Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Genetic parameters in skull...
3. Environmental factors in...
4. Molecular mechanisms of...
5. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(2):298-311. doi:10.7150/ijbs.29183 This issue Cite
Review
Signaling Mechanisms Underlying Genetic Pathophysiology of Craniosynostosis
Xiaowei Wu1,2, Yan Gu1,2 ![]()
1. Department of Orthodontics, Peking University School and Hospital of Stomatology, No. 22 Zhongguancun Avenue South, Haidian District, Beijing, 100081, PR. China
2. National Engineering Laboratory for Digital and Material Technology of Stomatology,Beijing Key Laboratory of Digital Stomatology, No. 22 Zhongguancun Avenue South, Haidian District, Beijing, 100081, PR. China
Received 2018-8-13; Accepted 2018-11-30; Published 2019-1-1
Abstract

Craniosynostosis, is the premature fusion of one or more cranial sutures which is the second most common cranial facial anomalies. The premature cranial sutures leads to deformity of skull shape and restricts the growth of brain, which might elicit severe neurologic damage. Craniosynostosis exhibit close correlations with a varieties of syndromes. During the past two decades, as the appliance of high throughput DNA sequencing techniques, steady progresses has been made in identifying gene mutations in both syndromic and nonsyndromic cases, which allow researchers to better understanding the genetic roles in the development of cranial vault. As the enrichment of known mutations involved in the pathogenic of premature sutures fusion, multiple signaling pathways have been investigated to dissect the underlying mechanisms beneath the disease. In addition to genetic etiology, environment factors, especially mechanics, have also been proposed to have vital roles during the pathophysiological of craniosynostosis. However, the influence of mechanics factors in the cranial development remains largely unknown. In this review, we present a brief overview of the updated genetic mutations and environmental factors identified in both syndromic and nonsyndromic craniosynostosis. Furthermore, potential molecular signaling pathways and its relations have been described.
Keywords: Craniosynostosis, Signaling Pathways, Genetic Mutations.
1. Introduction
The mammalian skull vault is composed of five bones: the paired frontal bones, the paired parietal bones, and the isolated occipital bone. The cranial bones are connected at the osteogenic edges via fibrocellular structures, which is termed as cranial sutures [1, 2]. The cranial sutures mainly consist of metopic suture, coronal suture, sagittal suture, and lambdoid suture (Fig. 1) [1]. The fusion of metopic suture, which is located between the paired frontal bones, starts as early as 3 months postnatal, and revealed completely fused pattern at the age of 9 months postnatal [3]. In addition, the coronal sutures, sagittal sutures and lambdoid suture fuse at the age of twenties [4]. The cranial sutures play as growth centers, which allow proportional amount of skull growth in accordance with the expansion of the developing brain.
(a) is a schematic photograph of normal cranial bones and sutures [1]. (b) is 3D reconstruction of sagittal suture premature fusion, which presented scaphocephalus; (c) is 3D reconstruction of metopic suture premature fusion, which presented trigonocephalus; (d) and (e) represent the synostosis of bilateral (brachycephalus) and unilateral (plagiocephalus anterior) coronal sutures, respectively; (f) exhibits the synostosis of unilateral lambdoid sutures, respectively.” [17].
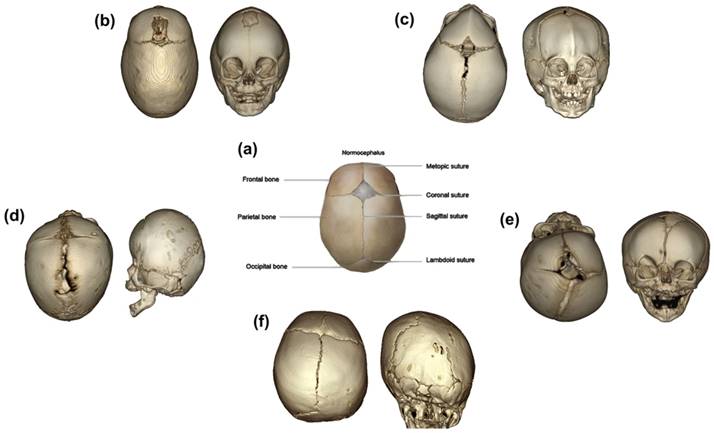
Craniosynostosis is the premature fusion of one or more cranial sutures, which is the second most common craniofacial anomalies, just listed behind oralfacial clefts [5, 6]. In a majority of cases, craniosynostosis expresses as an isolated and nonsyndromic disease, which composed about 85% of all cases [7]. The incidence of nonsyndromic craniosynostosis ranges from 0.7 to 6.4 per 10,000 live births [8, 9]. About 15-30% of cases occurs in association with additional clinical symptoms, which may include malformation of hand and feet, defection of skeletal and cardiac, and others. The prevalence of syndromic craniosynostosis is approximately 1 in 25,000 newborns [10-12]. The sagittal suture (47.1%) is mostly involved in the premature sutures, followed by metopic (21.5%), unilateral coronal (17.1%), bilateral coronal (3.3%), lambdoid (2.8%), and combinations (8.2%) [8]. To be different, some investigations revealed that the prevalence of coronal suture (20-25%) might be higher than metopic suture (5-15%) [13]. Also, higher frequency of the sagittal synostosis among males than females was observed [7, 14]. While, the prevalence of the coronal synostosis was higher in females than that of males [14, 15]. No significant gender predominance was observed in metopic and lambdoid suture synostosis [14, 16]. As the premature suture could restrict the growth perpendicular to the fused suture, it is difficult to create adequate space for the growth of brain, thus elicit compensatory overgrowth at other sutures [15]. The overgrowth could result in typical skull shapes according to the involvement of the special suture. The premature sagittal suture might exhibit compensatory growth in anteroposterior direction, which results in dolichocephaly or scaphocephaly. The bicoronal synostosis restricts the anteroposterior growth and results in brachycephaly. The unicoronal synostosis would result in flattened head of the affected side in accompany with compensatory growth of the contralateral side, which is termed as anterior plagiocephaly. The triangular shape of the skull from the top view is the typical feature of metopic synostosis, and such head shape is called trigocephaly. The synostosis of lambdoid is rare, and the unilateral lambdoid synostosis and bilateral synostosis would result in posterior plagiocephaly and turricephaly, respectively (Figure 1) [1, 5, 17]. In spite of the compensatory growth of uninvolved suture, the abnormalities may give rise to the intracranial pressure. If the high intracranial pressure was left without appropriate treatment, it could result in permanent injury of brain [12]. Moreover, craniosynostosis is also associated with other abnormalities, such as midfacial deformity, malformation of dentin, orbital deformation, hearing loss, breathing, and intellectual disability. These disabilities could both expressed in the nonsyndromic and syndromic craniosynostosis [12, 18, 19].
