Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(1):24-33. doi:10.7150/ijbs.28633 This issue Cite
Research Paper
PM2.5 Exposure Induces More Serious Apoptosis of Cardiomyocytes Mediated by Caspase3 through JNK/ P53 Pathway in Hyperlipidemic Rats
Qin Wang2*, Xiangdong Gan1*, Fei Li3, Yao Chen1, Wenliang Fu1, Xiaoming Zhu1, Dongqun Xu2, Minhui Long1,3 ![]() , Donggang Xu1
, Donggang Xu1 ![]()
1. Institute of Military Cognitive and Brain Sciences, Beijing, China.
2. National Institute of Environmental Health, Chinese center for disease control and prevention, Beijing, China
3. College of Life Science, South China Normal University, Guangzhou, China.
*Contributed equally.
Received 2018-7-19; Accepted 2018-9-26; Published 2019-1-1
Abstract

Exposure to airborne particulate matter with an aerodynamic diameter less than or equivalent to 2.5 microns (PM2.5) easily induces acute myocardial infarction in populations with high-risk cardiovascular diseases such as hyperlipidemia, but its mechanism remains unclear. In this study, hyperlipidemic rats were used to examine the effects of PM2.5 exposure on the cardiovascular system and the mechanism for its induction of cardiovascular events. We found that PM2.5 exposure resulted in bigger changes in the myocardial enzyme profile (cTnI, LDH, CK, CK-MB) in hyperlipidemic rats than that of control rats, as well as a significant increase in the C-reactive protein (CRP) level and a decrease in the superoxide dismutase (SOD) activity. It promoted a hypercoagulable state, significantly increased blood pressure and heart rate, while decreased the variability of heart rate in hyperlipidemic rats. In addition, pathological test showed that PM2.5 exposure more easily deteriorated myocardial injury in hyperlipidemic rats. It upregulated the phosphorylation levels of myocardial c-Jun NH2-terminal kinase (JNK) and P53, resulting in the elevated expression of downstream effector protein Bax and the decreased expression of Bcl-2, and then increased caspase3 level leading to cardiomyocyte apoptosis, while little change of caspase2 was observed. Taken together, PM2.5 exposure induced more serious inflammation and oxidative stress in the circulation system of hyperlipidemic rats, promoted a hypercoagulable state and triggered cardiomyocyte apoptosis, in which JNK/P53 pathway played a key role.
Keywords: PM2.5 exposure, hyperlipidemia, cardiacmyocyte apoptosis, JNK/P53 pathway, caspase3
Introduction
Particulate matter with an aerodynamic diameter less than or equivalent to 2.5 microns (PM2.5) is a major air pollutant that causes approximately seven million premature deaths worldwide [1]. The surface of PM2.5 absorbs a large quantity of toxic and hazardous substances (including organic, inorganic and biological components), which can not only remain in the air for a long period of time, subsequently enter and accumulate in the alveoli, but eventually enter the blood circulation system through respiration, thus affecting the health of organism [2-5]. PM2.5 exposure has a greater impact on the susceptible population of cardiovascular diseases than the healthy population, such as people with high cholesterol or diabetes [6-7]. PM2.5 is associated with cardiovascular diseases (CVD), and the CVD morbidity is also relative with PM2.5 exposure concentration. A study found that there was strong association between particulate and congestive heart failure, a 0.72% increase in risk per 10 μg/m3 elevation [8]. Brook reported that congestive heart failure increased 0.18% per 10 μg/m3 in the general population [9]. Annual average exposure to higher levels of PM (per 0.26 μg/m3 elevation), was closely correlated with a 1.1% increase in carotid intima media thickness (CIMT) in a cohort of elderly men [10]. It induces cardiovascular events by promoting inflammation and oxidative stress, leading to a hypercoagulable state, causing autonomic nervous tension and vasomotor changes, thus resulting in the lack of blood or oxygen supply to the cardiovascular system, ultimately leading to acute cardiovascular events such as myocardial injury or infarction [11-13]. Clinical diagnostic criteria of acute cardiovascular events include cardiac troponin (cTnI), creatine kinase (CK), creatine kinase-MB isoenzyme (CK-MB) and lactate dehydrogenase (LDH) levels, which are usually combined with blood pressure and electrocardiogram (ECG) for diagnosis [14-16]. Among them, cTnI is currently considered the most cardiac-specific and valuable indicator of myocardial injury [17]. In addition, several inflammatory markers also have important reference values during cardiovascular events. For example, C-reactive protein (CRP), a typical marker reflecting the body's inflammatory state, has an important reference value in evaluating acute myocardial infarction and cerebral infarction [18-20]. PM2.5 exposure can induce oxidative stress, in which superoxide dismutase (SOD) is an important factor for removing oxidative damage and for cell protection. Moreover, changes in SOD activity can reflect the body's oxidative stress status [21]. In addition, PM2.5 exposure can also affect the coagulation and fibrinolytic system, resulting in a hypercoagulable state, represented by elevating fibrinogen (FIB), reduced prothrombin time (PT) and activated partial thromboplastin time (APTT), as well as increased blood viscosity, which causes symptoms of ischemia or hypoxia [22-23]. Taken together, PM2.5 exposure can induce the body's acute inflammatory and oxidative stress reaction, as well as change the coagulation status, leading to cardiac ischemia, which causes disordered autonomous heart rhythm and myocardial apoptosis, eventually leading to myocardial injury and necrosis. Studies have shown that the C-Jun NH2-terminal protein kinase (JNK) or tumor suppressor factor P53 (P53) is the main pathway regulating the progression of cardiovascular diseases [24-25]. P53 induces cell apoptosis and plays a central role in regulating the apoptotic signaling pathway, which is important in regulating cardiomyocyte apoptosis in myocardial ischemia-reperfusion injury, myocardial infarction and hypertension [26-28]. Its downstream genes, such as Bcl-2 and Bax, are important effector molecules regulating apoptosis. In this study, by comparing the differences between hyperlipidemic rats and normal rats after exposure to PM2.5 collected in Beijing during the winter (October to December) of 2012, the toxic effects of PM2.5 on the cardiovascular system of rats with different pathological conditions, as well as its impact on inflammation and oxidative stress were analyzed. Our research would clarify the mechanism of PM2.5 exposure in inducing acute cardiovascular events in population with high-risk cardiovascular diseases.
