Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Functions of the Major...
The Effects of Stromal Cells on...
Conclusion and Future Aspects
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(14):2083-2093. doi:10.7150/ijbs.25720 This issue Cite
Review
Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation
Sen Guo, Chu-Xia Deng ![]()
Faculty of Health Sciences, University of Macau, Macau SAR, China
Received 2018-2-25; Accepted 2018-6-2; Published 2018-11-13
Abstract

The cellular environment where tumor cells reside is called the tumor microenvironment (TME), which consists of borders, blood vessels, lymph vessels, extracellular matrix (ECM), stromal cells, immune/inflammatory cells, secreted proteins, RNAs and small organelles. By dynamically interacting with tumor cells, stromal cells participate in all stages of tumor initiation, progression, metastasis, recurrence and drug response, and consequently, affect the fate of patients. During the processes of tumor evolution and metastasis initiation, stromal cells in TME also experience some changes and play roles in both the suppression and promotion of metastasis, while the overall function of stromal cells is beneficial for cancer cell survival and movement. In this review, we examine the effects of stromal cells in TME on metastasis initiation, including angiogenesis, epithelial-mesenchymal transition (EMT) and invasion. We also highlight functions of proteins, RNAs and small organelles secreted by stromal cells in their influences on multiple stages of tumor metastasis.
Keywords: TME, Stromal cells, Metastasis Initiation, Breast Cancer
Introduction
Tumor mass is very heterogeneous and resembles a complicated organ more than a simple accumulation of cells. The environment in which the tumor exists is called the tumor microenvironment (TME) and is composed of blood vessels, lymph vessels, ECM, stromal cells, immune/inflammatory cells, secreted proteins, RNAs and small organelles (Figure 1A) [1]. TME plays indispensable roles in tumor initiation, progression, metastasis, recurrence, and drug resistance.
Cells and Molecules (Grey) in TME. (A) TME and cells in TME: CAFs, MSCs, TAMs, Lymphocytes, Endothelial cells, Pericytes, Tumor cells in epithelial status and mesenchymal status. (B) The roles of CAFs in TME: Secret cytokines to affect tumor cells' fates; Remodel ECM; Immunosuppression. (C) The roles of MSCs in TME: Differentiate into other cell types; Secrete cytokines or miRNAs directly, or through exosomes; Transfer organelles through nanotubes; Are recruited by tumor cells. (D) The roles of TAMs in TME: Are recruited by other cells; Secrete cytokines or inflammatory signals to affect tumor cells' fate; Remodel ECM; Immunosuppression. (E) ECM in TME: ECM have many molecules and is remodeled by CAFs, MSCs and TAMs, while it affects the fates of tumors through integrins and other molecules.
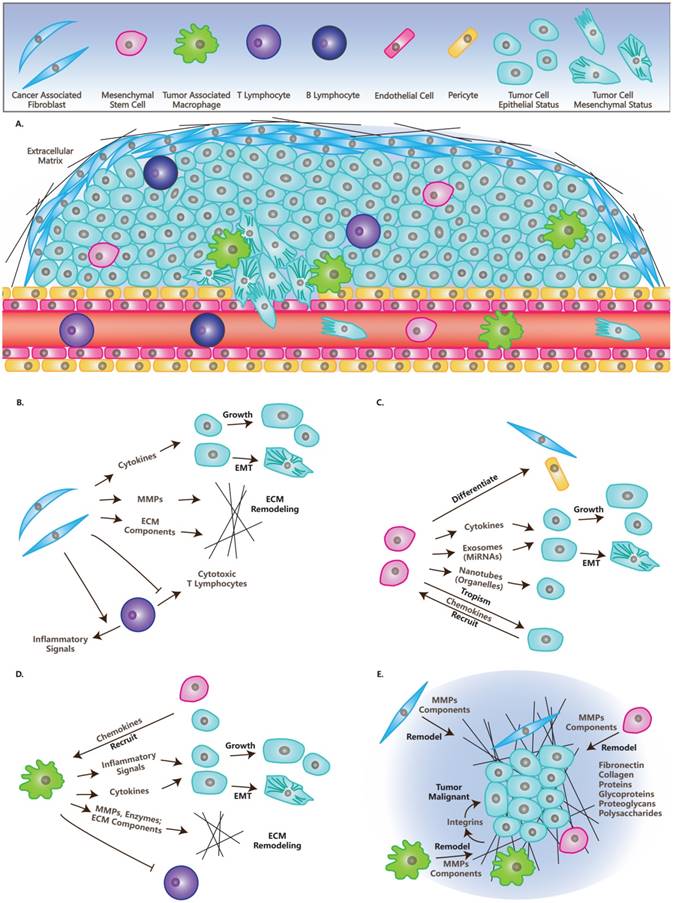
Metastasis can be separated into processes of initiation, progression and virulence according to the categories of metastatic genes. Initiation of metastasis mainly includes the processes that occur in preparation for malignant cells to invade and circulate into vessels in TME. Those processes are angiogenesis, epithelial-mesenchymal transformation (EMT) and invasion/intravasation [2].
Angiogenesis is essential for tumor and stromal cells to absorb nutrients and exchange air, and it provides a tunnel for cells to move [3]. Through EMT, tumor cells ordinarily become more stem-like, aggressive, invasive and have stronger resistance to multiple chemical therapies [4]. Invasion enables tumor cells to intravasate into the circulatory system and makes it possible to colonize at distant location after circulation [5]. The process of intravasation is essential for tumor cells to become circulating [5]. In this article, we review the effects of stromal cells in TME on metastasis initiation.
Functions of the Major Components in Tumor Microenvironment
Stromal Cells in TME
In a tumor, non-transformed cells, which include fibroblasts, mesenchymal stem cells, macrophages, lymphocytes, endothelial cells, and pericytes, participate in tumor progression and regression [1]. The targets that those cells have effects on and the mechanisms they are functioning through are summarized in table 1.
The Effects of Stromal Cells on Tumor in TME.
