Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Premalignant niche
Suitable microenvironment for...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(1):105-113. doi:10.7150/ijbs.28669 This issue Cite
Review
Remodeling the Microenvironment before Occurrence and Metastasis of Cancer
Xina Zhang1,2,3, Juanjuan Xiang1,2,3 ![]()
1. Hunan Cancer Hospital, The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, Hunan, PR China
2. Cancer Research Institute, School of Basic Medical Science, Central South University, Changsha, Hunan, China.
3. The Key Laboratory of Carcinogenesis of the Chinese Ministry of Health, Xiangya Hospital, Central South University, Changsha, Hunan, China.
Received 2018-7-21; Accepted 2018-10-24; Published 2019-1-1
Abstract

Tumorigenesis and progression of cancer are complex processes which transformed cells and stromal cells interact and co-evolve. Intrinsic and extrinsic factors cause the mutations of cells. The survival of transformed cells critically depends on the circumstances which they reside. The malignant transformed cancer cells reprogram the microenvironment locally and systemically. The formation of premetastatic niche in the secondary organs facilitates cancer cells survival in the distant organs. This review outlines the current understanding of the key roles of premalignant niche and premetastatic niche in cancer progression. We proposed that a niche facilitates survival of transformed cells is characteristics of senescence, stromal fibrosis and obese microenvironment. We also proposed the formation of premetastatic niche in secondary organs is critically influenced by primary cancer cells. Therefore, it suggested that strategies to target the niche can be promising approach to eradicate cancer cells.
Keywords: premalignant niche, premetastatic niche, co-evolution
Introduction
It is widely accepted that tumorigenesis is a multistage process during which molecular alterations in the genome of somatic cells accumulate. Gene mutations force normal cells to grow abnormally. Although most DNA replicates with fairly high fidelity, mistakes do happen[1]. However, the link between mutation and cancer incidence appears to be more complex[2]. People also observed that only a fraction of cells within tumors were capable of clonogenic growth. The heterogeneity among cancer cells can arise in multiple ways. Two theories have been proposed to explain this heterogeneity: extrinsic factors and intrinsic factors[3]. Evolutionary theories are always applied to understand how cancers develop and how heterogeneity exists. In this way, carcinogenesis is viewed as a Darwinian process of successive rounds of selection leading to the accumulation of mutations[4, 5]. Cells face diverse selective pressures as they react to changes in their environment[6]. The Darwin theory of evolution has been determined by the match between the current environmental demand and the phenotypic manifestation of mutations[5]. The competitive advantage of mutations during tumor initiation is dependent on the context in which they arise. Below, we will focus on cooperative relationship between transformed cells and microenvironment on the initiation and development of cancer (Figure 1).
Schematic diagram of formation of the premalignant niche and premetastatic niche. Mutations result either from DNA replication errors or from the damaging events. Accumulation of unrepaired mutations transforms normal cells. The survival of transformed cells critically depends on the circumstances which they reside. The niche at high risk of malignant transformation is associated with aging, fibrosis and obesity. Bone marrow-derived cells (BMDC) including haematopoietic progenitors, mesenchymal stem cells, endothelial progenitor cells comprise the main component in the premetastatic niche. Tumor-derived secreted factors (TDSFs) are crucial in creating a supportive microenvironment at the metastatic site. Chemokines or cytokines derived from the primary cancer cells reprogramming the distant organs and contribute to the establishment of premetastatic niche. Exosomes participate in cell-to-cell communication by the molecules enriched in their membrane, remodeling the microenvironment of target organs and help the formation of premetastatic niche.
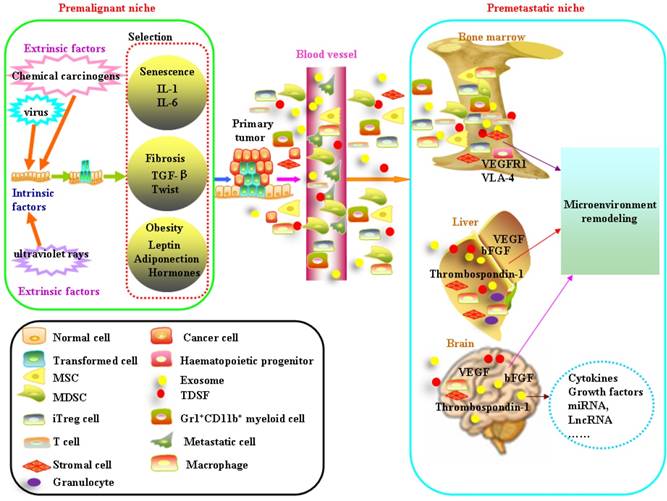
Premalignant niche
Mutations result either from DNA replication errors or from the damaging events. Accumulation of unrepaired mutations transforms normal cells. The survival of transformed cells critically depends on the circumstances which they reside. The niche at high risk of malignant transformation is associated with aging, fibrosis and obesity.
