jangamreddy JR, Grabarek J, Schweizer F, Klonisch T, Cieslar-Pobuda A. et al. Nuclear localized Akt enhances breast cancer stem-like cells through counter-regulation of p21(Waf1/Cip1) and p27(kip1). Cell Cycle. 2015;14:2109-20
24. Wu G-J, Pen J, Huang Y, An S, Liu Y, Yang Y. et al. KAP1 inhibits the Raf-MEK-ERK pathway to promote tumorigenesis in A549 lung cancer cells. Molecular carcinogenesis. 2018
25. Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307-20
26. Wagner EF, Nebreda ÁR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nature Reviews Cancer. 2009;9:537-49
27. Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nature reviews Cancer. 2001;1:194-202
28. Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nature reviews Drug discovery. 2013;12:917-30
29. Carol H, Gorlick R, Kolb EA, Morton CL, Manesh DM, Keir ST. et al. Initial testing (stage 1) of the histone deacetylase inhibitor, quisinostat (JNJ-26481585), by the Pediatric Preclinical Testing Program. Pediatric blood & cancer. 2014;61:245-52
30. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature reviews Drug discovery. 2006;5:769-84
31. Newbold A, Falkenberg KJ, Prince HM, Johnstone RW. How do tumor cells respond to HDAC inhibition? FEBS J. 2016;283:4032-46
32. Romanov VS, Abramova MV, Svetlikova SB, Bykova TV, Zubova SG, Aksenov ND. et al. p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle. 2010;9:3945-55
33. Cho J-H, Dimri M, Dimri GP. MicroRNA-31 Is a Transcriptional Target of Histone Deacetylase Inhibitors and a Regulator of Cellular Senescence. Journal of Biological Chemistry. 2015;290:10555-67
34. Miettinen TP, Peltier J, Hartlova A, Gierlinski M, Jansen VM, Trost M. et al. Thermal proteome profiling of breast cancer cells reveals proteasomal activation by CDK4/6 inhibitor palbociclib. EMBO J. 2018:37
35. Acevedo M, Vernier M, Mignacca L, Lessard F, Huot G, Moiseeva O. et al. A CDK4/6-Dependent Epigenetic Mechanism Protects Cancer Cells from PML-induced Senescence. Cancer research. 2016;76:3252-64
36. Witt O, Lindemann R. HDAC inhibitors: magic bullets, dirty drugs or just another targeted therapy. Cancer letters. 2009;280:123-4
37. Marques-Torrejon MA, Porlan E, Banito A, Gomez-Ibarlucea E, Lopez-Contreras AJ, Fernandez-Capetillo O. et al. Cyclin-dependent kinase inhibitor p21 controls adult neural stem cell expansion by regulating Sox2 gene expression. Cell stem cell. 2013;12:88-100
38. Ekholm SV, Reed SI. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Current Opinion in Cell Biology. 2000;12:676
39. J. SC. G1 phase progression: Cycling on cue. Cell. 1994;79:551-5
40. Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell stem cell. 2009;5:320-31
41. Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nature Biotechnology. 2007;25:84
42. Richon VM. Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. British Journal of Cancer. 2006;95:S2-S6
43. Fulda S. Histone deacetylase (HDAC) inhibitors and regulation of TRAIL-induced apoptosis. Experimental cell research. 2012;318:1208-12
44. Reddy RM, Yeow WS, Chua A, Nguyen DM, Baras A, Ziauddin MF. et al. Rapid and profound potentiation of Apo2L/TRAIL-mediated cytotoxicity and apoptosis in thoracic cancer cells by the histone deacetylase inhibitor Trichostatin A: the essential role of the mitochondria-mediated caspase activation cascade. Apoptosis: an international journal on programmed cell death. 2007;12:55-71
45. Ziauddin MF, Yeow WS, Maxhimer JB, Baras A, Chua A, Reddy RM. et al. Valproic acid, an antiepileptic drug with histone deacetylase inhibitory activity, potentiates the cytotoxic effect of Apo2L/TRAIL on cultured thoracic cancer cells through mitochondria-dependent caspase activation. Neoplasia (New York, NY). 2006;8:446-57
46. Zhang XD, Gillespie SK, Borrow JM, Hersey P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Molecular cancer therapeutics. 2004;3:425-35
47. Muhlethaler-Mottet A, Flahaut M, Bourloud KB, Auderset K, Meier R, Joseph JM. et al. Histone deacetylase inhibitors strongly sensitise neuroblastoma cells to TRAIL-induced apoptosis by a caspases-dependent increase of the pro- to anti-apoptotic proteins ratio. BMC cancer. 2006;6:214
48. Gillespie S, Borrow J, Zhang XD, Hersey P. Bim plays a crucial role in synergistic induction of apoptosis by the histone deacetylase inhibitor SBHA and TRAIL in melanoma cells. Apoptosis: an international journal on programmed cell death. 2006;11:2251-65
49. El Fajoui Z, Toscano F, Jacquemin G, Abello J, Scoazec JY, Micheau O. et al. Oxaliplatin sensitizes human colon cancer cells to TRAIL through JNK-dependent phosphorylation of Bcl-xL. Gastroenterology. 2011;141:663-73
50. Basu S, Rajakaruna S, Reyes B, Van Bockstaele E, Menko AS. Suppression of MAPK/JNK-MTORC1 signaling leads to premature loss of organelles and nuclei by autophagy during terminal differentiation of lens fiber cells. Autophagy. 2014;10:1193-211
51. Lin S, Hoffmann K, Gao C, Petrulionis M, Herr I, Schemmer P. Melatonin promotes sorafenib-induced apoptosis through synergistic activation of JNK/c-jun pathway in human hepatocellular carcinoma. Journal of Pineal Research. 2017;62:e12398
52. Ou DL, Shen YC, Yu SL, Chen KF, Yeh PY, Fan HH. et al. Induction of DNA Damage-Inducible Gene GADD45 Contributes to Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Cells. Cancer research. 2010;70:9309-18
53. Ge P, Cui Y, Liu F, Luan J, Zhou X, Han J. L-carnitine affects osteoblast differentiation in NIH3T3 fibroblasts by the IGF-1/PI3K/Akt signalling pathway. Bioscience trends. 2015;9:42-8
54. Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:2278-87
55. Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. The Biochemical journal. 2000;346(Pt 3):561-76
Author contact
![]() Corresponding authors: Jian Wu, E-mail: drwujianedu.cn; Shusen Zheng, E-mail: shusenzhengedu.cn. Address reprint requests to Jian Wu and Shusen Zheng, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University,79 QingChun Road, Hangzhou 310003, China.
Corresponding authors: Jian Wu, E-mail: drwujianedu.cn; Shusen Zheng, E-mail: shusenzhengedu.cn. Address reprint requests to Jian Wu and Shusen Zheng, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University,79 QingChun Road, Hangzhou 310003, China.