The abnormal fusion of suture could be caused by varieties of factors. The etiology of craniosynostosis includes both genetic factors and environmental interference. In recent years, as the application of exome and genome sequencing, mutations of several genes have been identified [20-22]. As for the syndromic craniosynostosis, up till now, over 180 syndromes have been identified to be related to the syndromic craniosynostosis, and specific gene mutations or chromosome abnormalities have been checked out over 20% of all cases [13, 23, 24]. Among them, mutations in FGFR1, FGFR2, FGFR3, Twist1, EFNB1 are frequently reported [25-29]. On the other hand, nonsyndromic craniosynostosis has been recognized as a multifactorial disorder but not a truly genetic condition. However, genome and exome sequencing results indicated strength correlations between genetic factors and nonsyndromic craniosynostosis [30]. Therefore, the understanding of genetic pathophysiological framework and its underlying molecular mechanisms are of vital importance for our comprehension of the etiology of craniosynostosis, and the development of effective pharmacological treatment. However, as only minority of craniosynostosis patients could identify definite mutations, the etiology of majorities is still unknown. Thus, some authors proposed that environmental interactions, including environmental exposure in utero and during juvenile stages, and the aberrant mechanistic forces during development, might play key roles in the etiology of craniosynostosis [5]. Unfortunately, up till now, the interaction of environmental factors and craniosynostosis is largely unknown and need to be further investigated. Even though, there still be some studies trying to figure out the significant roles of environmental exposure and abnormal mechanical forces during development, which would be reviewed in the later sections.
Throughout this review, the etiology of craniosynostosis, including genetic, environmental and mechanical factors, have been reviewed. Moreover, signaling pathways underling the disease were involved for a better understanding of the interplay between the factors mentioned above.
2. Genetic parameters in skull vault developmental biology
The pathophysiology of craniosynostosis is so complex, which could be influenced by both genetic and environmental factors. Moreover, the mechanical factor has also been considered as a vital factor [31]. As the development of high throughput sequencing technics, abnormalities of monogene or oligogene, and even the deletion or duplication of chromosomes have been identified in the involved cases, with a prevalence of at least 20% [24]. Syndromic craniosynostosis have been reckoned as having strong association with genetic factor, however, numbers of abnormalities have been identified in nonsyndromic craniosynostosis patients in recent years [30]. Moreover, considering the heterogeneous manifestations of both syndromic and nonsyndromic cases and the diversity of genes, genetic factor have been recognized as a nonnegligible etiology of craniosynostosis. In this chapter, the genetic parameters in skull bone and suture development are stated.
2.1. Specification and migration of stem cells
Three dimensional growth of developing cranial bones and craniofacial bone reparation are initiated from the specification and migration of stem cells. Some studies presumed that stem cell from periosteum participate in the generation of skull bones [32-35], whereas recent studies have identified mesenchyme stem cells distributed in the middle of sutures [36, 37]. Zhao et al. [36] reported a typical kind of Gli+ cells distributed within the cranial sutures, which could give rise to the cranial bones, periosteum, and dura mater. Selective ablation of Gli1+ cells of mice leads to craniosynostosis. Moreover, in Twist1 haploinsufficiency mice with craniosynostosis, the Gli1+ cells in the sutures reduced significantly. In addition, Maruyama et al. [37] recently identified Axin2+ cells, which presented long term self-renewal properties and could produce osteogenic cell types. Compared with the Gli1+ cells identified by Zhao and his co-workers, overlap of these two types of cells has been observed, whereas the author proposed that Axin2 is a more specific marker to isolate the suture stem cells. Moreover, the author demonstrated previously that knockout of Axin2, which is a regulator of Wnt signaling pathway, cooperated with FGFR disturbance result in the differentiation of stem cells into chondrocytes, which might participate in the process of craniosynostosis [38]. Therefore, the homeostasis of Axin2 might play critical roles in maintaining the functions of stem cells in cranial sutures. Mutations associated with the expression of such markers, such as hedgehog and Twist1, might disturb the function of suture stem cells, thus results in craniofacial disorders.
Studies have been processed for years to investigate the correlations between craniosynostosis and dysregulation of cellular proliferation and differentiation, while the role of apoptosis of cells during cranial development have been proposed by some authors [39-43]. Studies indicated that apoptosis is involved in the development of both nasal cartilage and calvarial bones [44, 45]. Premature fusion of anterior frontal suture has been depicted in BMPR1a mice, whose expression of BMP signaling has been specifically enhanced in neural crest derived tissues, and increased apoptosis was observed in cranial bones [45]. Meanwhile, the critical role of apoptosis in cranial sutures have also been illustrated [40, 42, 43]. Opperman et al. [42] found that additional of TGF- β3 might prevent suture obliteration with less apoptotic cell within sutures. Conversely, additional of TGF- β2 facilitate premature fusion of sutures with higher numbers of apoptotic cells in cranial sutures. Behr and co-workers [40] demonstrated impaired endochondral ossification within posterior frontal suture of Axin2-/- Mice, where higher level of chondrocyte apoptosis has been observed. As the identification of mesenchymal stem cells within cranial sutures, some authors hold the query that if apoptosis of mesenchymal stem cells might directly or indirectly induce craniosynostosis. A study published recently implied that, when enhancing the expression of BMP signaling in neural crest cells, loss of suture mesenchymal stem cells have been observed. Moreover, higher degree of cell death has also been illustrated in mutant mice [43]. Although studies published till now are far from making a conclusion, a speculation could be provided here is that depletion of mesenchymal stem cells within cranial sutures resulted from cell death /apoptosis might be a cellular mechanism of suture premature fusion.