Senescence-messaging secretome (SMS)
Aging is the biggest risk factor for cancer. By 2030, 70% of the tumor will occur in the population of 65 years and older[7]. In humans, cancer incidence rises with approximately exponential kinetics after 50 years of old[8], partly due to accumulation of oncogenic mutations over time. Cellular senescence, which is associated with aging, is a state of irreversible growth arrest[9]. It was previously assumed that senescence was functionally similar to apoptotic cell death[10]. However, senescent cells show marked and distinct changes in their pattern of gene expression [8, 11]. It is reported that tissue microenvironment is the main cause of the occurrence of age-related tumors [12-14]. The senescence-associated secretory phenotype (SASP; also known as the senescence-messaging secretome (SMS)) provides senescent cells with diverse functionality[10]. The nature of the SMS and its targets, and the overall downstream outcomes, vary considerably depending on the cellular context[10]. On the one hand, SMS can aid tissue repair, but on the other hand, the SMS can do great damage to normal tissue structures and function, promote malignant phenotypes in nearby cells[8]. The SMS contains several families of factors that can be divided into the following 3 major categories: soluble signaling factors (interleukins, chemokines, and growth factors), secreted proteases, and secreted insoluble components[9]. These SMS accumulate with age that may change the tissue microenvironment and promote the occurrence of chronic inflammatory diseases or tumors. The most prominent soluble factors are Interleukin-1 (IL-1) and Interleukin-6 (IL-6). IL-1 and IL-6 have been proposed as major upstream regulators of the senescence-associated cytokine network[10]. Circulating IL-1 and IL-6 promote a variety of chronic degenerative diseases, as well as cancer[15-23]. There is little measurable IL-6 in the circulation in the absence of inflammation. With advancing age, however, serum IL-6 becomes detectable even without evidence of inflammation. It is proposed that this reflects an age-associated loss in the normal regulation of gene expression for this molecule. There is also speculation that IL-6 may contribute to the pathogenesis of several diseases that are common in late-life including lymphoma, osteoporosis, and Alzheimer's disease[24, 25]. Senescent fibroblast cells also express IL-8, GROα, GROβ, MCP-2, MCP-4, MCP-3, MCP-1, MIP-3α, MIP-1α, CCL-1, IGFBPs, GCSF, GM-CSF[9]. Aside from soluble signaling cytokines and growth factors, senescent cells also secrete increased levels of matrix metalloproteinases (MMPs) and other molecules such as reactive oxygen species(ROS), nitric oxide and transported ions.
Fibrotic niche
Pathologic fibrosis is the feature of abnormal extracellular matrix (ECM) deposition caused by prolonged injury and deregulated processes of wound healing. Extracellular matrix proteins including collagen, elastin, fibronectin and laminin are abundant in ECM. Fibrotic diseases encompass a wide spectrum of entities including idiopathic pulmonary fibrosis (IPF),silicosis, asbestosis, ischemic heart disease, cirrhosis, splenic fibrous hyperplasia et al. It has long been observed that fibrosis is related to carcinogenesis. In some aspects, a tumor can be viewed as a fibrotic organ that contains cancer cells. A tumor cannot develop without the parallel expansion of a tumor stroma[26]. Fibroblasts are involved in tissue remodeling and repair. Physiologic fibroblasts maintain stromal homeostasis. Pathologic fibrosis always began with reaction to inflammation, characterized by pathologic fibroblasts and a stiff ECM[27]. Transforming growth factor-β (TGF-β) is the most predominant profibrotic growth factor, leading to an increase of collagen and fibronectin production by fibroblasts and the transition of fibroblasts into myofibroblasts. TGF-β also activates other stromal cells such as hepatic stellate cells (hStCs) to produce fibronectin (FN).
The increased rigidity of ECM is an important determinant for cell behavior[28, 29]. Substrate rigidity influences cell morphology, cell migration and cell growth[28]. Upon sensing force, cells react by active change in the actin cytoskeleton. The rigidity of ECM regulates localization and activity of YAP/TAZ[30]. The process of Epithelial-Mesenchymal-Transition (EMT) is known to result in a phenotype change in cells which are in a more invasive state[31]. Mechanical stress induces EMT by both physical forces and biochemical signals[32, 33]. Mechanical stress induces Twist expression in a manner dependent on β-catenin[34]. Recent studies highlight a link between EMT and cancer stem cell formation[35].
In denser, stiffer matrices (∼44 kPa), murine mammary epithelial cells displayed more invasive phenotypes, compared to the softer matrices of lower density (∼25 kPa)[36]. Pressure to osteocytes, the main mechanotransducing cells in bone, induces prostate cancer growth and invasion[29].The mechanical microenvironment may cause malignant transformation, possibly through stimulating intracellular signaling pathways that promote cancer cell survival or invasion[28, 37]. Matrix density-induced stiffness regulates epithelial cell phenotype, promotes cellular adhesion through a FAK-ERK signaling[36]. Transmembrane cell adhesion proteins, mostly integrins link the extracellular matrix to the cell's cytoskeleton. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages[38]. Besides the roles in cell adhesion, integins have an activating role in intracellular signaling events as signaling receptors. The different heterodimer with alpha and beta subunits cause the functional and molecular diversity of integrins. Upon integrin aggregation, a number of signal transduction pathways, such as the Raf-ERK/MAPK, PI3K-Akt, nuclear factor-kappa B (NF-κB), and c-Jun are activated[39].
In addition, substrate stiffness modulates genes expression, such as focal adhesion proteins (Itga6 and Parvb), cytoskeletal proteins (Dnah11 and Actb), and nuclear envelope protein Lbr and Nrm [40].The mechanisms how mechanical stresses affect the expression of genes that influence cell adhesion, migration can be explained by nucleus shape or LINC-mediated linking between cytoskeleton to the nucleoskeleton[40, 41]. Dynamic force-induced structural changes in Cajal bodies, a prominent nuclear body, may affect nuclear functions involved in gene expression[42]. Mechanical stresses can be transmitted from the cytoskeleton to the nucleus by LINC (linker of nucleoskeleton and cytoskeleton) complex that is composed by SUN1/2 proteins and regulates gene expression [40, 43].