| Cell Types | Mechanisms | Targets | Effects on Tumor | References | |
|---|---|---|---|---|---|
| CAFs | Secretion of Cytokines and Other Factors | Tumor Cells | Drug-Resistance, Proliferation, Metastasis | [6, 55, 107] | |
| Endothelial Cells | Promote Angiogenesis | ||||
| Secretion of ECM proteases and components | ECM | Promote ECM Remodeling | |||
| Suppression of Immune Activities | Cytotoxic T Lymphocytes | Function in Immuno-Suppression | |||
| Recruitment of T Lymphocytes | Progression Promoting T Lymphocytes | Promote Cancer Progression | |||
| Formation of Tumor Barriers | Tumor Cells | Provide Protection | |||
| MSCs | Differentiation | Fibroblast and Vascular Pericytes | Form Fibrovascular Network | [12-14] | |
| Other Stromal Cells | Maintain TME | ||||
| Secretion of Cytokines | Tumor Cells | Depends on Conditions | |||
| Transfer of Organelles Through Nanotubes | Tumor Cells and Stromal Cells | ||||
| Transfer of Proteins Through Exosomes | Tumor Cells and Stromal Cells | ||||
| Tumor Tropism | Tumor Cells | Promote Factors Delivery, TME Formation | |||
| TAMs | Immune Suppression | Cytotoxic Immune Cells | Promote Cancer Progression | [5, 15, 17, 74, 108, 109] | |
| Secretion of Cytokines (Including Inflammatory Factors) | Tumor Cells and Endothelial Cells | Promote Angiogenesis, EMT, Invasion, and Intravasation | |||
| Tropism Leading | Tumor Cells | Promote Intravasation | |||
| Secret ECM proteases and components | ECM | Promote ECM Remodeling | |||
| T Lympho-cytes | CD8+ | Cytotoxic T Lymphocytes | Tumor Cells | Kill Tumor Cells | [110-115] |
| CD4+ (Th1/Th2) | Secrection of Heterogeneous Cytokines | Lymphocytes (Mainly CD8+ T) | Active Antitumor Immunity | ||
| Th17 | Secrection of IL-17 Family Members | Lymphocytes and tumor cells | Regulate Antitumor Immunity and Angiogenesis | ||
| Treg | Suppression of Excessive Immune Activities | Lymphocytes (Mainly CD8+ T) | Mainly Suppress Antitumor Immunity | ||
| B Lymphocytes | Secretion of Antibodies | Other Lymphocytes and Tumor Cells | Active T and NK Cells; Kill Tumor Cells | [116-119] | |
| Suppression of Immune Activities | T Lymphocytes and NKs | Function in Immuno-Suppression | |||
| Secrection of IL-10 | T Lymphocytes and Tumor Cells | Convert T into Treg; Regulate Proliferation and Metastasis of Tumor Cells | |||
| Endothelial Cells | Line Vasculatures | Blood Vessels | Promote Angiogenesis | [18, 120-122] | |
| Diameter Extending | Blood Vessels | Promote Extravasation | |||
| Abnormal Growth | Blood Vessels | Produce Hypoxia in TME, Regulate Proliferation and Therapy Resistance of Tumor Cells | |||
| ECM Remodeling | ECM | promote ECM Remodeling | |||
| Immune Responses Altering (Lymphatic Vessels) | Immune System | Promote Lymphangiogenesis and cancer progression | |||
| Pericytes | Low Coverage around Vasculatures | Vasculatures | Promote Metastasis | [19, 123] | |
Fibroblasts are the predominant cell type in TME and are associated with all stages of the cancer. These activated fibroblasts in tumors are called myo-fibroblasts or Cancer-Associated-Fibroblasts (CAFs). CAFs enhance tumorigenesis, angiogenesis and metastasis by secreting growth factors and cytokines and promoting TME remodeling through the secretion of Matrix Metalloproteinases (MMPs), ECM components and other enzymes (Table 1) [6]. For immune activity, CAFs suppress the activity of cytotoxic T lymphocytes and recruit lymphocytes that produce inflammatory signals to promote cancer progression (Table 1). It has been reported that CAFs can reconstitute anti-metastatic TME into a pro-metastatic TME [7]. This might be due to the function of stroma derived factors. Fibrinogen-like protein 2 in TME activates CAFs and causes them to become pro-tumorigenic, which promotes the accumulation of myeloid-derived suppressor cells (MDSCs) through the secretion of CXCL12. This then affects tumorigenesis and cancer progression [8]. Additionally, CAFs facilitate resistance of anti-cancer drugs or therapies and provide protection or pro-proliferation factors in cancer cells (Figure 1B) [6]. Currently, more genes in fibroblasts inside TME have been shown to be potential marks for the cancer progression, like caveolin-1 [9], which can be developed as therapy targets.
Mesenchymal Stem Cells (MSCs) are a kind of multipotent cell that can differentiate into multiple mesenchymal cell lineages, including multipotent stromal cells or mesenchymal stromal cells [10]. MSCs have been reported to cure injured cells or tissues by differentiating into cells of the same type with injured cells. They interact with tumor cells via the secretion of growth factors or cytokines and by transferring mitochondria or microRNAs through tunneling nanotubes or exosomes (Table 1) [10]. MSCs are tumor tropisms and attract the attention of different groups to explore specific delivery vehicles for tumor therapy [11, 12]. Residing in tumors, MSCs form a fibrovascular network by differentiating into CAFs and vascular pericytes [13]. While the function of MSCs in cancer progression is diverse under different conditions, any changes can switch the functions of MSCs from tumor promotion into suppression (Figure 1C) [14].
Macrophages play important roles in inflammation, immunology, development and wound healing [15]. They can also be recruited to TME by tumor cells or MSCs through the secretion of specific factors, then they become polarized and are referred to as Tumor-Associated Macrophages (TAMs) [16]. TAMs suppress immune activity and promote cancer progression, which is similar to their functions in wound healing (Table 1) [17]. The balance of the inflammation significantly influences the effects TAMs have on tumors [15]. Also, macrophages remodel ECM by organizing ECM and secreting ECM components and enzymes (Figure 1D) [17].
In this article, we focus on three types of stromal cells including CAFs, MSCs and TAMs. Additional types of non-transformed cells in TME are summarized in Table 1. Lymphocytes have paradoxical functions in TME, although T cells and B cells can be found in invasive tumor margins in draining lymphoid organs or lymphoid structures adjacent to TME [1]. Endothelial cells in TME compose vasculatures and lymphatic vessels that branch to the ends of the tumor, which is needed for tumor growth and metastasis [18]. Similarly, pericytes or perivascular stromal cells function in structures in the support of blood vessels (Table 1) [19].
ECM in TME
ECM functions as a scaffold for the cells in TME and plays a dynamic role in cancer progression, especially as an essential regulator of invasive processes [1]. ECM is a complicated system of proteins, glycoproteins, proteoglycans and polysaccharides, which contains multiple growth factors and builds a tight interaction with cells in TME [20]. In healthy tissue, ECM plays a tumor suppressing role, while it becomes abnormal in tumors and plays a tumor promoting role [21]. The components functioning in tumor promoting roles in ECM are induced in TME [21, 22] and they influence tumor cells through interacting with integrins (Figure 1E) [23].
Recently, NK cells are reported to control metastasis formation through influencing tumor architecture by regulating the secretion of FN1, an important ECM component [24]. This highlights the function of interactions between stromal cells and ECM in TME.
Secreted Factors in TME
As summarized in table 2, secreted factors in TME function as signals between cells, or tools in ECM remodeling, including cytokines, integrins, proteases and microRNAs [25].