2.2. Lineage specification and boundary formation
The somites developed from paraxial mesoderm display physical and molecular metamerism, also form fate specific in the seven cranial somitomeres. As the neural crest cells subsequently migrate and spread around the loosely arranged mesoderm cells, the homeostasis of mesoderm-neural crest cells boundary is important for the smooth development of cranial structures [46]. Deckelbaum et al. [47] processed fate mapping experiments in mice and demonstrated significant roles of Engrailed1 in lineage specification and mesoderm-neural crest lineage boundary. Merrill et al. [48] demonstrated that mutations of Twist1 and Msx2 are associated with the defection of mesoderm-neural crest lineage boundary via disturbing the expression of ephrin-A2 and A4. Moreover, mutations of Engrailed1 also showed perturbations in the expression of both Twist1 and Msx2 [49]. Mutation of Jag1 has also been implicated to disturb the boundary of sutural lineage [49, 50]. In all, the transcription factors, including Engrailed1, Twist1, Msx2, ephrin-A2/A4, Jag1, are of vital importance in maintaining the boundary of mesoderm-neural crest lineage.
2.3. Osteogenic differentiation
After mesoderm cells and neural crest cells migrate to their target areas successfully, cells become condensed and differentiate into osteoblast cells [51]. The differentiated osteoblast cells then secrete extracellular matrix, and promote mineral deposition subsequently [52-54]. That results in the formation of cranial vault bones. Disruption of the osteogenic differentiation might lead to overgrowth of bone tissues in the primary patent sutures and results in craniosynostosis. Genetic studies of craniosynostosis cases have proposed plenty of genes, which involves in the process of osteogenic differentiation. The mostly identified mutations associated with osteogenic differentiation have been stated below.
Indian hedgehog (Ihh) is a member of hedgehog signaling pathway, and has been recognized as a master gene in the skeletal development and osteogenic differentiation. Ihh-/- mice exhibited alleviated-sizing frontal and parietal bones, attenuated BMP expression, and widened sutures. And such phenotypes have been recognized as a result from the defections of osteogenic differentiation [36, 55]. Moreover, Will et al. [20] identified nine enhancer clusters of Ihh, and demonstrated that deletion of the enhancer clusters of Ihh could result in delayed skull ossification. In addition, duplications of such clusters could lead to upregulation of Ihh expression and result in complete metopic suture fusion. The study above implied that disturbance of Ihh expression might influence the osteogenic differentiation of cells and result in craniosynostosis.
As for the high relevance ratio of Twist1 in craniosynostosis cases, studies associated with the functions of Twist1 have been processed for years [26, 56]. Results indicated that Twist1, as a basic helix-loop-helix transcription factor, plays as a negative regulator during osteogenic differentiation. Twist1+/- mice displayed increased skull bone formation within the cranial sutures, thus exhibited craniosynostosis phenotype [57]. In accordance with the results above, Bialek et al. [58] demonstrated that the decreased expression of Twist1 could elicit osteoblast differentiation via interfering with the functions of Runx2. The study indicated that Twist box is an antiosteogenic domain and could inhibit the transactivation function of Runx2. Moreover, Twist1 also attenuated the expressions of BSP and OCN [59]. Besides, interactions of TCF12 and Twist1 have been proposed [21, 60]. At the same time, mutations of TCF12 have been identified by Sharma et al. [21]. The study further revealed synergistic effects of TCF12 and Twist1 in the coronal synostosis. Furthermore, the authors proposed that the heterodimer of TCF12-Twist1 might both influences on the fate specification and mesoderm-neural crest cells boundary, and the osteogenic differentiation. Intragenic exon deletions and duplications of TCF12 have also been identified in individuals with craniosynostosis [60]. In all, Twist1 acts as a negative regulator for osteogenic differentiation by interacting with osteogenic factors such as Runx2 and thus mutations in Twist1 lead to increased osteogenesis resulting in premature fusion of the suture mesenchyme.
Fibroblast growth factors (FGF) are positive regulators of the process of osteogenic differentiation [61]. Most importantly, the receptors of FGF (FGFR), especially FGFR1, FGFR2, and FGFR3, are the most frequently identified mutations in craniosynostosis cases [62-65]. Thus, the studies of the functions of FGF/FGFR are of vital importance. Studies from Kim et al. [64] revealed that FGF2 could induce mesenchymal stem cells osteogenic differentiation via activating of protein kinase C (PKC) and the downstream Runx2. Also, Miraoui et al. [65] demonstrated that the positive regulation of FGFR2 in inducing osteogenic differentiation of mesenchymal stem cells is achieved via activation of PKC and Erk1/2. FGF18 has also been demonstrated to be closely related to the osteogenic differentiation of rat bone marrow-derived mesenchymal stem cells (BMSCs) [66]. Furthermore, PI3k/Akt signaling pathway also involved in the osteogenic differentiation of mesenchymal cells induced by FGF [67]. Taken together, FGF/FGFR signaling pathways might act as positive regulators in the process of osteogenic differentiation of stem cells, and many molecules, such as Runx2, PKC, etc., might act as downstream in this process. Runx2 is a positive regulator to osteogenic differentiation, and strongly interacted with many molecules, such as Twist1 and FGF stated above. Haploinsufficiency of Runx2 has been proved to be associated with widened sutures displayed both in mice and human [31]. In contrast, craniosynostosis exhibited closely relations with overexpression of Runx2 [68, 69].
Genetic studies have identified mutations of POR, who could bind to NADPH and transfers electrons from Flavine adenine dinucleotide (FAD) to flavin mononucleotide, and the electrons are donated to P450 enzymes subsequently. Such mutations are associated with Antley-Bixler syndrome [70]. Laue et al. [71] demonstrated that mutations of CYP26B1, encoding one of the P450 enzymes, exhibited long-bone fusion, differentiation of osteoblasts into osteocytes, and cranial suture fusion, which emphasized the vital roles of P450 enzymes in osteogenic differentiation of cells. Moreover, CYP26B1 is necessary for the degradation of retinoic acid, while high levels of retinoic acid have been observed in POR mutation cases [72, 73]. Taken together, mutations in POR might contribute to craniosynostosis via disturb the process of osteogenic differentiation. Altogether, osteogenic differentiation of stem cells is vital during the intramembranous ossification procedures, and abnormal expressions of associated genes might lead to the abnormity of bone formation in sutures.