Obesity niche
More and more evidence demonstrated obesity is linked to the increased risk of cancer incidence and mortality[44]. Obesity is excess fat in the body[45]. Obesity is closely related to type 2 diabetes, hyperlipidemia, hypertension and cardiovascular and cerebrovascular diseases[45-47]. Obesity is quickly overtaking tobacco as the leading preventable cause of cancer. Obesity contributes to the occurrence and development of cancer systemically or locally through affecting energy imbalance including insulin resistance, altered hormone signaling, and high circulating levels of proinflammatory mediators[44]. Hyperadiposity as a result of excess caloric intake or reduced caloric expenditure cause production of steroid hormones and adipokines. Adipose tissue has been considered to be the largest endocrine organ in the body, producing adipokines, cytokines and chemokines involved in metabolism and immune regulation[44-53]. A crosstalk between estrogen, insulin, insulin-like growth factor-1 (IGF-1) and adipokine signaling pathways plays an important role in the development of cancer[51]. Adipokines, including leptin and adiponectin, are hormones produced by adipocytes. There are conflicting data regarding the roles of adiponectin in the development of cancer. Circulating plasma concentrations of Adiponectin are inversely related to increased risks of malignancy. Decreased level of adiponectin are present in patients with breast cancer, prostate cancer, gastric cancer, et al[54]. Adiponectin inhibits cancer cell proliferation and promotes cancer cell apoptosis through inhibiting STAT3, PI3K/AKT, Wnt signaling[54]. However, increased adiponectin have been associated with increased risk of lung cancer and hepatic cancer[55, 56]. higher levels of adiponectin or higher adiponectin/leptin ratios in pancreatic cancer patients with positive or strongly positive expression of adiponectin receptor 1 and receptor 2[57, 58]. Adiponectin is also reported to have proliferative effect on cancer cells through enhancing ceramide catabolism and anti-apoptotic metabolite S1P[59]. Leptin and its receptor OB-R have been implicated in a number of malignancies through activating Janus kinase/Signal transducer and activator of transcription (JAK/STAT), PI3K/Akt and extracellular regulated protein kinases(ERK). Leptin increases the expressions of the tumor necrosis factor-alpha(TNF-α), interleukin-6(IL-6), vascular endothelial growth factor (VEGF) and hypoxia inducible factor-1alpha (HIF-α). It improves the ability of tumor cells to resist apoptosis, angiogenesis and hypoxia tolerance, which is beneficial to the progression and metastasis of tumor[60-62]. Besides, proinflammatory mediators such as C- reactive protein (CRP), TNF-α, IL-6, IL-8 in the circulation which is produced by adipocytes promote neoplasia and tumor progression locally and systemically[63]. Obesity-induced interstitial fibrosis promotes breast tumorigenesis by altering mammary ECM mechanics with important potential implications for anticancer therapies[64]. Insulin resistance is associated with worse prognosis in several cancers and insulin can stimulate the synthesis of IGF-1, which is linked to tumor progression. Insulin and IGF-1 activate the PI3K/Akt/mTOR and Ras/Raf/ MAPK pathways[65-70]. Obesity enhances local myofibroblast content in mammary adipose tissue and that these stromal changes increase malignant potential by enhancing interstitial ECM stiffness[64].
Suitable microenvironment for cancer progression
Premetastatic niche
Metastasis is the most life threatening event in cancer patients[71]. Metastasis can occur when cells break away from a primary tumor and travel through blood stream or through lymph vessels to other areas of the body, which is responsible for approximately 90% of cancer deaths[72]. The earlier theories regarded metastasis as a process of orderly anatomic spread[73]. In contrast, Fisher hypothesized that whether distant relapse occurs is predetermined from the onset of tumorigenesis[74]. It has been also noticed clinically that cancer metastasis does not scale with primary tumor size. Circulating cancer cells can be detected in varied cancers. However, metastasis is largely an inefficient process in which most circulating cancer cells fail to mature in a clinically meaningful fashion[75]. Researchers have identified dormant cancer cells in metastasis-free organs[76]. Disseminated tumor cells were detected in 30% of patients diagnosed with early stage (I-III) breast cancer, however, patients with disseminated tumor cells did not uniformly develop metastatic disease[77]. In 1889, Dr. Stephen Paget stated that metastasis did not occur randomly. His study which is published in The Lancet, demonstrated that metastasis only develop when the seed and soil are compatible. In this respect, tumors may have greater or lesser ability to colonize lymph nodes and distant organs, as driven by their match to the distant microenvironment[78]. Such permissive environment is always formed before arrival of cancer cells, which is called “premetastatic niche”[79].
Premetastatic niche is also composed of a heterogeneous mixture of stromal cells, vasculature, other supportive cells and extracellular matrix[75]. Bone marrow-derived cells (BMDC) including haematopoietic progenitors, mesenchymal stem cells, endothelial progenitor cells comprise the main component in the premetastatic niche. Bone marrow-derived cells (that is, macrophages and granulocytes) bind to FN-enriched hepatic sites, ultimately leading to liver pre-metastatic niche formation[80]. Metastatic cancer cells which overexpress Jagged activate both haematopoietic osteoclasts and mesenchymal osteoblasts by binding to Notch and promote both tumor cell growth and invasion in the bone. Tumor-derived secreted factors (TDSFs) are crucial in creating a supportive microenvironment at the metastatic site. Chemokines or cytokines derived from the primary cancer cells reprogramming the distant organs and contribute to the establishment of premetastatic niche[81]. Besides chronic inflammation, acute inflammation in the lung can foster metastatic seeding[82, 83]. Bacteria- and LPS-induced acute inflammation significantly enhanced lung metastasis. Acute lung infection dramatically increased cancer cell homing to the lung. A large number of the recent exosome literature highlights the roles of cancer cell-derived or stroma cell-derived exosomes on the reprogramming of microenvironment[80, 84, 85]. Exosomes are small, 30 to 100-nm membrane vesicles formed by the inward budding of late endosomes[86-88]. Exosomes contain cytokines, transcription factor, growth factor, and other bioactive molecules such as miRNA, LncRNA et al [89-91]. They participate in cell-to-cell communication by the molecules enriched in their membrane, remodeling the microenvironment of target organs and help the formation of premetastatic niche [80, 92-102](Table 1). Exosomes are widely distributed in various human body fluids, such as blood plasma/ serum, saliva, breast milk, cerebrospinal fluid and urine. Exosomes that are enriched with cancer-specific miRNAs, LncRNAs can be used as biomarkers for cancer progression [103-107].