Materials and Methods
PM2.5 collection and preparation
The ambient PM2.5 was sampled at flow rate of 77.59 L/min on a glass fiber filter (Whatman® 41 filters, Whatman Inc, Maidstone, UK) by a middle flowmeter (model:TSP/PM10/PM2.5-2, Beijing Geological Instrument-Dickel Co, Ltd) at the altitude of 40 meters between 2nd and 3rd ring road of Beijing city from October to December in the year 2012 (winter). The filter was immersed in deionized water after sampling, from which the sample was eluted out by ultrasonic cleaner (TP250B, Tianpeng Electronics Co. Ltd) and lyophilized, then stored at -20℃. A certain quantity of PM2.5 was weighed and suspended in 0.9% normal saline, homogenized for 15 min by ultrasonator, then stored at 4℃. The method for sampling PM2.5 is according to literature [29].
Chemical analysis of PM2.5
The inorganic components were detected by plasma emission spectroscopy (ICP-AES, Model: ULTIMA, JOBIN-YVON Company, France), and polycyclic aromatic hydrocarbon (PAHs) were detected by HPLC (Water® 2690 Separations Module, Water® 474 Scanning Fluorescence Detector and Symmetry® C1839 mm×150 mm, 65 μm column) using US EPA method (US EPA, 1998).
Animal grouping and exposure to PM2.5
All procedures used in this research were approved by the Institutional Animal Care and Use Committee of Institute of Military Cognitive and Brain Sciences. 70 Wistar male 8-weeks-old rats were purchased from the Animal Experiment Center of AMMS, and the code for the animals was SCXK(Jing)2012-0001, the inclusion criteria were as follows: 1. Body weight: 180-200g. 2. No abnormal appearance by a comprehensive observation. The selected rats were kept at constant temperature (20±2℃) and humidity (50%±5%), free drinking water, artificial lighting for 12 h a day, and adaptive feeding for one week, from which 35 rats were taken randomly and fed on high fat diet[30] (20% lard, 5% sugar, 3% cholesterol 0.5% sodium cholate, 0.2% propylthiouracil, 1% methionine, 1% vitamin D, 2% egg yolk powder, 68% the basic forage) for 24 weeks, 5 of which were utilized to evaluate the modeling of hyperlipemia. The other 35 rats were fed on the basic diet, 5 of which were used as the control. The remaining 30 with hyperlipemia and 30 normal rats were divided each into 3 groups (10 rats/group), labeled as the normal saline, low dose, and high dose group, and the biochemical indexes were determined before feeding on high fat diet and instilling the PM2.5. After setting up the model, the toxicant exposure to PM2.5 was performed by endotracheal instillation once per two days for continuous 3 times. The dose of endotracheal instilling PM2.5 suspension in the rat was 0, 4, 40 mg/kg respectively. The 10% chloral hydrate (ShangHai Hushi Laboratorial Equipment company) was used for anesthesia by intraperitoneal injection according to 0.27ml/100g weight at 24 h after the final exposure, SMUP/E biological signal processing system to monitor the rat's blood pressure and electrocardiogram, the blood samples were collected via abdominal aorta by the 2 ml specific anticoagulant vial (Institute of Medical Instruments, Beijing) and centrifuged at 4℃, 3000 r/min for 10 min, then the plasma was stored at -20℃.
Biochemical assay of blood
The serum hs-CRP was measured with a commercial kit (Rat CRP ELISA Kit, Invitrogen, CA, USA), serum cTn-I, LDH, CK and CK-MB were determined by mini vidas-IDAS full automatic fluorescence ELISA instrument (BioMerieux Company, France), All the assays were completed by the same physician strictly following the instruction manual. HITACHI-7060 Automatic Biochemical Analyzer (JAPAN) was used to detect the serum levels of total cholesterol (TC), triglyceride (TG), high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C) using the reagents supplied by RANDOX LABORATORIES LTD, UK. FIB, APTT and PT were assayed by Sysmex CA-530 coagulation analyzer (JAPAN), the reagents were supplied by Dade-Behring (GERMAN) and the serum SOD was determined according to the instruction manual provided by Nanjing Jiancheng Biotechnologies Co. Ltd. All the assays were repeated twice.
HE and TUNEL staining
The rat's left ventricle was collected and cut into the myocardial tissue block of 0.5 cm3. The tissue block was fixed by formaldehyde, dehydrated, embedded routinely with paraffin, cut into 5μm thick sections and stained with HE (hematoxylin-eosin) for light microscopy. Some sections were used for apoptotic assessment with a terminal deoxynucleotidyl transferase (TDT)-mediated dUTP nick-end labeling (TUNEL) assay. The morphological changes of myocardial tissue should be observed by optical microscopy, and the diagnosis of pathological changes should be confirmed by two pathologists.