Moreover, genetic mutations identified till now are mostly associated with osteogenic differentiation of stem cells, thus further evaluation about the roles of such genes in the etiology of craniosynostosis are of vital significance, which need to be deeply studied.
2.4. Bone remolding
The balance of osteogenesis and bone resorption is of vital importance in bone remolding to adapt the mechanical forces, and maintaining the patency of sutures. Thus, loss-of-function mutations of osteoclast cells might also influence suture patency. Nieminen et al. [74] identified missense mutations of IL11RA in craniosynostosis patients, which encodes interleukin 11 receptor alpha, and further investigations indicated that IL11RA mutations might elicit loss of gene function, and truncate the transduction of IL11 signal. The study proposed that IL11 signaling is critical for the development of cranial bones and the maintaining of suture patency. Mutations of IL11RA have also been identified recently. It is proved previously that IL11 could stimulate osteoblast differentiation [75-77]. Suga et al. [76, 77] indicated that IL-11 alone or accompany with BMP-2 could induce osteogenic differentiation of cells, and these studies implied the potential role of IL-11 signaling during osteogenesis. Furthermore, studies processed by Sims et al. [78] indicated that deletion of IL11 receptor alpha could also inhibit osteoclast differentiation, and IL11 signaling is essential for the bone remolding. Up till now, as most studies, including the studies stated above, focus on the role of IL11 in long bones remolding, the effects of IL11 signaling in cranial bone remolding are largely unknown. Considering that IL11RA mutations have been identified in the patients with craniosynostosis and the potential influence of such mutations on both osteoblast and osteoclast, the critical roles of IL11 signaling should not be ignored.
3. Environmental factors in craniosynostosis
Although the acknowledgement of genetic factors in the etiology of craniosynostosis becomes richer and more thorough, there still some manifestations could not closely connect with mutations of genes. The heterogeneous clinical features with parallel mutations indicted that other factors might take part in the pathogenic processes. Besides, only a small fraction of patients could be identified with genetic variations, while the etiology of approximately 70% patients is unknown. Therefore, the exploration of the pathophysiological roles of environmental risk factors, both environmental exposure and mechanistic force during development, is apparently significant.
3.1. Environmental exposure during development
Alderman et al. [79] estimated the relative odds between craniosynostosis cases and maternal smoking, and alcohol consumption. Results indicated strength relations between maternal smoking and craniosynostosis. The relative odds were significantly higher in those who smoked over one pack per day. However, no statistically significance was observed between alcohol consumption and premature synostosis. Similarly, Kallen [80] investigated the possible association between maternal smoking and craniosynostosis, in which 304 craniosynostosis infants without known genetic mutation were involved. The study revealed significant relationship between maternal smoking and craniosynostosis, while, to be different from the results of Alderman and his colleagues [79], intense association with sagittal synostosis but no correlation with coronal synostosis were detected. Increased sagittal and/or lambdoid premature fusion has been reported in infants, whose mothers have certain nitrosatable drugs intake histories, including chlorpheniramine, nitrofurantoin, and chlordiazepoxide [81]. Hyperthyroidism has also been recognized as the etiology of craniosynostosis. Higashino et al.[82] reported a secondary craniosynostosis with all skull sutures premature fusion, which is associated with juvenile hyperthyroidism. Moreover, craniosynostosis of squamosal suture due to hyperthyroidism has also been published [83]. In addition, male and parental education were also proposed to be associated with sagittal craniosynostosis [84]. Boulet et al. [9] also proposed higher risk of sagittal synostosis in male. The author also suggested that maternal age 35 years or older, multiple birth, and birth weight over 4,000g suffers a higher risk for craniosynostosis. Paternal occupations have also been estimated, and workers of agriculture and forestry, repairmen, and mechanics have been identified as higher risk to have an affected infant [85].
3.2. Mechanistic force during development
Among the multitudinous environmental risk factors that associated with craniosynostosis, extrinsic forces acting on cranial bones play vital importance, while the acknowledge of its functions and potential mechanisms are still unknown.
It is hypothesized that fetal head constraint might be associated with craniosynostosis. Sanchez-Lara et al. [86] estimated associations between craniosynostosis and plurality, macrosomia, post-germ gestational age, and nulliparity. The results indicated higher prevalence of metopic craniosynostosis in plurality and nulliparity groups. Meanwhile, higher risk of coronal premature fusion was observed in macrosomia cases. Moreover, early descent of a fetal head into the lower uterine, malformation of maternal uterine, and oligohydramnios could also generate head constraint, thus result in higher risks of craniosynostosis [12, 86]. Jacob et al. [87] investigated the roles of Indian hedgehog (Ihh), BMP-4, and Noggin in the process of craniosynostosis secondary to fetal constraint. Results indicated that intrauterine constraint might elicit partially premature fusion of coronal and squamosal sutures. The expression of Ihh and Noggin decreased within the fused sutures. To be different from that, BMP-4 expressed comparably in constrained and unconstrained sutures, which might be assigned to the suture specificity of BMP signaling. Besides, after 24 and 48 hours intrauterine constraint, the up-regulated expressions of TGF-β receptors type I and II, and FGFR2 have been detected in the area of osteogenic fronts, midsutural mesenchyme, and the dura beneath the fusing coronal sutures [88]. Except for the constraint generated by utero, microcephaly is also considered to be associated with premature fusion [89]. As the microcephaly exhibited reduced growth of brain, lower stretch might generate in the dura mater, which further reduced quasi-static tensile strain across the sutures. Similarly, ventriculoperitoneal shunting used for the treatment of hydrocephalus could also lead to craniosynostosis via alleviate the tensile strain of the sutures [90, 91]. These results indicated that the mechanical forces could change the tensile stress of the sutures, thus alter the regional microenvironment, and result in premature fusion of cranial sutures. Heller et al. [92] imposed forces on the cranial sutures with either oscillate or distract forces. Results indicated that, as mechanical stress was applied, increased expression of Runx2 and attenuated expression of Noggin were observed. In general, either the abnormal external constraints and/or absence of internal tensile stress might lead to craniosynostosis. Moreover, molecules, such as Ihh, Noggin, TGF-β, FGFR2, and Runx2, might be associated with the premature fusion of cranial sutures induced by mechanical stress. Even though results about BMP is not positive in Jacob's study [87], the function of BMP should be nonnegligible considering the suture specificity and sophisticated interactions of BMP with molecules stated above.