Exosomes and the formation of premetastatic niche
| Diseases | Exosomal moleculres | Type | Source cell | Target cell | Mechanisms | Reference |
|---|---|---|---|---|---|---|
| PDAC | MIF | protein | PDAC cells | macrophages | Fibrotic niche | [80] |
| BCa | miR-122 | miRNA | BCa cells | fibroblasts | Glucose metabolism niche | [92] |
| PC | miR-301a-3p | miRNA | PC cells | macrophages | Inflammatory niche | [93] |
| HCC | miR-103 | miRNA | Hepatoma cells | Endothelia | Angiogenesis niche | [94] |
| NPC | miR-23a | miRNA | NPC cells | endothelia | Angiogenesis niche | [95] |
| HCC | miR-1247-3p | miRNA | HCC cells | fibroblasts | Fibrotic niche | [96] |
| Gastric cancer | EGFR | protein | gastric cancer cells | stromal cells | Fibrotic niche | [97] |
| BCa | miR-23b | miRNA | MSCs | BCa cells | dormancy niche | [98] |
| HCC | miR-210 | miRNA | HCC cells | endothelial | Angiogenesis niche | [99] |
| Colon cancer | CEACAMs | protein | Colon cancer cells endothelial | T-cells | Immunosuppression niche | [100] |
Notes: PDAC: Pancreatic ductal adenocarcinomas; MIF: macrophage migration inhibitory factor; BCa: breast cancer; PC: pancreatic cancer; HCC: hepatocellular carcinoma.NPC:nasopharyngeal carcinoma;EGFR:epidermal growth factor receptor;MSC:mesenchymal stem cell; CEACAMs: carcinoembryonic antigen related cell adhesion molecules;
There are 6 characteristics and traits that define pre-metastatic niche including immunosuppression, inflammation, high angiogenesis and vascular permeability, active lymphangiogenesis, specific organotropism and high reprogramming efficiency[81].
Angiogenesis niche
Formation of vascular network is important to the proliferation and dissemination of cancer cells. Stable microvessels form a "dormant niche". Factors that sustain the homeostasis such as endothelial-derived thrombospondin-1 induce sustained cancer cells quiescence. When blood vessels begin to sprout, the new tips produce molecules that transform dormant cancer cells into metastatic tumors[108], in which process the thrombospondin-1 proteins give way to tumor necrosis factor (TNF) and periostin proteins in the neovasculature. Many proinflammatory chemokines generated by cancer cells support the development of vessels. TNF-α acts indirectly by inducing the production and release of VEGF and bFGF[109, 110]. CXCR4 promotes the migration of endothelial cells toward stromal cells derived factor SDF-1 to branch and develop new vessels. SDF-1-CXCR4 interaction increases VEGF production by endothelial cells, and VEGF and bFGF in turn enhances SDF-1[109, 110]. VEGFR1+VLA-4+ haematopoietic progenitors move from bone marrow and home to tumour-specific pre-metastatic sites and form cellular clusters before the arrival of tumour cells. These haematopoietic progenitors dictate organ-specific tumour spread through angiogenesis and chemotaxis.
Immunosuppression niche
Cancer cells somehow are like “foreign” material. Immune system recognizes and eliminates the cancer cells during the early phase of cellular transformation. The process of tumor immuno-editing includes the following three key phases: elimination, equilibrium, and escape[111]. During the escape phase, cancer cells resist the selective pressure from the immune system by acquiring mutations or undergoing other changes that allow for tumor progression in the face of an ongoing immune response[111-114]. The escaped cancer cells shape the immune system to be immunosuppressive to allow themselves to grow. Myeloid-derived suppressor cells (MDSC) and regulatory Treg cells are major components of the immune suppressive cells. These suppressor cells alter the microenvironment through the secretion of inflammatory and immunosuppressive cytokines to promote metastasis. There is an accumulation of metabolic enzymes that suppress T cell proliferation and activation, including IDO and arginase, and high expression of tolerance-inducing ligands like FasL, PD-1, CTLA-4, and B7[115-117]. Tumor-derived vesicles known as exosomes have also been implicated promoting differentiation of iTreg cells and myeloid derived suppressor cells (MDSCs)[118]. Suppressive immune cell populations such as Gr1+CD11b+ myeloid cells at secondary organ sites increase regional inflammatory cytokines such as S100A8 and S100A9 that promotes metastatic seeding[119, 120]. Primary tumor induction of S100A8 and S100A9 expression has also been shown to recruit Mac1+ myeloid cells via TLR4 to premetastatic sites[121, 122].