The Effects of Secreted Factors on Tumor Cells in TME.
| Factors | Classifications (Examples) | Mechanisms | Targets | Effects on Tumor | References | |
|---|---|---|---|---|---|---|
| Cytokines | Inflammatory Factor | Tumor Necrosis Factor (TNF-α) | Induce NF-κB, TGF-β and Apoptosis | Tumor and Stromal Cells | Regulate Growth, Angiogenesis, and Invasion | [124] |
| Interleukin (IL-6) | Induce JAK/STAT Pathway | Tumor and Endothelial Cells | Promote EMT, Proliferation and Inhibit Apoptosis | [125] | ||
| Growth Factor | TGF-β | Immune and Inflammatory Suppression, EMT Induction and Regulation of other Growth Factors | Tumor Cells and Stromal Cells | Early Stage: Tumor Suppressor; Late Stage: Promote Invasion and Metastasis | [56, 78, 126] | |
| VEGF | Promote the Migration and Proliferation of Endothelial Cells | Endothelial Cells | Promote EMT, Angiogenesis, and Intravasation | [127-129] | ||
| EGF | Promote Growth, Recruit Tumor Cells, Induce EMT TFs | Tumor Cells and Stromal Cells | Promote Proliferation, EMT and Intravasation | [128, 129] | ||
| FGF | Promote Proliferation, Angiogenesis | Endothelial Cells | Promote EMT, and Angiogenesis | [127, 128, 130] | ||
| PDGF | Induce EMT TFs, Stimulate VEGF and FGF while Stimulate Tsp-1 | Tumor Cells, Fibroblasts, Endothelial Cells and Pericytes | Affect Intravasation and Immune Surveillance; Promote Angiogenesis, Fibroblast recruitment, Tumor growth and Metastasis | [131, 132] | ||
| HGF | Induce EMT TFs | Tumor Cells | Promote EMT | [55] | ||
| Chemokines (CXCL12 and CXCR4) | Chemotaxis; Recruit Stromal Cells or Tumor Cells in TME | Tumor Cells and Stromal Cells | Influence Growth, Proliferation and Migration of Tumor Cells and Stromal Cells | [133, 134] | ||
| Integrins | Noncovalently Linked α and β Subunits (like α4β1 and αvβ3, α5β1) | Regulate Cytoskeleton; Cell-Cell/Cell-Matrix Signal Transferring; Maintaining of Cell-Matrix Adhesions; Tissue Remodeling | Cell-Cell and Cell-Matrix Contact in TME | Mostly Promote Angiogenesis, Intravasation and Metastasis | [28, 83, 135] | |
| Proteases | Matrix Metalloproteinases (MMPs) | Degrade ECM, Basement Membrane and Cell-Cell/Cell-Matrix Junctions; Release Factors from ECM | ECM, and Junctions | Promote Invasion, Metastasis, Growth and Angiogenesis while Regulate Apoptosis | [33, 34, 59] | |
| urokinase-type Plasminogen Activator (uPA) | Activate Plasminogen into Plasmin | Endothelial Cells | Promote Intravasation | [5, 83] | ||
Cytokines are kind of small proteins that have low molecular weights, which mediate the communication between cells [26]. Cytokines in TME, including tumor necrosis factor, interleukins, growth factors and chemokines, regulate the progression of cancer and determine the fate of stromal cells. This is depended on the balance of their pro- and anti-functions in inflammation, proliferation, tumorigenesis, migration and apoptosis (Table 2) [27].
Integrins are integral membrane proteins and a large family of cell surface receptors [28]. Integrins are essential in the signaling and transfer of information between cells or between a cell and the ECM. They are also essential in maintaining cell-matrix adhesions. Irregular cell-cell adhesions, caused by disrupted integrins, are signs of cancer (Table 2). In TME, the expressions of metastasis-promoting integrins are enhanced, while those suppressing proliferation, survival and migration are repressed [28]. Recently, some groups are focusing on exosome derived integrins as the researches on exosome are increasing [29]. Also, integrins are reported to affect internal activities of tumor, like nuclear alteration [30].
Extracellular proteolysis plays an essential role in tissue homeostasis and TME [31, 32]. Among these proteinases that mediate the proteolysis of ECM, matrix metalloproteinases (MMPs) have the closest relationship with cancer progression (Table 2) [33]. It is already known that ECM degradation that is mediated by MMPs promotes the invasion and metastasis of cancer [33]. Furthermore, recent research indicates that MMPs promote tumor growth, angiogenesis and regulate apoptosis and some MMPs function in tumor suppression (Table 2). Thus, MMPs are also a family of proteins with paradoxical role in TME [34].
MicroRNAs are endogenous, small non-coding RNAs, each of which has 18-24 nucleotides. They negatively regulate target mRNAs, post-transcriptionally by disrupting transcription or translation [35]. MicroRNAs participate in several pathways and function in the regulation of different components of TME [36]. Besides microRNA, lncRNA or long non-coding RNA is also a potent participant that is secreted in TME. Some lncRNAs function in the communication between TME and stromal cells, like transforming fibroblasts to be tumor-promoting [37].
The Effects of Stromal Cells on Metastasis Initiation
The Effects of Stromal Cells on Angiogenesis
Angiogenesis plays an initial role in the process of metastasis, which provides a way for aggressive tumor cells to leave primary tumors and move to distant metastatic sites. In the process of angiogenesis, stromal cells in TME play promoting roles. We introduce these regulatory pathways involving stromal cells in the following section [38].
Stromal cells affect the fate of TME through the secretions of these angiogenesis-promoting factors and angiogenesis-inhibiting factors. Among these factors, VEGF is the most potent. VEGF promotes the migration and proliferation of endothelial cells and the permeability of vessels [39, 40]. In cells, activated Ras and PI3K signal pathways trigger increased transcription of VEGF and other factors that regulate VEGF, like TGF-β, PDGF and bFGF [41].
Thrombospondin-1 (Tsp-1) is a potent endogenous antiangiogenic protein. It functions by binding to CD36 on the surface of endothelial cells and makes them insensitive to VEGF and bFGF. Tsp-1 also inactivates MMP9 through binding; this decreases the release of VEGF and bFGF from ECM. Thus, in aggressive tumors, repression of Tsp-1 is required and common [42-44]. The PI3K-Rho-ROCK-Myc pathway is active in several types of human breast cancer cells and it represses Tsp-1 via phosphorylation. This pathway is called the “angiogenic switch” due to its cell-autonomous character [45]. Some tumor cells could overcome the inhibitory function of Tsp-1 by producing increased levels of VEGF [38].
CAFs are the predominant stromal cells in TME and function as a scaffold along with ECM [6]. ECM secreted by CAFs results in a hypoxic environment inside the tumor. This effect results in the recruitment of TAMs and triggers TAMs and tumor cells to produce more HIF-1α and VEGF [38]. HIF-1α induces tumor and stromal cells to produce more VEGF and other pro-angiogenesis factors. VEGF contributes to the recruitment and activation of endothelial cells [46]. Cytokines secreted by CAFs also contribute to angiogenesis. CXCL12 (Stromal Cell-Derived Factor 1, SDF1), a chemokine secreted by CAFs, promotes angiogenesis through the recruitment of endothelial progenitor cells (EPCs) into TME [47] and its receptor, CXCR4, has already become a novel target for drug delivery [48, 49]. CAFs also can secrete VEGF directly, as well as other growth factors that promote angiogenesis by inhibiting the angiogenesis-suppressing role of TSP1 [6].
Injection of tumor cells with MSCs into mice produces twice the vessels than injecting tumor cells alone. MSCs promote angiogenesis directly through the secretion of VEGF and indirectly, through the secretion of other growth factors, like TGF-β, to influence the effect of VEGF and the functions of other cells [50]. In a recent clinical therapy, researchers found that radiation therapy stimulates the secretion of inflammatory mediators, like SDF-1α and PDGF-B, from tumor cells. These signals increase the recruitment of MSCs through the binding of CXCR4 or PDGFR-β and enhancement of vasculogenesis by triggering MSCs to differentiate into pericytes [51].