Western blotting
The heart tissue should be homogenized with RIPA lysis buffer (Applygen Technologies Inc, China) in ice-bath, from which the total protein should be extracted and quantified by the kit for detection of protein content with Lowry method (KeyGEN Biotechnology Co, China). The total protein was separated by electrophoresis on 12% sodium dodecyl sulfate sodium salt polyacrylamide gelelectrophoresis (SDS-PAGE), then transferred onto polyvinylidene fluoride (PVDF) membrane of 0.2 µm aperture (ImmobilonTM Millipore, USA). The membranes were incubated with anti-Bax, anti-Bcl-2, anti-phospho-p53, anti-phospho-JNK (all at 1:1000 ), (Cell Signaling Technology, Inc, USA) antibodies overnight at 4℃ and the corresponding secondary antibodies for 1 h at room temperature. Finally, the membranes should be treated with SuperSignal West Femto Substrate (Thermo Scientific, USA) for developing in dark room, and the result was analyzed using the housekeeping gene GAPDH as the internal reference (Cwbiotech, Inc, China).
Statistical analysis
Statistical analysis of all data was performed by using SPSS 13.0. Differences between means were assessed by using one-way analysis of variance (ANOVA) followed by Dunnett's test for multiple comparisons. The symbol* and ** were used to show the statistically significant difference (P<0.05) and very significant difference (P<0.01). All the data were shown as mean±SD and the obeyed data to be checked by t-test.
Results
Assessment of the model of rats with hyperlipidemia and analysis of myocardial injury
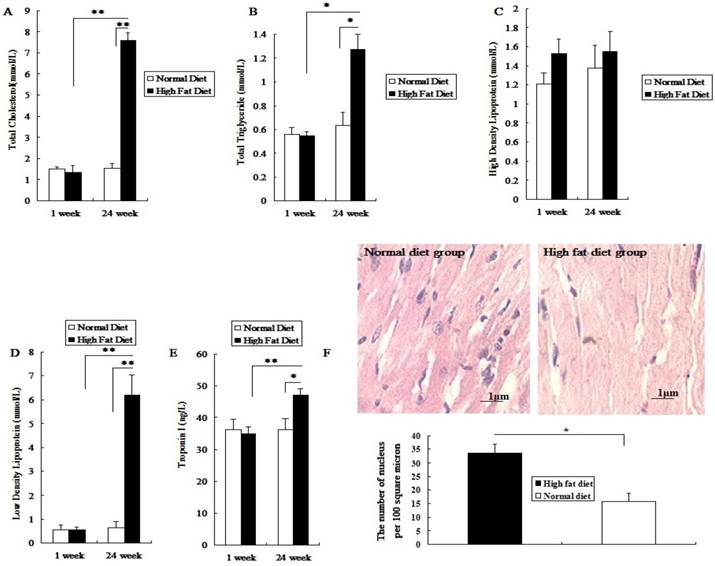
Before being fed a high fat diet, the total cholesterol (TC), triglyceride (TG), high density lipoprotein(HDL), low density lipoprotein(LDL) and cTnI levels in rat serum did not show a significant difference between the two groups (P>0.05; Figure 1A-1E). After being fed different diets for 24 weeks, the levels of serum TC, LDL, TG and cTnI in the high fat diet group significantly increased (P<0.05; Figure 1A, 1B, 1D, 1E). However, the HDL level was not significantly changed (P>0.05; Figure 1C). Meanwhile, after 24 weeks of high fat diet, the number of myocardial nuclei in these rats was higher than that in the normal diet group (P<0.05; Figure 1F). The results showed that rats fed with a high fat diet for 24 weeks presented symptoms of hyperlipidemia, with mild myocardial injury, which made them a susceptible animal model for the evaluation of toxic effects of PM2.5 exposure on the cardiovascular system.
Establishment and assessment of hyperlipemic rat model. The serum was collected after high-fat diet for 24 weeks. A, the value of TC in serum; B, the value of TG in serum; C, the value of HDL-C in serum; D, the value of LDL-C in serum; E, the value of cTn-I in serum; F, The HE staining of myocardial tissue. The data are expressed as mean ± SD (n=7), * P <0.05 and ** P <0.01 vs the normal diet group. The pathological tissue block was sampled from the rat's left ventricle, and the sampling site for each rat should be same.
Effects of PM2.5 exposure on the coagulation system
With increasing exposure doses of PM2.5, the coagulation status index which included FIB, PT and APTT in hyperlipidemic rats were changed obviously, while the changes of coagulation indexes could be observed only at dose of 40mg/kg weight body in normal rats (P<0.05; Figure 2A, 2B, 2C). Besides that, there were significant differences in coagulation status index between two different concentration of PM2.5 in hyperlipidemic rats (P<0.05; Figure 2A, 2B, 2C). In both groups, pairwise comparison between treatments of same dose also showed significant difference (P<0.05; Figure 2A, 2B, 2C). No vascular pathological abnormalities were found in rats fed with normal diet at different PM2.5 exposure concentrations( Figure 2D1, Figure 2D2, Figure 2D3), while thrombosis was found in rats fed with high-fat diet when they were exposed to PM2.5 at dose of 40 mg/kg body weight( Figure 2D6).
Influence of exposure to PM2.5 on the rat's blood coagulation system under the different physiological status. The plasma was collected from the rat fed on high fat diet at the time point of 24 h after the last dripping of PM2.5. A, the value of FIB in plasma; B, the value of PT in plasma; C, the value of APTT in plasma; D, HE staining of rat blood vessels. The date are shown as mean ± SD (n=7), * P<0.05 indicating statistically significant difference; ** P<0.01 indicating very significant difference. The blood vessels were taken from the aortic arch.