Directional movement of cancer cells to the premetastatic niche
Homing to and seeding in the secondary sites are the most important steps in the process of metastasis. Homing is a rapid process in which cancer cells actively cross the blood/endothelium barrier by adhesion interaction. Cell adhesion molecules (CAMs) are glycoproteins synthesized by cells, which are involved in interaction between cells and cells or cells and matrix, and involve in cell signal transduction and cell migration. CAMs can be divided into four main groups: selectins, integrins, Ig superfamily and cadherins. Integrins are heterodimeric transmembrane adhesion receptors that bind to ECM ligands outside a cell to the actin cytoskeleton inside the cell[123]. There are several types of integrins on the cell surface. The binding specificity allows cells expressing certain integrin heterodimers pass through an ECM containing specific components[124]. Cells sense and respond to their environment through spatio-temporal patterns of integrin versus ligand expression[124]. Ligands of integrins are fibronectin, vitronectin, collagen, and laminin. Integrins play physiological or pathological roles depending on the components of ECM. Integrin αvβ5, α5β1, α6β4, α9β1 on the cell surface help to maintain normal homeostasis through binding to fibronectin TSP-1. Stromal compartment initially inhibit cancer progression by maintaining architecture[125]. Alterations in integrin including aberrant expression and activation of downstream effectors are involved in carcinogenesis[39]. Most circulating cancer cells die in circulation as a result of shear stress and/or anoikis[126]. Activated αvβ3 can keep circulating cancer cells from shear stress by binding to leukocytes and platelets[127]. When circulating cancer cells once arrive at distant organs, integrin-ligand interactions help cancer cells colonize to the metastatic environment[39]. Cancer cell invasion are heavily dependent on integrin-mediated adhesion to the ECM[128]. Intergrin α4β1 permits cancer cell engagement of fibrinogen, ICAM and VCAM are expressed by the vascular and stromal cells of bone marrow[129]. Integrin α5β3 on cancer cells not only helps the cancer cells home to bone marrow through adhering to the vitronectin, osteopontin, bone sialoprotein, fibronectin and thrombospondin in bone marrow but also serves as physical anchors permitting metastatic cells to establish footholds in the bone marrow. α6β4, α6β1 and αvβ5 have been showed to be expressed in exosomes and mediated lung cancer cell metastasis[130]. Exosomal integrins can activate the phosphorylation of Src and the expression of the pro-inflammatory S100[130]. Cancer cell invasion can occur as individual cell migration or collective cell migration, both of which are dependent on downregulation of E-cadherin induced loosening of cell junction[128]. The crosstalk between integrins and E-cadherin mediate epithelial cell-cell adhesion and cell-matrix adhesion signaling. The balance between E-cadherin-mediated adhesion junctions and integrin-mediated cell-matrix contacts determine the metastasis process. α-catenin has a pivotal role in the crosstalk between E-cadherin adhesions and integrin-mediated cell-ECM interactions[131].
Tumor cell homing to secondary organs is regulated by cytokines, chemokines, and their receptors [78]. The chemokines, such as CXCL12, have been demonstrated a driving role in the directional movement of cancer cells which overexpressed CXCR4[132]. Lungs, bone, liver, brain, and regional lymph nodes that express high levels of stromal cell-derived factor-1 (SDF-1α/CXCL12) are the most common sites for residence of breast cancer cells expressing CXCR4[133, 134]. CXCL12 is also a ligand that promotes chemotaxis of endothelial cells and hematopoietic progenitors to bone marrow from circulation[135, 136]. Factors including VEGF-A, TGF-β, and TNF-α attract tumor cells by upregulating the expression of S100A8 and S100A9[137].
Conclusion
Tumorigenesis and progression of cancer are complex processes which transformed cells and stromal cells interact and co-evolve. Intrinsic and extrinsic factors cause the mutations of cells. A niche in high-risk localized cancer cells is characteristics of senescence, stromal fibrosis and obese microenvironment which contribute on the survival of mutated cells. Cancer cells reprogram the microenvironment locally and systemically. The formation of premetastatic niche in the secondary organs facilitate the cancer cells survival in the distant organs. Strategies to target the niche can be promising approach to eradicate cancer cells.
Abbreviations
SASP: Senescence-associated secretory phenotype; SMS: Senescence-messaging secretome; IL-1: Interleukin-1; IL-6: Interleukin-6; IL-8: Interleukin-8; GROα:Growth regulating oncogene alpha;GROβ: Growth regulating oncogene beta;MCP-2: Monocyte chemoattractant protein 2;MCP-4: Monocyte chemoattractant protein 4; IGFBPs: Insulin-like growth factor binding proteins; G-CSF: Granulocyte colony-stimulating factor; GM-CSF: Granulocyte-macrophage colony-stimulating factor; MMPs: Matrix metalloproteinases; ROS: Reactive oxygen species; ECM: Extracellular matrix; IPF: Idiopathic pulmonary fibrosis; FAK: Focal adhesion kinase; ERK: Extracellular signal-regulated protein kinase; TGF-β: Transforming growth factor-β; hStCs: Hepatic stellate cells; FN: Fibronectin; LOXL2: Lysy1 oxidase-like2; EMT: Epithelial mesenchymal transition; ER: Endoplasmic reticulum; YAP: Yes-associated protein; TAZ: Transcriptional co-activator with PDZ-binding motif; MSCs: Mesenchymal stem cells; BMSCs: Bone marrow mesenchymal stem cells; IGF-1: Insulin-like growth factor-1; OB-R: Leptin receptor; CRP: C- reactive protein; TNF-α: Tumor necrosis factor-alpha; PI3K: Phosphatidylinositol 3 kinase; Akt: Protein kinase B; mTOR: Mammalian target of rapamycin; MAPK: Mitogen-activated protein kinase; JAK: Janus kinase; STAT: Signal transducer and activator of transcription; ERK: Extracellular regulated protein kinases; VEGF: Vascular endothelial growth factor; HIF-α: Hypoxia inducible factor-1alpha; SDF-1α: Stromal cell-derived factor-1; CXCR4: Chemokine receptor type 4; BMDCs: Bone marrow-derived cells; TDSFs: Tumor-derived secreted factors; DTC: Disseminated tumor cell; HSC: Hematopoietic stem cell; HPC: Hematopoietic progenitor cell; MDSCs: Myeloid derived suppressor cells; TSP-1: Thrombospondin-1; PDGF: Platelet-derived growth factor; bFGF: basic fibroblast growth factor.