Hypoxia produced by tumors may recruit TAMs to those hypoxic sites, and then induce TAMs to produce HIF1-α and enhance its function on transcription factors. This increases the production of pro-angiogenesis factors like VEGF, basic Fibroblast Growth Factor (bFGF), TNFα, and CXCL12 [3, 38]. Conversely, Colony-Stimulating Factor (CSF) increases the production of metalloelastase via macrophages [52]. Metalloelastase cleaves plasminogen into several small proteins, including the antiangiogenic protein, angiostatin [53].
The Effects of Stromal Cells on EMT
EMT is the transition of tumor cells from epithelial status to mesenchymal status and, therefore, gives migrating and invasive abilities to these tumor cells [54]. Stromal cells in TME also play crucial roles in EMT, together with multiple factors that are illustrated in Figure 2 and are discussed below.
The Effects of TME on Epithelial-Mesenchymal Transition. In TME, stromal cells, like CAFs, MSCs and TAMs, secrete growth factors like TGF-β, PDGF, EGF and HGF, along with miRNAs like miR21 and miR200 family, to regulate EMT through EMT transcription factors or RAS/RAF/MEK/ERK/MAPK pathway. They also secret MMPs to degrade ECM and junctions between cells and cells with ECM to “free” tumor cells. Inflammatory signals secreted by TAMs, like TNF-α and IL-6, trigger EMT through TGF-β or NF-κB pathways. Meanwhile, hypoxia produced in TME up-regulate HIFα-1 and trigger EMT through the NF-κB pathway.
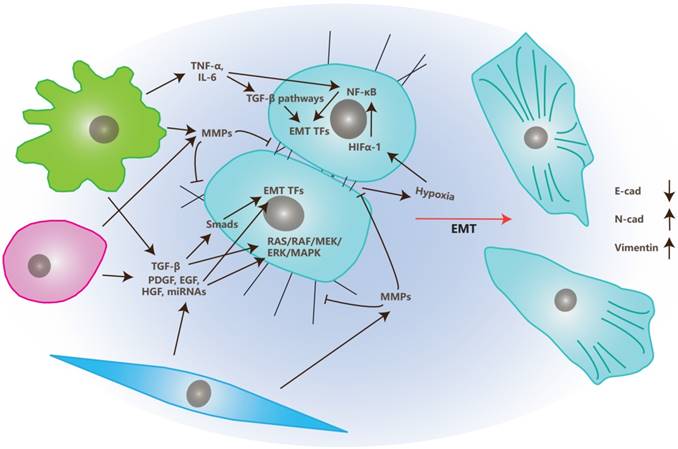
In TME, CAFs, MSCs, and TAMs affect EMT primarily through the secretion of growth factors like TGF-β, PDGF, EGF, VEGF, HGF or MMPs like MMP1, MMP2, and MMP9 [4, 55].
There are several classes of growth factors functioning in the process of EMT. The key regulators among them are TGF-β family members, which may be secreted by stromal cells and tumor cells [56, 57]. TGF-β induces EMT mainly through two pathways: Smads dependent and Smads independent pathways. In the Smads dependent pathway, TGF-β induces phosphorylation and heterodimer formation of Smad2 and Smad3 by binding to membrane-bound TGF-β receptors [58]. The Smad2/Smad3 complex then interacts with Smad4 and is transferred into the nucleus where they induce EMT Transcription Factors (EMT TFs) such as snail, twist, and ZEB [58]. In the Smads independent pathway, TGF-β induces EMT by activating RAS/ERK, JNK, p38 MAPK pathways that are cooperated with integrins or simply through integrin interactions [4].
Growth factors besides TGF-β (like EGF, FGF, HGF, VEGF and PDGF), also induce EMT via the induction of EMT TFs and activation of the RAS/RAF/MEK/ERK/MAPK pathway [55].
Matrix metalloproteinases (MMPs) also play important roles in the process of EMT. MMPs can degrade cell-matrix adhesions as well as cell-cell junctions that assist the morphological transition from epithelial to mesenchymal. MMPs also release EMT regulators by degrading ECM into TME [59].
Besides secreting growth factors and MMPs, TAMs also participate in this process through the secretion of inflammatory factors like TNF-α and IL-6 [60-62]. Those inflammatory factors trigger EMT by inducing TGF-β or NF-κB pathways. Additionally, TNF-α also induce EMT through the triggering of ROS. IL-6 does it by activating the JAK/STAT3 pathway [63, 64]. While, enhanced expression levels of COX2 in TAM is positively correlated to the secretion of COX2, IL-6, PGE2 and MMP9, and promotes the process of EMT of breast cancer cells by activating the Akt pathway [65].
Recently, Cédric Blanpain reported that tumor cells in the EMT process show progressive states from epitheial to mesenchymal [66]. Further, they determined the microenvironmental changes company with the progressing EMT states. They also found that the number and density of endothelial cells and lymphatic cells (especially macrophages) were increased in the progressing of EMT process. This suggests interactions between blood vessel formation and inflammatory with EMT [66]. Stromal cells also can release MicroRNAs like miR21 and miR200 family members through exosomes to communicate with cancer cells and induce EMT [67-69].
The Effects of Stromal Cells on Invasion and Intravasation
Invasion is a process in which tumor cells migrate from one place to another by breaking the ECM or basement membrane [70]. For example, the migration of tumor cells from primary tumors into sites near vessels is a typical invasion [71].
In the process of invasion, MMPs are secreted by stromal cells or tumor cells and degrade the ECM and basement membrane in the path of tumor cell migration. MMPs also regulate the skeleton of tumor cells that is related to cell motility and invasion [72].
Conversely, intravasation is the process by which tumor cells migrate into blood or lymph vessels through Trans-Endothelial Migration (TEM). There are two kinds of intravasation: paracellular and transcellular. Most tumor cells undergo paracellular intravasation. In the process of paracellular TEM (Figure 3), tumor cells migrate across the vessel walls composed of endothelial cells by opening the junctions between endothelial cells. In this process, endothelial cells undergoing retraction to make space for tumor cells [5]. In transcellular intravasation, tumor cells migrate into the vessels directly across the body of endothelial cells. Endothelial cells undergo degradation of skeletons and contraction in this process [73].
The Effects of TME on Intravasation. Tumor cells in mesenchymal status is leaded by Vessel Associated/Tumor Associated Macrophages and invade toward the vessel wall, tumor cells in turn, recruit macrophages. Growth factors, chemokines, proteases and inflammatory factors that are secreted by stromal or tumor cells, promote the opening of junctions between endothelial cells. Factors promoting angiogenesis enhance intravasation due to the loose junction between endothelial cells and low coverage of pericytes in newly formed blood vessels.