Effects of PM2.5 exposure on the blood pressure and heart rate of hyperlipidemic rats
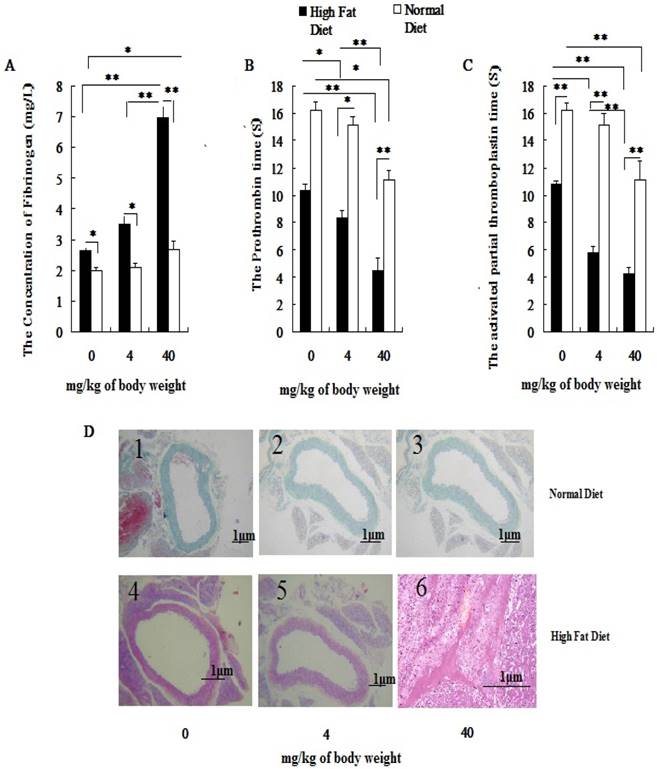
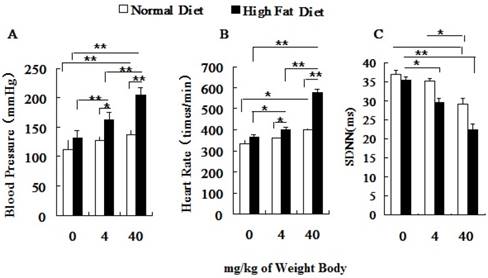
Both the blood pressure (BP) and the heart rate (HR) of hyperlipidemic rats were significantly higher than those of the normal rats before PM2.5 exposure (Figure 3A, 3B). After PM2.5 exposure, with an increase of its concentration, the blood pressure, heart rate of hyperlipidemic rats significantly increased and standard deviation of NN intervals (SDNN) decreased greatly, and all indexes were significantly different from the control rats (P<0.01). Pairwise comparison between the two different doses in hyperlipidemic rats also showed significant differences (P<0.01). Changes in BP, HR and SDNN only happened at dose of 40 mg/kg body weight in normal rats. In hyperlipidemic rats, blood pressure and heart rate showed a more obvious raise with the increasing doses of PM2.5, while the heart rate variability index, represented by the SDNN showed a significant reduction (Figure 3C).
Influence of PM2.5 on the variation of blood pressure and heart rate. ECG was measured at 24h after the last dripping. A, blood pressure; B, heart rate; C, SDNN; The data are presented as mean ± SD (n=7); * P <0.05 indicating statistically significant difference; ** P <0.01 indicating very significant difference.
Effects of PM2.5 exposure on myocardial enzymes, oxidative stress, inflammation
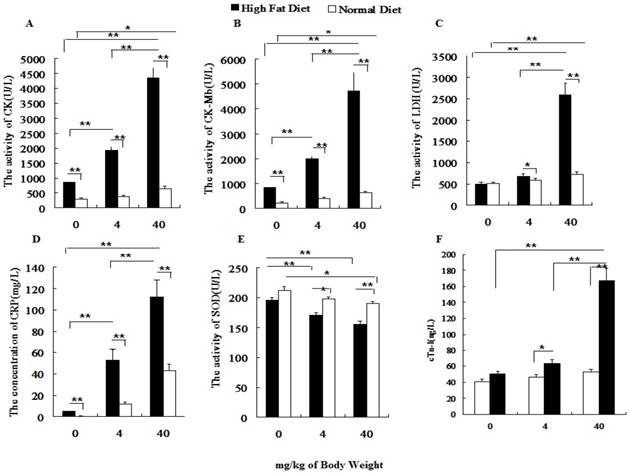
Before PM2.5 exposure, hs-CRP, CK, CK-MB in normal rats were significantly lower than those of hyperlipidemic rats. After the PM2.5 exposure, the hs-CRP, CK, CK-MB, LDH and cTnI levels significantly increased in hyperlipidemic rats at different doses of PM2.5, while, all indexes were changed greatly only at dose of 40 mg/kg weight body (P<0.01) in normal rats. In addition, under the same exposure concentration, the serum hs-CRP, cTnI, CK, CK-MB and LDH levels of hyperlipidemic rats were significantly higher than those of the normal rats (P<0.01; Figures 4A-4D, 4F), whereas the SOD level was significantly lowered (P<0.01; Figure 4E).