Acknowledgements
This work was supported by National Natural Science Foundation, China (grant number 81472695, 81773147, 81272255); Strategic Priority Research Program of Central South University (ZLXD2017004); National Training and Research Base for Talents of principles of carcinogenesis foundation (111 project: 111-2-12).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ostby I, Oyehaug L, Steen HB. A stochastic model of cancer initiation including a bystander effect. J Theor Biol. 2006;241:751-64
2. Steen HB. The origin of oncogenic mutations: where is the primary damage? Carcinogenesis. 2000;21:1773-6
3. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283-96
4. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23-8
5. Rozhok AI, DeGregori J. Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations. Proc Natl Acad Sci U S A. 2015;112:8914-21
6. Goymer P. Natural selection: The evolution of cancer. Nature. 2008;454:1046-8
7. Balducci L. Epidemiology of cancer and aging. J Oncol Manag. 2005;14:47-50
8. Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011;21:354-9
9. Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273-83
10. Perez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. 2014;14:547-58
11. Blagosklonny MV. Cell cycle arrest is not senescence. Aging (Albany NY). 2011;3:94-101
12. Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513-20
13. McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol. 2010;28:4022-8
14. Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375-9
15. Kiecolt-Glaser JK, Preacher KJ, MacCallum RC, Atkinson C, Malarkey WB, Glaser R. Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc Natl Acad Sci U S A. 2003;100:9090-5
16. Maggio M, Guralnik JM, Longo DL, Ferrucci L. Interleukin-6 in aging and chronic disease: a magnificent pathway. J Gerontol A Biol Sci Med Sci. 2006;61:575-84
17. Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109-19
18. Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714-9
19. Sun Q, Liu Q, Zheng Y, Cao X. Rapamycin suppresses TLR4-triggered IL-6 and PGE(2) production of colon cancer cells by inhibiting TLR4 expression and NF-kappaB activation. Mol Immunol. 2008;45:2929-36
20. Guo Y, Nemeth J, O'Brien C, Susa M, Liu X, Zhang Z. et al. Effects of siltuximab on the IL-6-induced signaling pathway in ovarian cancer. Clin Cancer Res. 2010;16:5759-69
21. Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N. et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28:2940-7
22. Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3'-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002;62:1087-92
23. Lee S, Yoon S, Kim DH. A high nuclear basal level of ERK2 phosphorylation contributes to the resistance of cisplatin-resistant human ovarian cancer cells. Gynecol Oncol. 2007;104:338-44
24. Ershler WB, Sun WH, Binkley N, Gravenstein S, Volk MJ, Kamoske G. et al. Interleukin-6 and aging: blood levels and mononuclear cell production increase with advancing age and in vitro production is modifiable by dietary restriction. Lymphokine Cytokine Res. 1993;12:225-30
25. Ershler WB, Sun WH, Binkley N. The role of interleukin-6 in certain age-related diseases. Drugs Aging. 1994;5:358-65
26. Torres S, Garcia-Palmero I, Herrera M, Bartolome RA, Pena C, Fernandez-Acenero MJ. et al. LOXL2 Is Highly Expressed in Cancer-Associated Fibroblasts and Associates to Poor Colon Cancer Survival. Clin Cancer Res. 2015;21:4892-902
27. Parker MW, Rossi D, Peterson M, Smith K, Sikstrom K, White ES. et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124:1622-35
28. Nagelkerke A, Bussink J, Rowan AE, Span PN. The mechanical microenvironment in cancer: How physics affects tumours. Semin Cancer Biol. 2015;35:62-70
29. Sottnik JL, Dai J, Zhang H, Campbell B, Keller ET. Tumor-induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer Res. 2015;75:2151-8
30. Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol. 2012;13:591-600
31. Ferrao PT, Behren A, Anderson RL, Thompson EW. Editorial: Cellular and Phenotypic Plasticity in Cancer. Front Oncol. 2015;5:171
32. Sekyrova P, Ostblom J, Andang M. Blebbing as a physical force in cancer EMT - parallels with mitosis. Semin Cancer Biol. 2012;22:369-73
33. Gjorevski N, Boghaert E, Nelson CM. Regulation of Epithelial-Mesenchymal Transition by Transmission of Mechanical Stress through Epithelial Tissues. Cancer Microenviron. 2012;5:29-38
34. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178-96
35. Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22:396-403
36. Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326-43
37. Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP. et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis. 2017;6:e352
38. Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 1997;88:39-48
39. Moschos SJ, Drogowski LM, Reppert SL, Kirkwood JM. Integrins and cancer. Oncology (Williston Park). 2007;21:13-20
40. Alam SG, Zhang Q, Prasad N, Li Y, Chamala S, Kuchibhotla R. et al. The mammalian LINC complex regulates genome transcriptional responses to substrate rigidity. Sci Rep. 2016;6:38063
41. Thomas CH, Collier JH, Sfeir CS, Healy KE. Engineering gene expression and protein synthesis by modulation of nuclear shape. Proc Natl Acad Sci U S A. 2002;99:1972-7
42. Poh YC, Shevtsov SP, Chowdhury F, Wu DC, Na S, Dundr M. et al. Dynamic force-induced direct dissociation of protein complexes in a nuclear body in living cells. Nat Commun. 2012;3:866
43. Simon DN, Wilson KL. The nucleoskeleton as a genome-associated dynamic 'network of networks'. Nat Rev Mol Cell Biol. 2011;12:695-708
44. Iyengar NM, Hudis CA, Dannenberg AJ. Obesity and cancer: local and systemic mechanisms. Annu Rev Med. 2015;66:297-309
45. Divella R, De Luca R, Abbate I, Naglieri E, Daniele A. Obesity and cancer: the role of adipose tissue and adipo-cytokines-induced chronic inflammation. J Cancer. 2016;7:2346-59
46. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
47. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436-44
48. Iyengar NM, Kochhar A, Morris PG, Morris LG, Zhou XK, Ghossein RA. et al. Impact of obesity on the survival of patients with early-stage squamous cell carcinoma of the oral tongue. Cancer. 2014;120:983-91
49. Berger NA. Obesity-associated gastrointestinal tract cancer: from beginning to end. Cancer. 2014;120:935-9
50. Villarroya F, Cereijo R, Villarroya J, Giralt M. Brown adipose tissue as a secretory organ. Nat Rev Endocrinol. 2017;13:26-35
51. Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, Inflammation, and Cancer. Annu Rev Pathol. 2016;11:421-49
52. Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body Fatness and Cancer-Viewpoint of the IARC Working Group. N Engl J Med. 2016;375:794-8