4. Molecular mechanisms of craniosynostosis
4.1. FGF/FGFR and related signaling
Fibroblast growth factor (FGF) pathway has been recognized as playing fundamental roles during the processes of embryonic development and organ formation. FGFs are composed of 23 ligands, and studies implied that at least 18 ligands, including FGF1-FGF10 and FGF16-FGF23, could interact with FGFR1-4 and heparin. However, FGF11-14 displayed highly homologies with other FGFs but low heparin binding affinity [93]. The activation of FGFRs further elicit the cascade signals to mediate the processes of cellular proliferation, migration, differentiation, and angiogenesis [94]. FGFRs are highly conserved transmembrane receptors, which includes three extracellular immunoglobulins like domains (D1, D2, and D3), a transmembrane domain, and a cytoplasmic tyrosine kinase domain [95, 96] (Figure 2(a)). The D1 domain and the acid box, which is identified as the linker of D1 and D2 domains, are proposed to participate into the autoinhibitory process of receptors [97, 98]. The D2 domain exhibit binding affinity with heparin sulphate or heparin. The N-terminal of D3 domain is encoded by exon IIIa, and the C-terminal is encoded by either exon IIIb or exon IIIc [99, 100]. As the D3 region experienced alternative splicing, different types of FGFRs exhibited distinct affinities and specificities to different FGFs [101]. Up till now, 5 types of FGFRs have been identified in human. Among them, FGFR5 was identified in recent years and also be designated as FGFR like 1 [102-104]. The initial of FGF/FGFR signaling pathway is the binding of FGFs with FGFRs. Afterward, in the presence of heparin sulphate, a dimerization event would occur. The dimers consisted of two FGFRs, two heparin sulphate, and two FGFs [98].
FGFRs signaling pathway could be elicited by FGFs with the existence of heparin. The conformation of dimerized FGFRs could phosphorylate intracellular domain of FGFRs (a), which could further interact with MAPK (b), PI3K/Akt (d), PLCγ (c) signaling pathways [1, 145].
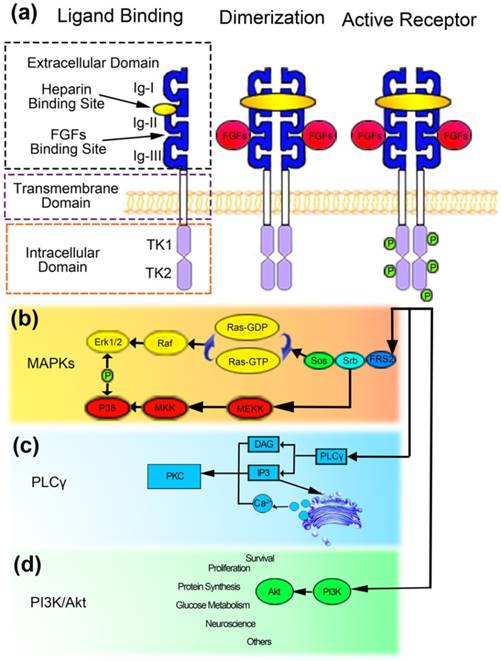
The dimerization might phosphorylate each other at the intracellular tyrosine kinase domains, and serve as a docking region for adaptor proteins to regulate the cascade signals [105]. The downstream signaling pathways of FGF/FGFRs includes PI3K/Akt, Ras/MAPK, and phospholipase C γ (PLCγ) signaling pathways [106]. The Ras/MAPK signaling pathway, which is associated with the proliferation and differentiation of cells, is the most common thread resulted from the mutations of FGFRs [107] (Figure 2(b)). The PI3K/Akt pathway has also been implied to be involved in the fate determination and cellular polarity [96, 108] (Figure 2(d)). Besides, the binding of PLCγ might generate inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 could elicit the release of calcium from endoplasmic reticulum and, together with DAG, activate protein kinase C (PKC), thus influence the migration, adhesion, and morphology of cells [96, 106, 108] (Figure 2(c)). The majority of mutations occurred in FGFRs were gain-of-function mutations, which would excessive activate the downstream signals, thus disturb the physiological processes and induce craniosynostosis. It is worthy to note that the perturbation of TGF-β also influenced the expression of Erk1/2 and resulted in craniosynostosis [109]. The convergence of multi-signals on the Erk1/2 signaling pathway indicated the vital role of Erk1/2 in the pathogenic processes of craniosynostosis. Over the past years, researchers tried to interfere the processes of suture fusion via molecule mechanisms, especially FGFs and related signaling [110-112]. A soluble form of FGFR2IIIcS252w, which could competitively bind to FGFs avoiding their binding to FGFRs, has been introduced into Apert mutant mice, and the syndromic manifestations, including craniosynostosis, have been partially rescued [111]. Besides, Erk1/2 plays a role as the downstream signaling of FGFs, which have attracted enormous attention. Studies implied that genetic inhibition of the Erk1/2 expression with hairpin RNA might prevent the craniosynostosis. Moreover, U0126 has also been used to pharmacologically inhibit the activation of MEK1/2 both pre- and post-natal, and results revealed partially rescue of the suture premature fusion [110]. Being consistent with the results above, Yin et al. [112] proved the potential application of MEK1 inhibitor PD98059. The molecule mechanisms-based treatments of craniosynostosis processed above gave potential strategy and new acknowledges to the pharmacological treatment of craniosynostosis, while we still confront some difficulties, such as the side-effects of drugs, the unwillingly blockage of other tissues development, and the rest.