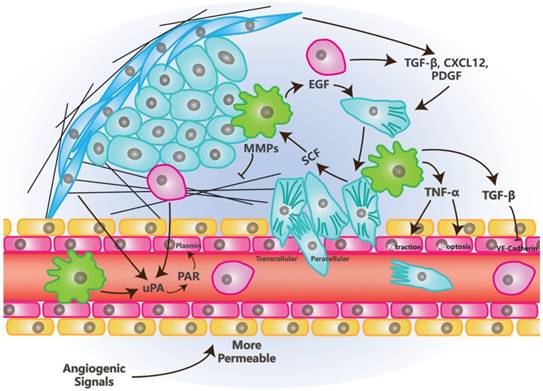
There are also multiple stromal cells and factors that function in the process of intravasation in TME [5, 74]. Besides endothelial cells, Vessel Associated Macrophages (VAMs) or Tumor Associated Macrophages (TAMs) are crucial to this process. VAMs attract tumor cells to invade toward vessels through the secretion of EGF, meanwhile, tumor cells secret SCF to recruit macrophages. These processes build EGF/SCF paracrine interaction which depends on the contact with each other [75]. TNF-α, which is secreted by macrophages, may trigger the retraction of endothelial cells by binding to its receptors while it also can make vessels more permeable for tumor cells by inducing an apoptosis signal, along with other apoptotic ligands [76].
Other stromal cells like MSCs and CAFs regulate the process of intravasation through the secretion of TGF-β, PDGF, CXCL12/CXCR4 and MMPs [5].
TGF-β promotes EMT and invasion [57] while plays a paradoxical role in intravasation. Long-term exposure of TGF-β promotes the proliferation of endothelial cells and inhibits the crossing of tumor cells into vessels. Transient signals induced by TGF-β promote intravasation through the down-regulation of CSF1, LHX2/PDGFβ and Twist [5, 77, 78]. Similarly to VEGF, TGF-β also promotes the opening of junctions between endothelial cells by inhibiting the complex of VE-Cadherin and β-catenin [74]. Besides TGF-β itself, Latent TGF-β Binding Protein 3 (LTBP3) promotes the formation of intravasation involved angiogenesis [79] and suggests that more investigation on this factor is needed.
Urokinase-type Plasminogen Activator (uPA) is the soluble portion of the protease system named uPA/PAR. This soluble portion can be secreted by CAFs, MSCs and macrophages [80-82]. Its function depends on its binding to the PAR receptor and this binding is promoted by MMP1. After uPA/PAR binding, these proteins may cleave plasminogen into plasmin, which cleaves CDCP1. Cleaved CDCP1 promotes the intravasation via the FAK/PI3K pathway that cooperates with β1-integrin [5, 83].
Angiogenesis is highly related to the process of intravasation. Those angiogenic stimulators, like VEGF, bFGF and PDGF, promote intravasation by promoting angiogenesis to provide larger and more permeable vessels. These factors also promote the invasion of tumor cells toward vessels [74, 77]. There are also several microRNAs that facilitate intravasation of breast cancer cells, like miR10b and miR200c [84, 85].
Conclusion and Future Aspects
Stromal cells are indispensable in tumors and TME. They are recruited by tumor cells and affect metastasis initiation through the regulation of tumor cells and themselves.
In TME, CAFs mainly play promoting roles in metastasis initiation (Table 1). This might be due to its functions in immune suppression, ECM remodeling and the secretion of pro-inflammatory and pro-metastatic cytokines, enzymes and microRNAs [67]. MSCs in TME affect metastasis initiation by differentiating into other stromal cells or secreting multiple factors. Currently, many labs are focused on “cell therapy”, based on the research on MSCs. TAMs are also potent stromal cells in TME. In metastasis initiation, they function with at least three major features. First, they secret inflammatory signals like TNF-α and IL-6, then activate angiogenic or EMT-promoting pathways. A recent report showed that COX2 in TAMs is essential to their function with this feature [65]. Second, they regulate the process of intravasation by forming EGF/SCF paracrine interaction with tumor cells. Third, TAMs also secret multiple kinds of cytokines like IGF [86]. Other than those three types, TAMs also transfer iron to tumor and promote tumor cell proliferation [87], which open a new door for research on TAMs.
Other than these types of stromal cells, there are still several types of non-transformed cells in TME like osteoblasts and osteoclasts in bone metastasis [88], platelets and monocytes in angiogenesis and tumor progression [89, 90], and neutrophils in metastatic tumors [91]. The participation of stromal cells in TME not only gives researchers more targets and avenues, but also results in more uncontrolled factors in cancer research.
Recently, a group put forward a method that defined the sites of metastatic tumor microenvironment by staining three parts: migrative cancer cells, TAMs and endothelial cells [92]. They named these sites TMEM (Tumor Microenvironment of Metastasis) and found the number and status of these sites correlate with the metastasis of the tumor. Furthermore, they explored that TMEM mediated neoadjuvant chemotherapy induced the metastasis of breast cancer [93], which is crucial in the clinical application and still a hot-point in current drug development [94].
Exosomes in TME have been showed to participate in multiple stages of cancer progression, including metastasis initiation [95]. They are good mediators of information transfer between cells and their cargos include miRNAs, mRNAs, DNAs and proteins [96]. Exosomes could also be released by stromal cells and participate in the regulation of cancer progression, especially those from MSCs [97-99].
Stromal cells also undergo metabolic changes in TME, which contributes to the fates of tumor cells. Reports have shown that TAMs and CAFs undergo metabolic reprograming of glucose, lipids and amino acids in TME [100, 101]. While, tumor cells probably enhance the aerobic glycolysis of TAMs and CAFs and contribute to their activation [102, 103], and these stromal cells should reshape the metabolism of TME and convert nutrients into forms that are absorbable for tumor cells [102]. Metabolism also changes the pH value in TME. One group showed that acidification of extracellular fluids promotes the activation of CAF from MSC [104]. It would be a future aspect to explore whether and how metabolism functions in the metastasis of tumor cells in TME.
Currently, stromal cells could also be used in combining therapies with immune therapy. Firstly, immune therapy modifies TME to enrich preferred phenotypes and increase immune effector cells, while decreasing immune suppressive cells. Secondly, immune therapy activates, triggers and regulates stromal cells, while stromal cells release chemokines or other factors to recruit or regulate immune cells [105, 106]. These processes are all potential targets in the future combination of therapies.