Influence of PM2.5 exposure on serum biochemical indexes at different status. Blood sample was collected from abdominal aorta at 24h after the last exposure to PM2.5. A, the activity of CK in serum; B, the activity of CK-MB in serum; C, the activity of LDH in serum; D, the concentration of CRP in serum; E, the activity of SOD in serum; F, the concentration of cTn-I in serum; The data are presented as mean ± SD (n=7)
Cardiomyocyte apoptosis caused by PM2.5 exposure
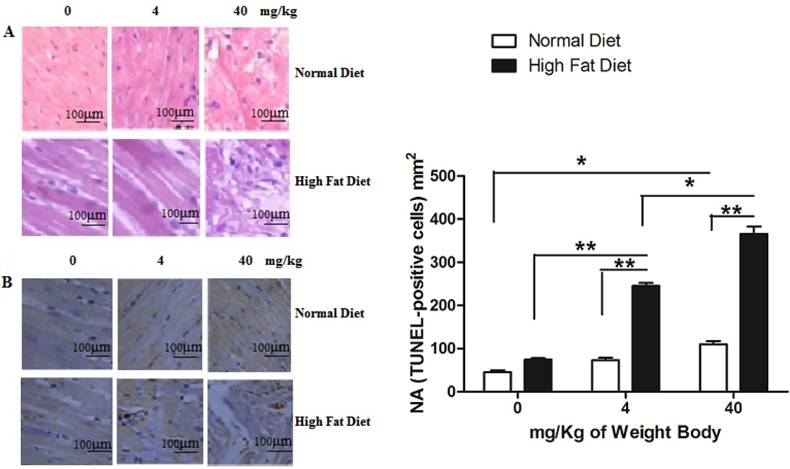
Clinical evidences have proved that PM2.5 exposure can initiate susceptible population myocardial infarction. In our study, we found that instillation with saline and low concentrations of PM2.5 did not significantly affect the structure of myocardial tissues in the normal rats, and their myocardial fibers were well organized with no pathological changes, such as myocardial fibrosis or necrosis. Only treated with PM2.5 at 40 mg/kg weight body, mild injury was observed in cardiomyocytes (Figure 5A). In hyperlipidemic rats instilled with saline, the myocardial tissue showed normality (Figure 5A). However, when instilled with low concentrations of PM2.5, they showed some pathological changes in the myocardial tissue (Figure 5A), while with high concentrations of PM2.5, extensive myocardial fibrosis, necrosis, telangiectasia and myocardial interstitial edema were observed (Figure 5A). TUNEL analysis indicated that with the increase of PM2.5 concentration, the apoptotic cardiomyocytes increased correspondingly. Moreover, the number of TUNEL-positive cells of hyperlipidemic group were higher than that of the normal group (Figure 5B).
Pathological changes of myocardial tissue after PM2.5 exposure. A, HE staining of rat myocardial tissue; B, The TUNEL staining of rat myocardial tissue. The data are shown as mean ± SD (n=7), * P<0.05 indicating a statistically significant difference, and ** P<0.01 indicating a very significant difference. Pathological samples were obtained from the rat's left ventricle, and the sampling site for each rat was the same.
PM2.5 exposure activated the JNK/P53 pathway
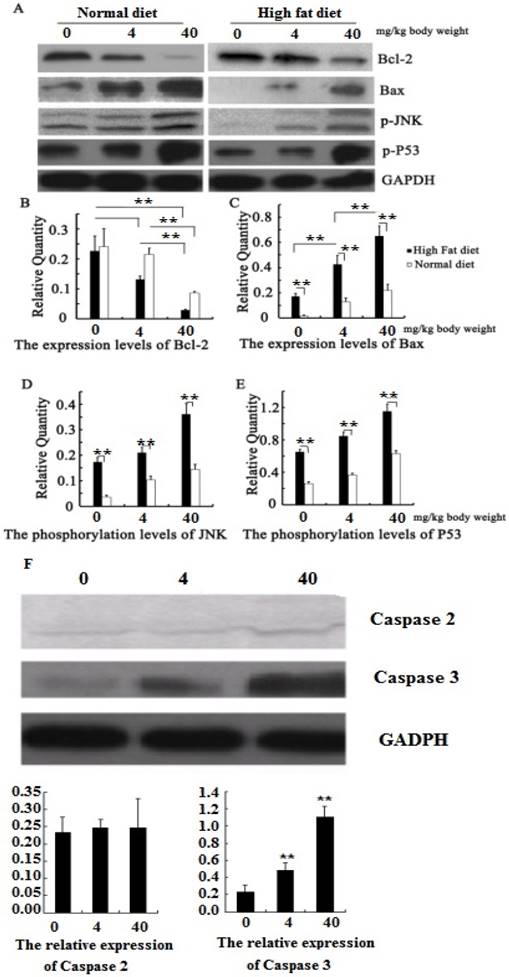
We have found that PM2.5 exposure could induce cardiomyocyte apoptosis, but the mechanism is unclear. Our further study found that the phosphorylation levels of JNK and P53 in the myocardial tissue increased significantly in hyperlipidemic rats or in normal rats after PM2.5 exposure (P<0.01; Figure 6 AB), with a corresponding increase in the pro-apoptotic Bax levels. At the same exposure concentration, the expression levels of p-JNK, p-p53 and Bax in the cardiomyocytes of hyperlipidemic rats were far higher than those of the control rats (P<0.01). However, the Bcl-2 expression level showed the opposite trend (Figure 6 AB). To understand how PM2.5 triggered cardiomyocyte apoptosis via JNK or P53, we further detected the change of its downstream effective protein to evaluate which pathways are important. The result showed that both JNK and P53 expression were increased with the concentration of PM2.5 exposure, and also found that the expression of caspase2 mediated by P53 was not significantly changed while the expression of caspase3 regulated by JNK were increased with the concentration of PM2.5 exposure (Figure 6 F).
Quantitative analysis of the effects of PM2.5 on p53 / JNK and its dowmstream proteins. A, the expression of proteins by Western blotting; B, the protein expression levels of Bcl-2; C, the protein expression levels of Bax; D, the phosphorylation levels of JNK; E, the phosphorylation levels of P53; F, the expression of caspase 2 and 3 after PM2.5 exposure in hyperlipemic rats. The data are presented as mean ± SD (n=5); * P<0.05 indicating a statistically significant difference, and ** P<0.01 indicating a very significant difference.