4.2. Twist1 and related signaling
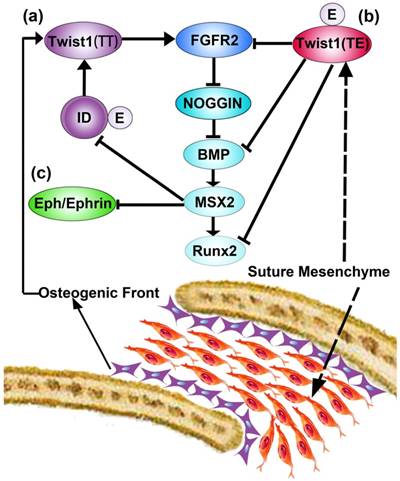
Twist1 is a basic helix-loop-helix transcription factor, whose mutations were mostly identified in the patients of Saethre-Chotzen syndrome [21]. Up till now, there are two proposals pathways associated with the coronal synostosis induced by Twist1 mutations. The first speculation is established by Connerney et al. [113]. The author proposed that Twist1 could either generate homodimers (T/T) or interact with E protein to generate heterodimers (T/E). The homodimers could activate FGFR2 expression in the osteogenic fronts and facilitate the osteogenic differentiation of cells. However, the heterodimers could inhibit the expression of FGFR2 in the midsutural mesenchyme. The regulation of the dimerization is achieved via inhibitor proteins of DNA binding/differentiation, which is called ID proteins. The ID proteins are preferentially existed in the osteogenic fronts, and competitively binding to E proteins, which lead to the homodimerization of Twist1 (Figure 3(a)). However, in the midsutural mesenchyme where ID proteins are absent, Twist1 dimerize with E proteins and inhibited the osteogenic differentiation of cells, thus maintain the patency of sutures (Figure 3(b)). The ID proteins could be up-regulated by BMP, and BMP are upregulated by FGFR2 via downregulation of Noggin [56, 113]. In addition, TCF12, whose mutations have been identified in the craniosynostosis cases, is one of the E proteins. The phenotypes of TCF12 and Twist1 mutations are in accordance with the speculation stated here. Thus, it is proposed that the homeostatic of BMP, Noggin, FGFR2, ID proteins, and E proteins are of vital importance in the development of cranial sutures. Another signaling pathway being implied to be associated with Twist1 pathophysiology is Eph/Ephrin signaling pathway. Eph/Ephrin signals have been proposed to interact with Twist1 and Msx2, and disrupt the mesoderm-neural crest lineage boundary. Eph/Ephrin signaling pathway plays downstream signals of Twist1 and Msx2 [48, 49] (Figure 3(c)). Studies implied that haploinsufficiency of Twist1 could augment the expression of Msx2, which further alleviate the expressions of EphrinA2 and A4. The downregulation of Ephrin leads to the invasion of neural crest cells into mesenchymal cells of coronal sutures, thus displayed coronal suture synostosis [48]. Furthermore, mutations of Msx2, EphrinB1, EphrinA4 have been identified in coronal synostosis cases [48, 114, 115]. Taken together, the speculation implied the vital role of Twist1 in the maintainability of mesoderm-neural crest lineage boundary. Moreover, EphrinA4 could regulate both Erk1/2 and BMP pathways via interaction with Twist1 [49].
Schematic diagram of gene interactions associated with Twist1, which is modified from Ref. [145]. The diagram is proposed by Connerney et al. [113] and Merrill et al. [48]. (a) represents the homodimers of Twist1, and (b) represents the heterodimers of Twist1. The interactions of Eph/Ephrin with Twist1and MSX2 have been illustrated as (c).
4.3. TGF signaling pathway
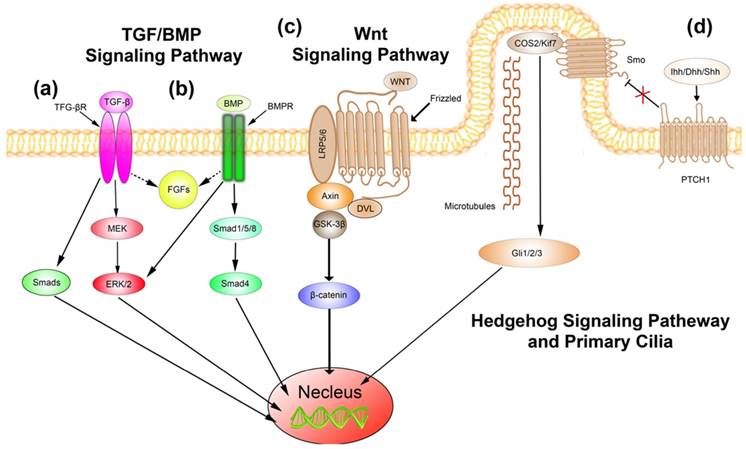
Even though no TGF mutation has been identified in craniosynostosis cases, mutations of its receptors have been published before [116, 117]. Therefore, the vital roles of TGF in maintaining the patency of cranial sutures have been illuminated [41, 118]. Studies revealed that additional of TGF‐β2 might facilitate the obliteration of sutures, whereas TGF‐β3 exhibited reverse effects [118]. Opperman et al. [42] demonstrated that additional of TGF-β2 could induce the premature fusion of sutures via enhancing the proliferation of cells, whereas removal of TGF-β2 did not downregulate the proliferation of cells but inhibit cells differentiation. In addition, high levels of apoptotic cells have been observed in the TGF-β2 additive groups, whose sutures suffer from premature fusion. To be different from the results observed in TGF-β2 additive groups, the study exhibited converse results in TGF-β3 additive studies. The molecular mechanisms involved in the TGF signaling-induced craniosynostosis attracted enormous attentions, and have been well investigated. Studies implied that TGF-β2 might stimulate the phosphorylation of Erk1/2, and resulted in the closure of sutures [41] (Figure 4(a)). Besides, as Smad proteins are of vital importance downstream mediators of TGF signaling, the roles of Smads in suture patency have been discussed. Transient up-regulated expressions of phosphorylated Smad2 and Smad3 have been observed in patent sutures, while, as the decrease of phosphorylated Smad2 and Smad3, the expression of Smad7 increased. However, in fusing sutures, continuous elevations of phosphorylated Smad2 and Smad3 have been observed, and no significant changes of Smad7 could be found. To investigate the role of Smad7, siRNA of Smad7 was applied. Interestingly, downregulation of Smad7 might elicit suture fusion and increase the expressions of phosphorylated Smad2 and Smad3. Moreover, upregulation of FGF10 and phosphorylation of Erk1/2 have also been observed [119] (Figure 4(a)). These results indicated that TGF signaling plays critical roles during the development of cranial suture, and perturbance of associated molecules might result in craniosynostosis. In addition, the interactions of TGFs with Erk1/2, Smads, and FGF are significant during the processes of craniosynostosis, which need to be thoroughly investigated.