Acknowledgements
We thank the members of the Deng laboratory for critical discussions. This work is supported by the Chair Professor Grant (CPG2017-00026-FHS), MYRG2016-00132-FHS and MYRG2016-00139 of University of Macau and FDCT grants (065/2015/A2 and 094/2015/A3) to Chu-Xia Deng. The authors declare no conflict of interest.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125:5591-6
2. Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341-52
3. Mittal K, Ebos J, Rini B. Angiogenesis and the tumor microenvironment: vascular endothelial growth factor and beyond. Semin Oncol. 2014;41:235-51
4. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-8
5. Chiang SP, Cabrera RM, Segall JE. Tumor cell intravasation. Am J Physiol Cell Physiol. 2016;311:C1-C14
6. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582-98
7. Murata T, Mekada E, Hoffman RM. Reconstitution of a metastatic-resistant tumor microenvironment with cancer-associated fibroblasts enables metastasis. Cell Cycle. 2017;16:533-5
8. Zhu Y, Zhang L, Zha H, Yang F, Hu C, Chen L. et al. Stroma-derived Fibrinogen-like Protein 2 Activates Cancer-associated Fibroblasts to Promote Tumor Growth in Lung Cancer. Int J Biol Sci. 2017;13:804-14
9. Shen XJ, Zhang H, Tang GS, Wang XD, Zheng R, Wang Y. et al. Caveolin-1 is a modulator of fibroblast activation and a potential biomarker for gastric cancer. Int J Biol Sci. 2015;11:370-9
10. Spees JL, Lee RH, Gregory CA. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res Ther. 2016;7:125
11. Hata N, Shinojima N, Gumin J, Yong R, Marini F, Andreeff M. et al. Platelet-derived growth factor BB mediates the tropism of human mesenchymal stem cells for malignant gliomas. Neurosurgery. 2010;66:144-56 discussion 56-7
12. Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A. et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614-23
13. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B. et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4:e4992
14. Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F 3rd. Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011;29:11-9
15. Brady NJ, Chuntova P, Schwertfeger KL. Macrophages: Regulators of the Inflammatory Microenvironment during Mammary Gland Development and Breast Cancer. Mediators Inflamm. 2016;2016:4549676
16. Shaked Y, McAllister S, Fainaru O, Almog N. Tumor dormancy and the angiogenic switch: possible implications of bone marrow- derived cells. Curr Pharm Des. 2014;20:4920-33
17. Liguori M, Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers (Basel). 2011;3:3740-61
18. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298-307
19. Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193-215
20. Mongiat M, Andreuzzi E, Tarticchio G, Paulitti A. Extracellular Matrix, a Hard Player in Angiogenesis. Int J Mol Sci. 2016:17
21. Robertson C. The extracellular matrix in breast cancer predicts prognosis through composition, splicing, and crosslinking. Exp Cell Res. 2016;343:73-81
22. Insua-Rodriguez J, Oskarsson T. The extracellular matrix in breast cancer. Adv Drug Deliv Rev. 2016;97:41-55
23. Singh C, Shyanti RK, Singh V, Kale RK, Mishra JPN, Singh RP. Integrin expression and glycosylation patterns regulate cell-matrix adhesion and alter with breast cancer progression. Biochem Biophys Res Commun. 2018;499:374-80
24. Glasner A, Levi A, Enk J, Isaacson B, Viukov S, Orlanski S. et al. NKp46 Receptor-Mediated Interferon-gamma Production by Natural Killer Cells Increases Fibronectin 1 to Alter Tumor Architecture and Control Metastasis. Immunity. 2018;48:107-19 e4
25. Mbeunkui F, Johann DJ Jr. Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemother Pharmacol. 2009;63:571-82
26. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185
27. Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci. 2011;7:651-8
28. Alphonso A, Alahari SK. Stromal cells and integrins: conforming to the needs of the tumor microenvironment. Neoplasia. 2009;11:1264-71
29. Paolillo M, Schinelli S. Integrins and Exosomes, a Dangerous Liaison in Cancer Progression. Cancers (Basel). 2017:9
30. Madrazo E, Conde AC, Redondo-Munoz J. Inside the Cell: Integrins as New Governors of Nuclear Alterations?. Cancers (Basel). 2017:9
31. Craddock RJ, Hodson NW, Ozols M, Shearer T, Hoyland JA, Sherratt MJ. Extracellular matrix fragmentation in young, healthy cartilaginous tissues. Eur Cell Mater. 2018;35:34-53
32. Kumar S, Kulkarni R, Sen S. Cell motility and ECM proteolysis regulate tumor growth and tumor relapse by altering the fraction of cancer stem cells and their spatial scattering. Phys Biol. 2016;13:036001
33. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
34. Noel A, Gutierrez-Fernandez A, Sounni NE, Behrendt N, Maquoi E, Lund IK. et al. New and paradoxical roles of matrix metalloproteinases in the tumor microenvironment. Front Pharmacol. 2012;3:140
35. Manasa VG, Kannan S. Impact of microRNA dynamics on cancer hallmarks: An oral cancer scenario. Tumour Biol. 2017;39:1010428317695920
36. Kuninty PR, Schnittert J, Storm G, Prakash J. MicroRNA Targeting to Modulate Tumor Microenvironment. Front Oncol. 2016;6:3
37. Ding L, Ren J, Zhang D, Li Y, Huang X, Hu Q. et al. A novel stromal lncRNA signature reprograms fibroblasts to promote the growth of oral squamous cell carcinoma via LncRNA-CAF/interleukin-33. Carcinogenesis. 2018
38. Watnick RS. The role of the tumor microenvironment in regulating angiogenesis. Cold Spring Harb Perspect Med. 2012;2:a006676
39. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC. et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB J. 2015;29:227-38
40. Liu S, Jiang M, Zhao Q, Li S, Peng Y, Zhang P. et al. Vascular endothelial growth factor plays a critical role in the formation of the pre-metastatic niche via prostaglandin E2. Oncol Rep. 2014;32:2477-84
41. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int J Mol Sci. 2018:19
42. Jeanne A, Schneider C, Martiny L, Dedieu S. Original insights on thrombospondin-1-related antireceptor strategies in cancer. Front Pharmacol. 2015;6:252
43. Radziwon-Balicka A, Santos-Martinez MJ, Corbalan JJ, O'Sullivan S, Treumann A, Gilmer JF. et al. Mechanisms of platelet-stimulated colon cancer invasion: role of clusterin and thrombospondin 1 in regulation of the P38MAPK-MMP-9 pathway. Carcinogenesis. 2014;35:324-32
44. Zhang X, Kazerounian S, Duquette M, Perruzzi C, Nagy JA, Dvorak HF. et al. Thrombospondin-1 modulates vascular endothelial growth factor activity at the receptor level. FASEB J. 2009;23:3368-76
45. Kazerounian S, Lawler J. Integration of pro- and anti-angiogenic signals by endothelial cells. J Cell Commun Signal. 2018;12:171-9
46. Hoeppner LH, Sinha S, Wang Y, Bhattacharya R, Dutta S, Gong X. et al. RhoC maintains vascular homeostasis by regulating VEGF-induced signaling in endothelial cells. J Cell Sci. 2015;128:3556-68
47. Zhang M, Qiu L, Zhang Y, Xu D, Zheng JC, Jiang L. CXCL12 enhances angiogenesis through CXCR7 activation in human umbilical vein endothelial cells. Sci Rep. 2017;7:8289