Characteristics of PM2.5 components and mass concentration in winter from Beijing urban area
Table 1 showed the concentration range of PM2.5, its adsorbed metal elements and polycyclic aromatic hydrocarbons (PAHs) from Oct. to Dec. 2012 in Beijing.
Air quality monitoring parameters
| Component | Low concentration (μg/m3 ) | High concentration (μg/m3 ) |
|---|---|---|
| particle | 81.800 | 173.800 |
| Al | 2.050 | 2.160 |
| Ca | 7.800 | 13.600 |
| Fe | 1.510 | 1.820 |
| Mg | 4.800 | 5.600 |
| Na | 3.800 | 4.400 |
| As | 0.009 | 0.0230 |
| Zn | 0.270 | 3.130 |
| Cu | 0.0460 | 0.160 |
| Cd | 0.003 | 0.0230 |
| Pb | 0.093 | 0.350 |
| PAH(4) | 0.072 | 0.2010 |
| PAH(5,6) | 0.082 | 0.2920 |
Discussion
PM2.5 exposure causes great harm to human health, resulting in various diseases such as asthma, bronchitis and cardiovascular diseases [4,6]. Epidemiological survey and toxicological experiments show that prolonged exposure to PM2.5 may induce oxidative stress and inflammatory response in lung and whole body, increasing the risk of death caused by atherosclerosis (AS) and ischemic heart disease [12-13]. Short term exposure to the high concentrations of PM2.5 may trigger cardiovascular complications, such as vulnerable plaque rupture, thrombosis and acute ischemic heart and cerebrovascular events [31]. Epidemiological data also suggest that people with obesity and hyperlipidemia, children, elderly, people with heart and lung diseases, as well as immunocompromised patients are more susceptible to PM2.5 exposure than healthy people [32-33]. Hyperlipidemia, a type of metabolic disorder, has had a high prevalence rate in recent years, as well as being a high risk factor for other cardiovascular diseases. In Jilin Province, northeast China, the prevalence of hyperlipidemia among participants reached 51.09% [34], and the prevalence of hyperlipidemia among subjects from the southwestern China was 49.3% [35], and these people were easy suffered from CAD when PM2.5 pollution happened. Rats induced with high fat diet present typical symptoms, making them suitable for studying the toxic effects of PM2.5 exposure in animals that are susceptible to cardiovascular diseases. And the cardiovascular events resulting from PM2.5 exposure can be diagnosed by ECG and related physiological and biochemical indexes [36]. Besides that, studies found that SD rats inhaled concentrated ambient particles, their myocardial LDH level increased by 5%, while their serum LDH level increased by 80%, and CK, CK-MB and CTnI levels also showed different degrees of elevation [37-38]. In this study, we employed these indexes to detect the effect of PM2.5 exposure on hyperlipidemic rats. In addition, the damage to human health due to PM2.5 is associated not only with its own characteristics and pollution levels, but also with the tolerance of the exposed population [39-41]. Beijing is typical PM2.5 pollution region for susceptible population, so we utilized PM2.5 collected from Beijing to study its impact on hyperlipidemic rats.
This study showed that exposure to high concentrations of PM2.5 resulted in significant change in ECG and blood pressure in hyperlipidemic rats. Their heart rates and blood pressures increased with the increase of PM2.5 concentration, indicating a dose effect relationship. Simultaneously, their blood biochemical indexes, such as myocardial enzymes, acute-phase proteins and coagulation parameters also changed obviously. We also verified that after PM2.5 exposure, the CK, CK-MB, LDH, cTnI, CRP, SOD, PT, APTT and FIB levels were significantly different from the corresponding control groups in both groups of rats under different pathological states. In particular, hyperlipidemic rats showed more drastic changes compared to normal rats at the same exposure concentration, causing remarkable changes in the myocardial enzyme profile. The pathological assay also showed that the degree of myocardial injury in hyperlipidemic rats exposed to PM2.5 was significantly higher than that in normal rats which did not show significant changes in the cardiac structure and function after exposure to the high concentrations of PM2.5. In hyperlipidemic rats, exposure to low PM2.5 concentration did not result in severe consequences, but exposure to high concentration of PM2.5 caused thrombosis and blockage of blood vessels in some rats (Fig.2). We also found that PM2.5 exposure could induce more severe reactions of acute inflammation and oxidative stress in hyperlipidemic rats, which could lead to changes in the blood composition and coagulation system, indicating that the toxicological effects caused by PM2.5 exposure could vary under different pathological conditions.
During myocardial ischemia, a large amount of cardiomyocyte apoptosis occurs. With Bcl-2 and Bax being the main genes regulating cardiomyocyte apoptosis, both of them are regulated by the JNK and P53 signaling pathway [26-28]. We also found that the phosphorylation levels of JNK and P53 raised with the increase in concentration of PM2.5 exposure. The expression level of Bax was much higher in hyperlipidemic rats than that in normal rats, while Bcl-2 was significantly reduced, which may be the reason for the high occurrence of cardiomyocyte apoptosis in hyperlipidemic rats. Caspases are executors of apoptosis, and are mediated by different signal molecules. In the process of apoptosis, the caspase2 can be regulated by P53 only to execute the death of cell, and caspase3 can be adjusted by JNK/P53 or JNK to achieve the cell death. In this study, we found that PM2.5 exposure could not influence the expression of caspase2, but caspase3 increased with the concentration of PM2.5 exposure. These results indicated that PM2.5 exposure could easily affect JNK/P53/Caspase3 or JNK/Caspse3 to induce the cardiomyocyte apoptosis, which may be the main reason for cardiovascular diseases induced by PM2.5 exposure.