The figure illustrates the signal transduction cascades of TGF/BMP, Wnt, Hedgehog signaling pathway in the development of cranial sutures. Any possible impairment might evoke craniosynostosis[121]. (a) TGF signaling; (b) BMP signaling; (c)Wnt signaling; (d) Hedgehog signaling.
4.4. BMP signaling pathway
Bone morphogenetic proteins (BMP) are members of TGF superfamily, which are essential for the development of multi-tissues, including craniomaxillofacial development [44, 45, 120]. BMPs could bind to transmembrane receptors. The activation of receptors could phosphorylate Smad1/5/8, which interact with Smad4 subsequently. The Smad complex then translocate into the nucleus and stimulate the expression of downstream genes [121]. Studies revealed that enhanced expression of BMP signaling via activating its receptors resulted in premature suture fusion. The expressions of phosphorylated Smad1/5/8 were upregulated (Figure 4(b)). Meantime, enhanced expression of FGF2, FGFR1, FGFR2, and phosphorylated Erk1/2 have also been illuminated as the activation of BMP receptors (Figure 4(b)). While, studies indicated that FGF and Erk1/2 signaling were not the direct cause of BMP receptor mutation induced craniosynostosis. In other words, FGF and Erk1/2 might partially be involved in the craniosynostosis induced via activating BMP signaling pathway, while the critical roles of Smads dependent pathway might be significant [45]. In addition to Smad1/5/8 and Smad4, Smad6, an inhibitory member of Smads, is believed to function preventing premature fusion of cranial sutures. Timberlake and colleagues reported rare mutations in Smad6 and all individuals having such mutations develop a midline craniosynostosis when those individuals carry a high risk allele for BMP2 [122]. It is speculated that the high risk allele for BMP2 may produce higher amount of BMP2, thus combined with a loss of function of SMAD6, BMP signaling levels should become higher to lead to midline craniosynostosis. In combination with the knowledge from animal models showing that increased BMP signaling results in premature fusion of the metopic suture [44, 45], those findings strongly suggest that increased BMP signaling is at least one of the molecular mechanisms for human midline craniosynostosis. A re-synostosis case after operation has also been published recently, and the authors proposed that the mutations of Smad6 together with TCF12 exacerbate the severe manifestations and accelerate the process of re-synostosis [123]. Besides, a study implied that the antagonist of BMP, Noggin, is associated with the patency of sutures. Warren et al.[124] demonstrated that, in the patent sagittal and coronal sutures, continuous expression of Noggin have been observed, but in fused sutures, Noggin was absent and high levels of BMP signaling activity was observed. Furthermore, recent studies also indicated that Msx1 and Msx2 mutations could result in ectopic bone formation in the frontal foramen via activating BMP signaling [125]. Also, the disturbance of Msx1 and Msx2 were implied in the migration of neural crest cells and abnormalities of frontal bones and parietal bones [126]. Considering the critical roles of BMP signaling during the processed of craniosynostosis, some researchers hold the assumption that targeted intervention of the biological processes might be potential therapy for craniosynostosis. Zhou et al. [127] fabricated Glypican-loaded titania implants, which could regulate the expression of BMP2, and demonstrated that the delivery system could efficiently transfect C2C12 cells with favorable release periods. Moreover, Gremlin1, an attractive BMP antagonist, was encapsulated in hydrogel for the prevention of re-synostosis after surgery. The study revealed that the re-synostosis has been delayed [128]. Although the biomaterials did not achieve prevention of re-synostosis, the approach still enrich new sights for the treatment of craniosynostosis. As the interactions between BMP and other molecules are elusive, and a plenty of problems about the architecture of delivery carrier need to be overcome, the investigation of the molecular mechanisms should be processed thoroughly and deeply.
4.5. Wnt signaling pathway
Wnt is a conserved signaling pathway, which plays vital roles in the processes of cell proliferation, differentiation, migration, and patterning [129]. Wnt signaling pathway could be further classified as canonical β-catenin pathway and non-canonical pathways, which includes Wnt/Ca2+ pathway and planar cell polarity pathway. In the canonical β-catenin pathway, Wnt binds to transmembrane receptors, Frizzled and LRP5/6, and leads to the release of β-catenin into the cytoplasm. The accumulated β-catenin translocate into nucleus and stimulate the target genes expression (Figure 4(c)). The characteristics of Wnt/Ca2+ pathway are the intracellular Ca2+ release and activation of PKC. The planar cell polarity pathway is implicated within the palatal shelves [130]. Considering the assignable role of Wnt signaling pathway in skeletal development, studies have been processed to identify the possibly genetic mutations, associated with Wnt signaling pathway, in craniosynostosis patients. Potter et al.[131] identified genetic mutations associated with Wnt signaling pathway, including downregulation of both SFRP2 and DKK2, in nonsyndromic craniosynostosis patients. The study also implied that both genes played as negative regulators of Wnt signaling. In addition, negative regulators of Wnt, including Axin1, DVL, Mesp1, PMSC, and NPHP4, have also been illuminated in nonsyndromic craniosynostosis cases [132]. Genetic mutations of Wnt signaling have also been identified in syndromic craniosynostosis. Getinkaya et al. [133] demonstrated FRZB, SFRP2, and Wnt2 mutations in Apert syndrome patients. Furthermore, Axin2 acts as a pronouncing member of degradation complex, which phosphorylate β-catenin and promote the degradation of β-catenin [134]. Studies proposed that aberrant expression of Axin2 could accelerate the osteogenic processes and lead to craniosynostosis [135]. As a plenty of genetic mutations of Wnt signaling have been identified, the critical roles of Wnt signaling in the etiology and pathophysiology of craniosynostosis should be thoroughly studied. Even though, some studies based on animal models have been processed [40, 135, 136], there still have largely unknown and need to be further studied.