53. Wang JB, Gu MJ, Shen P, Huang QC, Bao CZ, Ye ZH. et al. Body Mass Index and Mortality: A 10-Year Prospective Study in China. Sci Rep. 2016;6:31609
54. Obeid S, Hebbard L. Role of adiponectin and its receptors in cancer. Cancer Biol Med. 2012;9:213-20
55. Kerenidi T, Lada M, Tsaroucha A, Georgoulias P, Mystridou P, Gourgoulianis KI. Clinical significance of serum adipokines levels in lung cancer. Med Oncol. 2013;30:507
56. Wang SN, Yang SF, Tsai HH, Lee KT, Yeh YT. Increased adiponectin associated with poor survival in hepatocellular carcinoma. J Gastroenterol. 2014;49:1342-51
57. Phelip JM, Bageacu S, Baconnier M, Barabino G, Del Tedesco E, Benhamou PY. et al. Comparison of adiponectin concentration between pancreatic cancer and colorectal cancer. J Gastrointest Oncol. 2011;2:232-9
58. Krechler T, Zeman M, Vecka M, Macasek J, Jachymova M, Zima T. et al. Leptin and adiponectin in pancreatic cancer: connection with diabetes mellitus. Neoplasma. 2011;58:58-64
59. Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH. et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55-63
60. Booth A, Magnuson A, Fouts J, Foster M. Adipose tissue, obesity and adipokines: role in cancer promotion. Horm Mol Biol Clin Investig. 2015;21:57-74
61. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219-46
62. Vansaun MN. Molecular pathways: adiponectin and leptin signaling in cancer. Clin Cancer Res. 2013;19:1926-32
63. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498-503
64. Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K. et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med. 2015;7:301ra130
65. Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505-18
66. Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y. et al. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol. 2002;20:42-51
67. Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL. et al. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol. 2008;9:1039-47
68. Barone BB, Yeh HC, Snyder CF, Peairs KS, Stein KB, Derr RL. et al. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. JAMA. 2008;300:2754-64
69. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87-90
70. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21-35
71. Chin AR, Wang SE. Cancer Tills the Premetastatic Field: Mechanistic Basis and Clinical Implications. Clin Cancer Res. 2016;22:3725-33
72. Aljohani HM, Aittaleb M, Furgason JM, Amaya P, Deeb A, Chalmers JJ. et al. Genetic mutations associated with lung cancer metastasis to the brain. Mutagenesis. 2018;33:137-45
73. Kim SH, Redvers RP, Chi LH, Ling X, Lucke AJ, Reid RC. et al. Identification of brain metastasis genes and therapeutic evaluation of histone deacetylase inhibitors in a clinically relevant model of breast cancer brain metastasis. Dis Model Mech. 2018
74. Robertson DJ, Stukel TA, Gottlieb DJ, Sutherland JM, Fisher ES. Survival after hepatic resection of colorectal cancer metastases: a national experience. Cancer. 2009;115:752-9
75. Comen EA. Tracking the seed and tending the soil: evolving concepts in metastatic breast cancer. Discov Med. 2012;14:97-104
76. Suzuki M, Mose ES, Montel V, Tarin D. Dormant cancer cells retrieved from metastasis-free organs regain tumorigenic and metastatic potency. Am J Pathol. 2006;169:673-81
77. Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC. et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med. 2005;353:793-802
78. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239-52
79. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285-93
80. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK. et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816-26
81. Liu Y, Cao X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell. 2016;30:668-81
82. Smith HA, Kang Y. Acute infection induces a metastatic niche: a double menace for cancer patients. Clin Cancer Res. 2013;19:4547-9
83. Yan L, Cai Q, Xu Y. The ubiquitin-CXCR4 axis plays an important role in acute lung infection-enhanced lung tumor metastasis. Clin Cancer Res. 2013;19:4706-16
84. Sceneay J, Smyth MJ, Moller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev. 2013;32:449-64
85. Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013;32:623-42
86. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569-79
87. van Niel G, Porto-Carreiro I, Simoes S, Raposo G. Exosomes: a common pathway for a specialized function. J Biochem. 2006;140:13-21
88. Schorey JS, Bhatnagar S. Exosome function: from tumor immunology to pathogen biology. Traffic. 2008;9:871-81
89. Epple LM, Griffiths SG, Dechkovskaia AM, Dusto NL, White J, Ouellette RJ. et al. Medulloblastoma exosome proteomics yield functional roles for extracellular vesicles. PLoS One. 2012;7:e42064
90. Li L, Li C, Wang S, Wang Z, Jiang J, Wang W. et al. Exosomes Derived from Hypoxic Oral Squamous Cell Carcinoma Cells Deliver miR-21 to Normoxic Cells to Elicit a Prometastatic Phenotype. Cancer Res. 2016;76:1770-80
91. Hosseini-Beheshti E, Pham S, Adomat H, Li N, Tomlinson Guns ES. Exosomes as biomarker enriched microvesicles: characterization of exosomal proteins derived from a panel of prostate cell lines with distinct AR phenotypes. Mol Cell Proteomics. 2012;11:863-85
92. Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J. et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17:183-94
93. Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T. et al. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kgamma to promote pancreatic cancer metastasis. Cancer Res. 2018
94. Fang JH, Zhang ZJ, Shang LR, Luo YW, Lin Y, Yuan Y. et al. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology. 2018
95. Bao L, You B, Shi S, Shan Y, Zhang Q, Yue H. et al. Metastasis-associated miR-23a from nasopharyngeal carcinoma-derived exosomes mediates angiogenesis by repressing a novel target gene TSGA10. Oncogene. 2018;37:2873-89
96. Fang T, Lv H, Lv G, Li T, Wang C, Han Q. et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9:191
97. Zhang H, Deng T, Liu R, Bai M, Zhou L, Wang X. et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat Commun. 2017;8:15016
98. Ono M, Kosaka N, Tominaga N, Yoshioka Y, Takeshita F, Takahashi RU. et al. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci Signal. 2014;7:ra63
99. Lin XJ, Fang JH, Yang XJ, Zhang C, Yuan Y, Zheng L. et al. Hepatocellular Carcinoma Cell-Secreted Exosomal MicroRNA-210 Promotes Angiogenesis In Vitro and In Vivo. Mol Ther Nucleic Acids. 2018;11:243-52
100. Muturi HT, Dreesen JD, Nilewski E, Jastrow H, Giebel B, Ergun S. et al. Tumor and endothelial cell-derived microvesicles carry distinct CEACAMs and influence T-cell behavior. PLoS One. 2013;8:e74654