In addition, the toxic effects of PM2.5 exposure are closely associated with its composition and concentration. Beijing, as the representative metropolis in developing countries, has serious PM2.5 pollution. From 2010 to 2012, its daily average PM2.5 value was 96.2 μg/m3, which is 3-7 times of the WHO standard [42]. Its highest value exceeds 600 μg/m3 on a haze day. As the minute tidal volume of rat is about 0.16 L [43], the PM2.5 exposure dose for rat in the haze day could be about 1.7 mg/d, which is equivalent to the PM2.5 exposure dose (4 mg/kg) used in our study. We selected the exposure concentrations of 4-40 mg/kg, in order to better illustrate the impact of PM2.5 exposure on the cardiovascular system. The possible differences in the impact of PM2.5 collected from other regions and the toxic effects of exposure at other concentration levels on cardiomyocyte apoptosis need to be further studied.
Furthermore, we found that PM2.5 exposure could more easily induce oxidative stress and inflammation in hyperlipidemic rats, promoted a hypercoagulable state, which may affect the cardiac function.
Conclusions
This study first identified that JNK or JNK/p53 was involved in the cardiomyocyte apoptosis induced by PM2.5 exposure, which regulated its downstream effector genes, Bcl-2 and Bax, and then increased the expression of caspas3 leading to the myocardial injury. PM2.5 exposure had bigger effect on JNK/p53 pathway in hyperlipidemic rats than that in normal rats, which may be the mechanism for hyperlipidemic rats being more susceptible to PM2.5 exposure. It provides the basis for further exploring the toxic effects of PM2.5 exposure on the cardiovascular system and the mechanism of acute cardiovascular event induced in susceptible populations.
Acknowledgements
This work was supported by National Natural Science Foundation of China (NO. 81170255) (NO.81770857) and National 973 Project (2011CB503803). The authors are very grateful to the Pathological Department of 307 Hospital and Professor Lu Yinglin for their guidance, and Doctor Wang Sun-Tao of Beijing Armed Police Hospital for blood biochemical assays.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lary D J, Lary T, Sattler B. Using Machine Learning to Estimate Global PM2.5 for Environmental Health Studies. Environ Health Insights. 2015;9(Suppl 1):41-52
2. Ning L, Sioutas C, Cho A. et al. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environmental Health Perspectives. 2003;111(4):455-460
3. Ferin J, Oberdorster G, Penney D P. Pulmonary retention of ultrafine and fine particles in rats. Am J Respir Cell Mol Biol. 1992;6(5):535-542
4. Simkhovich B Z, Kleinman M T, Kloner R A. Air pollution and cardiovascular injury epidemiology, toxicology, and mechanisms. Journal of the American College of Cardiology. 2008;52(9):719-726
5. Nemmar A, Hoet P H M, Vanquickenborne B. et al. Passage of Inhaled Particles Into the Blood Circulation in Humans. Circulation. 2002;105(4):411-414
6. Brook R D, Franklin B, Cascio W. et al. Air Pollution and Cardiovascular Disease A Statement for Healthcare Professionals From the Expert Panel on Population and Prevention Science of the American Heart Association. Circulation. 2004;109(21):2655-2671
7. Sun Q, Wang A, Jin X. et al. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. Jama the Journal of the American Medical Association. 2005;294(23):3003-3010
8. Dominici F, Peng R D, Bell M L. et al. Fine Particulate Air Pollution and Hospital Admission for Cardiovascular and Respiratory Diseases. Jama the Journal of the American Medical Association. 2006;295(10):1127-1134
9. Brook R D, Rajagopalan S, Rd P C. et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331-2378
10. Wilker E H, Mittleman M A, Coull B A. et al. Long-term exposure to black carbon and carotid intima-media thickness: the normative aging study. Environmental Health Perspectives. 2013;121(9):1061-1067
11. Seaton A, Macnee W, Donaldson K. et al. Particulate air pollution and acute health effects. Lancet. 1995;345(8943):176-178
12. Ostro B, Malig B, Broadwin R. et al. Chronic PM2.5 exposure and inflammation: determining sensitive subgroups in mid-life women. Environmental Research. 2014;132:168-175
13. Neophytou A M, Hart J E, Cavallari J M. et al. Traffic-related exposures and biomarkers of systemic inflammation, endothelial activation and oxidative stress: a panel study in the US trucking industry. Environmental Health. 2013;12(1):105-114
14. Hamm C W, Goldmann B U, Heeschen C. et al. Emergency room triage of patients with acute chest pain by means of rapid testing for cardiac troponin T or troponin I. N Engl J Med. 1997;337(23):1648-1653
15. Antman E, Bassand J P, Klein W. et al. Myocardial infarction redefined—a consensus document of The Joint European Society of Cardiology/American College of Cardiology committee for the redefinition of myocardial infarction: The Joint European Society of Cardiology/ American College of Cardio. Journal of the American College of Cardiology. 2000;36(3):959-969
16. Katus H A, Diederich K W, Hoberg E. et al. Circulating cardiac myosin light chains in patients with angina at rest: identification of a high risk subgroup. Journal of the American College of Cardiology. 1988;11(3):487-493
17. Smith S C, Ladenson J H, Mason J W. et al. Elevations of Cardiac Troponin I Associated With Myocarditis Experimental and Clinical Correlates. Circulation. 1997;95(1):163-168