48. Miller EJ, Jecs E, Truax VM, Katzman BM, Tahirovic YA, Wilson RJ. et al. Discovery of Tetrahydroisoquinoline-Containing CXCR4 Antagonists with Improved in vitro ADMET Properties. J Med Chem. 2018
49. Ziegler ME, Hatch MM, Wu N, Muawad SA, Hughes CC. mTORC2 mediates CXCL12-induced angiogenesis. Angiogenesis. 2016;19:359-71
50. Coffelt SB, Marini FC, Watson K, Zwezdaryk KJ, Dembinski JL, LaMarca HL. et al. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc Natl Acad Sci U S A. 2009;106:3806-11
51. Wang HH, Cui YL, Zaorsky NG, Lan J, Deng L, Zeng XL. et al. Mesenchymal stem cells generate pericytes to promote tumor recurrence via vasculogenesis after stereotactic body radiation therapy. Cancer Lett. 2016;375:349-59
52. Tsuji T, Kelly NJ, Takahashi S, Leme AS, Houghton AM, Shapiro SD. Macrophage elastase suppresses white adipose tissue expansion with cigarette smoking. Am J Respir Cell Mol Biol. 2014;51:822-9
53. Aharinejad S, Paulus P, Sioud M, Hofmann M, Zins K, Schafer R. et al. Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer Res. 2004;64:5378-84
54. Liu Y, Liu B, Zhang GQ, Zou JF, Zou ML, Cheng ZS. Calpain inhibition attenuates bleomycin-induced pulmonary fibrosis via switching the development of epithelial-mesenchymal transition. Naunyn Schmiedebergs Arch Pharmacol. 2018
55. Jing Y, Han Z, Zhang S, Liu Y, Wei L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011;1:29
56. Morrison CD, Parvani JG, Schiemann WP. The relevance of the TGF-beta Paradox to EMT-MET programs. Cancer Lett. 2013;341:30-40
57. Ma M, He M, Jiang Q, Yan Y, Guan S, Zhang J. et al. MiR-487a Promotes TGF-beta1-induced EMT, the Migration and Invasion of Breast Cancer Cells by Directly Targeting MAGI2. Int J Biol Sci. 2016;12:397-408
58. Zhao M, Mishra L, Deng CX. The role of TGF-beta/SMAD4 signaling in cancer. Int J Biol Sci. 2018;14:111-23
59. Nistico P, Bissell MJ, Radisky DC. Epithelial-mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect Biol. 2012:4
60. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361-71
61. Wang S, Yan Y, Cheng Z, Hu Y, Liu T. Sotetsuflavone suppresses invasion and metastasis in non-small-cell lung cancer A549 cells by reversing EMT via the TNF-alpha/NF-kappaB and PI3K/AKT signaling pathway. Cell Death Discov. 2018;4:26
62. Wu ST, Sun GH, Hsu CY, Huang CS, Wu YH, Wang HH. et al. Tumor necrosis factor-alpha induces epithelial-mesenchymal transition of renal cell carcinoma cells via a nuclear factor kappa B-independent mechanism. Exp Biol Med (Maywood). 2011;236:1022-9
63. Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F. et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26:7445-56
64. Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104:733-44
65. Gan L, Qiu Z, Huang J, Li Y, Huang H, Xiang T. et al. Cyclooxygenase-2 in tumor-associated macrophages promotes metastatic potential of breast cancer cells through Akt pathway. Int J Biol Sci. 2016;12:1533-43
66. Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S. et al. Identification of the tumour transition states occurring during EMT. Nature. 2018
67. Wang Z, Tan Y, Yu W, Zheng S, Zhang S, Sun L. et al. Small role with big impact: miRNAs as communicators in the cross-talk between cancer-associated fibroblasts and cancer cells. Int J Biol Sci. 2017;13:339-48
68. Markopoulos GS, Roupakia E, Tokamani M, Chavdoula E, Hatziapostolou M, Polytarchou C. et al. A step-by-step microRNA guide to cancer development and metastasis. Cell Oncol (Dordr). 2017;40:303-39
69. Xia H, Hui KM. MicroRNAs involved in regulating epithelial-mesenchymal transition and cancer stem cells as molecular targets for cancer therapeutics. Cancer Gene Ther. 2012;19:723-30
70. Truong D, Puleo J, Llave A, Mouneimne G, Kamm RD, Nikkhah M. Breast Cancer Cell Invasion into a Three Dimensional Tumor-Stroma Microenvironment. Sci Rep. 2016;6:34094
71. Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER. et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649-56
72. Chabottaux V, Ricaud S, Host L, Blacher S, Paye A, Thiry M. et al. Membrane-type 4 matrix metalloproteinase (MT4-MMP) induces lung metastasis by alteration of primary breast tumour vascular architecture. J Cell Mol Med. 2009;13:4002-13
73. Khuon S, Liang L, Dettman RW, Sporn PH, Wysolmerski RB, Chew TL. Myosin light chain kinase mediates transcellular intravasation of breast cancer cells through the underlying endothelial cells: a three-dimensional FRET study. J Cell Sci. 2010;123:431-40
74. Reymond N, d'Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858-70
75. Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ. et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005;65:5278-83
76. Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc Natl Acad Sci U S A. 2012;109:13515-20
77. Tsuji T, Ibaragi S, Hu GF. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009;69:7135-9
78. Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11:1287-96
79. Deryugina EI, Zajac E, Zilberberg L, Muramatsu T, Joshi G, Dabovic B. et al. LTBP3 promotes early metastatic events during cancer cell dissemination. Oncogene. 2018
80. Breznik B, Motaln H, Vittori M, Rotter A, Lah Turnsek T. Mesenchymal stem cells differentially affect the invasion of distinct glioblastoma cell lines. Oncotarget. 2017;8:25482-99
81. Sugioka K, Mishima H, Kodama A, Itahashi M, Fukuda M, Shimomura Y. Regulatory Mechanism of Collagen Degradation by Keratocytes and Corneal Inflammation: The Role of Urokinase-Type Plasminogen Activator. Cornea. 2016;35(Suppl 1):S59-S64
82. Tian B, Chen X, Zhang H, Li X, Wang J, Han W. et al. Urokinase plasminogen activator secreted by cancer-associated fibroblasts induces tumor progression via PI3K/AKT and ERK signaling in esophageal squamous cell carcinoma. Oncotarget. 2017
83. Casar B, Rimann I, Kato H, Shattil SJ, Quigley JP, Deryugina EI. In vivo cleaved CDCP1 promotes early tumor dissemination via complexing with activated beta1 integrin and induction of FAK/PI3K/Akt motility signaling. Oncogene. 2014;33:255-68
84. Croset M, Goehrig D, Frackowiak A, Bonnelye E, Ansieau S, Puisieux A. et al. TWIST1 expression in breast cancer cells facilitates bone metastasis formation. J Bone Miner Res. 2014;29:1886-99
85. Sigloch FC, Burk UC, Biniossek ML, Brabletz T, Schilling O. miR-200c dampens cancer cell migration via regulation of protein kinase A subunits. Oncotarget. 2015;6:23874-89
86. Ireland L, Santos A, Campbell F, Figueiredo C, Hammond D, Ellies LG. et al. Blockade of insulin-like growth factors increases efficacy of paclitaxel in metastatic breast cancer. Oncogene. 2018
87. Mertens C, Mora J, Oren B, Grein S, Winslow S, Scholich K. et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology. 2018;7:e1408751
88. Wu JB, Yin L, Shi C, Li Q, Duan P, Huang JM. et al. MAOA-Dependent Activation of Shh-IL6-RANKL Signaling Network Promotes Prostate Cancer Metastasis by Engaging Tumor-Stromal Cell Interactions. Cancer Cell. 2017;31:368-82