101. Alderton GK. Metastasis Exosomes drive premetastatic niche formation. Nat Rev Cancer. 12: 447.
102. Liu Y, Cao X. Organotropic metastasis: role of tumor exosomes. Cell Res. 2016;26:149-50
103. Alvarez ML, Khosroheidari M, Kanchi Ravi R, DiStefano JK. Comparison of protein, microRNA, and mRNA yields using different methods of urinary exosome isolation for the discovery of kidney disease biomarkers. Kidney Int. 2012;82:1024-32
104. Rabinowits G, Gercel-Taylor C, Day JM, Taylor DD, Kloecker GH. Exosomal microRNA: a diagnostic marker for lung cancer. Clin Lung Cancer. 2009;10:42-6
105. Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B. et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24:766-9
106. Thind A, Wilson C. Exosomal miRNAs as cancer biomarkers and therapeutic targets. J Extracell Vesicles. 2016;5:31292
107. Yang H, Fu H, Xu W, Zhang X. Exosomal non-coding RNAs: a promising cancer biomarker. Clin Chem Lab Med. 2016;54:1871-9
108. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H. et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15:807-17
109. Patel LR, Camacho DF, Shiozawa Y, Pienta KJ, Taichman RS. Mechanisms of cancer cell metastasis to the bone: a multistep process. Future Oncol. 2011;7:1285-97
110. Salcedo R, Oppenheim JJ. Role of chemokines in angiogenesis: CXCL12/SDF-1 and CXCR4 interaction, a key regulator of endothelial cell responses. Microcirculation. 2003;10:359-70
111. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329-60
112. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125:5591-6
113. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793-800
114. Paul P, Rouas-Freiss N, Khalil-Daher I, Moreau P, Riteau B, Le Gal FA. et al. HLA-G expression in melanoma: a way for tumor cells to escape from immunosurveillance. Proc Natl Acad Sci U S A. 1998;95:4510-5
115. Lu Z, Hunter T. Metabolic Kinases Moonlighting as Protein Kinases. Trends Biochem Sci. 2018;43:301-10
116. Phan AT, Goldrath AW, Glass CK. Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity. 2017;46:714-29
117. Blazar BR, Carreno BM, Panoskaltsis-Mortari A, Carter L, Iwai Y, Yagita H. et al. Blockade of programmed death-1 engagement accelerates graft-versus-host disease lethality by an IFN-gamma-dependent mechanism. J Immunol. 2003;171:1272-7
118. Wang W, Wang L, Ruan L, Oh J, Dong X, Zhuge Q. et al. Extracellular vesicles extracted from young donor serum attenuate inflammaging via partially rejuvenating aged T-cell immunotolerance. FASEB J. 2018 fj201800059R
119. Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P. et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res. 2011;71:5101-10
120. Klassen LMB, Chequin A, Manica GCM, Biembengut IV, Toledo MB, Baura VA. et al. MMP9 gene expression regulation by intragenic epigenetic modifications in breast cancer. Gene. 2018;642:461-6
121. Marshall NB, Lukomska E, Nayak AP, Long CM, Hettick JM, Anderson SE. Topical application of the anti-microbial chemical triclosan induces immunomodulatory responses through the S100A8/A9-TLR4 pathway. J Immunotoxicol. 2017;14:50-9
122. Ma L, Sun P, Zhang JC, Zhang Q, Yao SL. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int J Mol Med. 2017;40:31-8
123. Iwamoto DV, Calderwood DA. Regulation of integrin-mediated adhesions. Curr Opin Cell Biol. 2015;36:41-7
124. Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25:234-40
125. Strand DW, Hayward SW. Modeling stromal-epithelial interactions in disease progression. Discov Med. 2010;9:504-11
126. Vanharanta S, Massague J. Origins of metastatic traits. Cancer Cell. 2013;24:410-21
127. Felding-Habermann B, O'Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH. et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A. 2001;98:1853-8
128. Gkretsi V, Stylianopoulos T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front Oncol. 2018;8:145
129. Schneider JG, Amend SR Weilbaecher, KN. Integrins and bone metastasis: integrating tumor cell and stromal cell interactions. Bone. 2011;48:54-65
130. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M. et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329-35
131. Borghi N, Lowndes M, Maruthamuthu V, Gardel ML, Nelson WJ. Regulation of cell motile behavior by crosstalk between cadherin- and integrin-mediated adhesions. Proc Natl Acad Sci U S A. 2010;107:13324-9
132. Long X, Li L, Zhou Q, Wang H, Zou D, Wang D. et al. Long non-coding RNA LSINCT5 promotes ovarian cancer cell proliferation, migration and invasion by disrupting the CXCL12/CXCR4 signalling axis. Oncol Lett. 2018;15:7200-6
133. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-6
134. Weigelt B, Wessels LF, Bosma AJ, Glas AM, Nuyten DS, He YD. et al. No common denominator for breast cancer lymph node metastasis. Br J Cancer. 2005;93:924-32
135. Nagasawa T. [Bone and Stem Cells. Bone marrow microenvironment niches for hematopoietic stem and progenitor cells]. Clin Calcium. 2014;24:517-26
136. Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN. et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227-30
137. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8:1369-75
Author contact
![]() Corresponding author: Prof. Juanjuan Xiang (xiangjjedu.cn) Tel: +86-731-82355401
Corresponding author: Prof. Juanjuan Xiang (xiangjjedu.cn) Tel: +86-731-82355401