18. Wu S, Deng F, Jing H. et al. Blood Pressure Changes and Chemical Constituents of Particulate Air Pollution: Results from the Healthy Volunteer Natural Relocation (HVNR) Study. Environmental Health Perspectives. 2013;121(1):66-72
19. Wu S, Deng F, Wei H. et al. Chemical constituents of ambient particulate air pollution and biomarkers of inflammation, coagulation and homocysteine in healthy adults: A prospective panel study. Particle & Fibre Toxicology. 2012;9(1):49
20. Lim L S, Haq N, Mahmood S. et al. Atherosclerotic Cardiovascular Disease Screening in Adults: American College of Preventive Medicine Position Statement on Preventive Practice. American Journal of Preventive Medicine. 2011;40(3):381.e1
21. Yang S R, Rahman I, Trosko J E. et al. Oxidative stress-induced biomarkers for stem cell-based chemical screening. Preventive Medicine. 2012;54(3):S42-S49
22. Rd P C. Epidemiology of fine particulate air pollution and human health: biologic mechanisms and who's at risk? Environmental Health Perspectives. 2000;108(Suppl 4):713-723
23. Schicker B, Kuhn M, Fehr R. et al. Particulate matter inhalation during hay storing activity induces systemic inflammation and platelet aggregation. European Journal of Applied Physiology. 2009;105(5):771-778
24. Foster C R, Daniel L L, Daniels C R. et al. Deficiency of Ataxia Telangiectasia Mutated Kinase Modulates Cardiac Remodeling Following Myocardial Infarction: Involvement in Fibrosis and Apoptosis. Plos One. 2013;8(12):e83513
25. Jiang H K, Wang Y H, Sun L. et al. Aerobic Interval Training Attenuates Mitochondrial Dysfunction in Rats Post-Myocardial Infarction: Roles of Mitochondrial Network Dynamics. International Journal of Molecular Sciences. 2014;15(4):5304-5322
26. Cisternas P, Moreno R D. Comparative analysis of apoptotic pathways in rat, mouse, and hamster spermatozoa. Molecular Reproduction & Development. 2006;73(10):1318-1325
27. Long X, Boluyt MO, Hipolito ML. et al. p53 and the hypocia-induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest. 1997;99(11):2635-2643
28. Watanabe H, Ohta S, Kumon Y. et al. Increase in p53 protein expression following cortical infarction in the spontaneously hypertensive rat. Brain Research. 1999;837(1-2):38-45
29. Zhu X M, Wang Q, Xing W W. et al. PM2.5 induces autophagy-mediated cell death via NOS2 signaling in human bronchial epithelium cells. International Journal of Biological Sciences. 2018;14(5):557-564
30. Zhang Q, Wang G, Jiye A. et al. Application of GC/MS-based metabonomic profiling in studying the lipid-regulating effects of Ginkgo biloba extract on diet-induced hyperlipidemia in rats. Acta Pharmacologica Sinica. 2009;30(12):1674-1687
31. Bai N, Khazaei M, Van S E. et al. The pharmacology of particulate matter air pollution-induced cardiovascular dysfunction. Pharmacology & Therapeutics. 2007;113(1):16-29
32. Colais P, Faustini A, Stafoggia M. et al. Particulate Air Pollution and Hospital Admissions for Cardiac Diseases in Potentially Sensitive Subgroups. Epidemiology. 2012;23(3):473-481
33. Khot U N, Khot M B, Bajzer C T. et al. Prevalence of conventional risk factors in patients with coronary heart disease. JAMA. 2003;290:898-904
34. Rao W, Su Y, Yang G. et al. Cross-Sectional Associations between Body Mass Index and Hyperlipidemia among Adults in Northeastern China. International Journal of Environmental Research & Public Health. 2016;13(5):516-525
35. Deng B, Luo T, Huang Y. et al. Prevalence and determinants of hyperlipidemia in moderate altitude areas of the Yunnan-Kweichow plateau in Southwestern China. High Altitude Medicine & Biology. 2012;13(1):13-21
36. Roger V L, Weston S A, Gerber Y. et al. Trends in incidence, severity, and outcome of hospitalized myocardial infarction. Circulation. 2010;121(7):863-869
37. Gurgueira S A, Lawrence J, Coull B. et al. Rapid increases in the steady-state concentration of reactive oxygen species in the lungs and heart after particulate air pollution inhalation. Environ Health Perspective. 2002;110(8):749-755
38. Carlsten C, Kaufman J A, Trenga C. et al. Coagulation markers in healthy human subjects exposed to diesel exhaust. Thrombosis Research. 2007;120(6):849-855
39. Graham J J. Fine Particulate Matter National Ambient Air Quality Standards: Public Health Impact on Populations in the Northeastern United States. Environmental Health Perspectives. 2005;113(9):1140-1147
40. Shiraki R, Holmen B A. Airborne Respirable Silica near a Sand and Gravel Facility in Central California: XRD and Elemental Analysis to Distinguish Source and Background Quartz. Environmental Science & Technology. 2002;36(23):4956-4961
41. Sacks J D, Stanek L W, Luben T J. et al. Particulate Matter-Induced Health Effects: Who Is Susceptible? Environmental Health Perspectives. 2011;119(4):446-454
42. Xie W, Li G, Zhao D. et al. Relationship between fine particulate air pollution and ischaemic heart disease morbidity and mortality. Heart. 2015;101(4):257-263
43. Crosfill M L, Widdicombe J G. Physical characteristics of the chest and lungs and the work of breathing in different mammalian species. J Physiol. 1961;158(1):1-14
Author contact
![]() Corresponding authors: Donggang Xu and Minhui Long; xudgac.cn and longminhui2006com
Corresponding authors: Donggang Xu and Minhui Long; xudgac.cn and longminhui2006com