89. Qi C, Li B, Guo S, Wei B, Shao C, Li J. et al. P-Selectin-Mediated Adhesion between Platelets and Tumor Cells Promotes Intestinal Tumorigenesis in Apc(Min/+) Mice. Int J Biol Sci. 2015;11:679-87
90. Sidibe A, Ropraz P, Jemelin S, Emre Y, Poittevin M, Pocard M. et al. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat Commun. 2018;9:355
91. Felix K, Gaida MM. Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer -Active Players in Tumor Progression. Int J Biol Sci. 2016;12:302-13
92. Robinson BD, Sica GL, Liu YF, Rohan TE, Gertler FB, Condeelis JS. et al. Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin Cancer Res. 2009;15:2433-41
93. Karagiannis GS, Pastoriza JM, Wang Y, Harney AS, Entenberg D, Pignatelli J. et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci Transl Med. 2017:9
94. Lv Y, Xu C, Zhao X, Lin C, Yang X, Xin X. et al. Nanoplatform Assembled from a CD44-Targeted Prodrug and Smart Liposomes for Dual Targeting of Tumor Microenvironment and Cancer Cells. ACS Nano. 2018
95. Suchorska WM, Lach MS. The role of exosomes in tumor progression and metastasis (Review). Oncol Rep. 2016;35:1237-44
96. Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl). 2013;91:431-7
97. Bliss SA, Sinha G, Sandiford OA, Williams LM, Engelberth DJ, Guiro K. et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016;76:5832-44
98. Zhang Z, Li X, Sun W, Yue S, Yang J, Li J. et al. Loss of exosomal miR-320a from cancer-associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 2017;397:33-42
99. Zheng P, Chen L, Yuan X, Luo Q, Liu Y, Xie G. et al. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J Exp Clin Cancer Res. 2017;36:53
100. Shan T, Chen S, Chen X, Lin WR, Li W, Ma J. et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol Rep. 2017;37:1971-9
101. Rabold K, Netea MG, Adema GJ, Netea-Maier RT. Cellular metabolism of tumor-associated macrophages - functional impact and consequences. FEBS Lett. 2017
102. Reina-Campos M, Moscat J, Diaz-Meco M. Metabolism shapes the tumor microenvironment. Curr Opin Cell Biol. 2017;48:47-53
103. Dehne N, Mora J, Namgaladze D, Weigert A, Brune B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr Opin Pharmacol. 2017;35:12-9
104. Zhu H, Guo S, Zhang Y, Yin J, Yin W, Tao S. et al. Proton-sensing GPCR-YAP Signalling Promotes Cancer-associated Fibroblast Activation of Mesenchymal Stem Cells. Int J Biol Sci. 2016;12:389-96
105. Giuliani M, Janji B, Berchem G. Activation of NK cells and disruption of PD-L1/PD-1 axis: two different ways for lenalidomide to block myeloma progression. Oncotarget. 2017;8:24031-44
106. Nicodemus CF. Antibody-based immunotherapy of solid cancers: progress and possibilities. Immunotherapy. 2015;7:923-39
107. Liao Z, Tan ZW, Zhu P, Tan NS. Cancer-associated fibroblasts in tumor microenvironment - Accomplices in tumor malignancy. Cell Immunol. 2018
108. Poh AR, Ernst M. Targeting Macrophages in Cancer: From Bench to Bedside. Front Oncol. 2018;8:49
109. Knutsdottir H, Condeelis JS, Palsson E. 3-D individual cell based computational modeling of tumor cell-macrophage paracrine signaling mediated by EGF and CSF-1 gradients. Integr Biol (Camb). 2016;8:104-19
110. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298-306
111. Campbell DJ, Koch MA. Treg cells: patrolling a dangerous neighborhood. Nat Med. 2011;17:929-30
112. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12:5423-34
113. Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer. 2011;105:93-103
114. Lee GR. Phenotypic and Functional Properties of Tumor-Infiltrating Regulatory T Cells. Mediators Inflamm. 2017;2017:5458178
115. Qi W, Huang X, Wang J. Correlation between Th17 cells and tumor microenvironment. Cell Immunol. 2013;285:18-22
116. Shen M, Wang J, Ren X. New Insights into Tumor-Infiltrating B Lymphocytes in Breast Cancer: Clinical Impacts and Regulatory Mechanisms. Front Immunol. 2018;9:470
117. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K. et al. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res. 2011;71:3505-15
118. Schioppa T, Moore R, Thompson RG, Rosser EC, Kulbe H, Nedospasov S. et al. B regulatory cells and the tumor-promoting actions of TNF-alpha during squamous carcinogenesis. Proc Natl Acad Sci U S A. 2011;108:10662-7
119. Horikawa M, Minard-Colin V, Matsushita T, Tedder TF. Regulatory B cell production of IL-10 inhibits lymphoma depletion during CD20 immunotherapy in mice. J Clin Invest. 2011;121:4268-80
120. Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011;17:1371-80
121. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D. et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071-121
122. Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat Rev Cancer. 2012;12:210-9
123. Harrell CR, Simovic Markovic B, Fellabaum C, Arsenijevic A, Djonov V, Volarevic V. Molecular mechanisms underlying therapeutic potential of pericytes. J Biomed Sci. 2018;25:21
124. Qu Y, Zhao G, Li H. Forward and Reverse Signaling Mediated by Transmembrane Tumor Necrosis Factor-Alpha and TNF Receptor 2: Potential Roles in an Immunosuppressive Tumor Microenvironment. Front Immunol. 2017;8:1675
125. Gasche JA, Hoffmann J, Boland CR, Goel A. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int J Cancer. 2011;129:1053-63
126. Santibanez JF, Quintanilla M, Bernabeu C. TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clin Sci (Lond). 2011;121:233-51
127. Ai S, Cheng XW, Inoue A, Nakamura K, Okumura K, Iguchi A. et al. Angiogenic activity of bFGF and VEGF suppressed by proteolytic cleavage by neutrophil elastase. Biochem Biophys Res Commun. 2007;364:395-401
128. Aljada A, O'Connor L, Fu YY, Mousa SA. PPAR gamma ligands, rosiglitazone and pioglitazone, inhibit bFGF- and VEGF-mediated angiogenesis. Angiogenesis. 2008;11:361-7
129. Zhao D, Tu Y, Wan L, Bu L, Huang T, Sun X. et al. In vivo monitoring of angiogenesis inhibition via down-regulation of mir-21 in a VEGFR2-luc murine breast cancer model using bioluminescent imaging. PLoS One. 2013;8:e71472
130. Lei H, Deng CX. Fibroblast Growth Factor Receptor 2 Signaling in Breast Cancer. Int J Biol Sci. 2017;13:1163-71
131. Ostman A. PDGF receptors in tumor stroma: Biological effects and associations with prognosis and response to treatment. Adv Drug Deliv Rev. 2017;121:117-23
132. Papadopoulos N, Lennartsson J. The PDGF/PDGFR pathway as a drug target. Mol Aspects Med. 2017
133. Nasser MW, Elbaz M, Ahirwar DK, Ganju RK. Conditioning solid tumor microenvironment through inflammatory chemokines and S100 family proteins. Cancer Lett. 2015;365:11-22
134. Hirbe AC, Morgan EA, Weilbaecher KN. The CXCR4/SDF-1 chemokine axis: a potential therapeutic target for bone metastases? Curr Pharm Des. 2010;16:1284-90
135. Longmate W, DiPersio CM. Beyond adhesion: emerging roles for integrins in control of the tumor microenvironment. F1000Res. 2017;6:1612
Author contact
![]() Corresponding author: Chu-Xia Deng, Faculty of Health Sciences, University of Macau, Macau SAR, China. cxdengmo
Corresponding author: Chu-Xia Deng, Faculty of Health Sciences, University of Macau, Macau SAR, China. cxdengmo