4.6. Hedgehog signaling and primary cilia
Mutations of Hedgehog signaling associated proteins have been identified for years. Jenkins et al.[137] identified loss-of-function mutations in RAS oncogene family 23 (RAB23), which is an inhibitor of Hedgehog signaling. In addition, mutations in Gli3, a downstream protein of Hedgehog signaling, displayed synostosis of metopic suture [138]. Moreover, Hedgehog signaling has close relationships with primary cilia. Hedgehog ligand binds to patched1, which presents in cilia, and then the transmembrane protein smoothened moves into cilia. The smoothened plays key role in activation of Gli1-3 downstream, and regulate the functions of cells subsequently [139] (Figure 4(d)). Primary cilia are microtubule based structures of cells, whose function is perceiving the extracellular stimulations and transducing signals. The intraflagellar transport (IFT) machinery is essential for the trafficking of proteins [140]. IFT mutations, such as IFT140, IFT122, and WDR35, are mainly identified in craniosynostosis patients with cranioectodermal dysplasia [141-143]. The mutations disrupted the transport functions of primary cilia, and lead to abnormalities of skeletal development. Otherwise, studies indicated that FGF/FGFR signaling pathway could regulate the functions of cilia [144].
5. Conclusion
Genetic factors have been implied to have close relationships with craniosynostosis. Multiple mutations of genes have been identified during the last two decades, both in the syndromic and nonsyndromic craniosynostosis. The most common variants that identified in patients including FGFR1-3, Twist1, EFNB, Msx2, etc. Studies indicated that FGF/FGFR signaling plays vital roles during the embryonic development of cranial, and interacts with PI3K/Akt, Ras/MAPK, and PLCγ signaling pathways. Besides, Twist1, whose mutation mainly identified in Saethre-Chotzen syndrome and focus on coronal sutures, displays close correlations with FGFR2, EFNB, and BMP signaling pathways. Besides, Wnt, BMP and Hedgehog signaling pathways are also involved in the potential mechanisms beneath the genetic mutations. The complex and interacted signaling pathways make it difficult to manipulate the pathogenic processes via targeted regulate typical molecular. In addition, considering the overlapping phenotypes and heterogeneous penetrance of analogous cases, environment factors have been implied to be associated with suture synostosis. The stimulus generated from growing brain may produce tensile stress across the sutures and manipulate the osteogenic processes regionally. In summary, the etiology of craniosynostosis is complicated and further studies are need to clarify the specific roles of each parameter. The identification of new mutations should be continuous processed for providing evidences to prenatal screening and counselling. Furthermore, up till now, most studies focus on the syndromic craniosynostosis, as syndromic cases exhibit severer manifestations and stronger genetic associations. However, the studies about nonsyndromic cases, which occupied over 70% of all cases, are insufficient. Further investigations for illuminating disease-specific cellular signaling pathways are necessary in order to provide thorough insights into the development of craniosynostosis and other developmental disorders.
Acknowledgements
The research is supported by National Natural Science Foundation of China 81771027.
Competing Interests
The authors declare no potential conflicts of interest with respect to the authorship and publication of this article.
References
1. Moosa S, Wollnik B. Altered FGF signalling in congenital craniofacial and skeletal disorders. Semin Cell Dev Biol. 2016;53:115-25
2. Barreto S, Gonzalez-Vazquez A, Cameron AR. et al. Identification of stiffness-induced signalling mechanisms in cells from patent and fused sutures associated with craniosynostosis. Sci Rep. 2017;7:11494
3. Vu HL, Panchal J, Parker EE. et al. The timing of physiologic closure of the metopic suture: a review of 159 patients using reconstructed 3D CT scans of the craniofacial region. J Craniofac Surg. 2001;12:527-32
4. Cohen MM Jr. Sutural biology and the correlates of craniosynostosis. Am J Med Genet. 1993;47:581-616
5. Kutkowska-Kazmierczak A, Gos M, Obersztyn E. Craniosynostosis as a clinical and diagnostic problem: molecular pathology and genetic counseling. J Appl Genet. 2018;59:133-47
6. Esteve-Altava B, Valles-Catala T, Guimera R. et al. Bone Fusion in Normal and Pathological Development is Constrained by the Network Architecture of the Human Skull. Sci Rep. 2017;7:3376
7. Heuze Y, Holmes G, Peter I. et al. Closing the Gap: Genetic and Genomic Continuum from Syndromic to Nonsyndromic Craniosynostoses. Curr Genet Med Rep. 2014;2:135-45
8. Lee HQ, Hutson JM, Wray AC. et al. Changing epidemiology of nonsyndromic craniosynostosis and revisiting the risk factors. J Craniofac Surg. 2012;23:1245-51
9. Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989-2003. Am J Med Genet A. 2008;146A:984-91
10. Reardon W. Craniosynostosis. Diagnosis, evaluation and management. J Med Genet. 2000;37:727
11. Cohen MM Jr. Craniosynostosis and syndromes with craniosynostosis: incidence, genetics, penetrance, variability, and new syndrome updating. Birth Defects Orig Artic Ser. 1979;15:13-63
12. Al-Rekabi Z, Cunningham ML, Sniadecki NJ. Cell Mechanics of Craniosynostosis. Acs Biomater Sci Eng. 2017;3:2733-43
13. Kimonis V, Gold JA, Hoffman TL. et al. Genetics of craniosynostosis. Semin Pediatr Neurol. 2007;14:150-61
14. Patel A, Terner J, Travieso R. et al. On Bernard Sarnat's 100th birthday: pathology and management of craniosynostosis. J Craniofac Surg. 2012;23:105-12
15. Lajeunie E, Le Merrer M, Bonaiti-Pellie C. et al. Genetic study of nonsyndromic coronal craniosynostosis. Am J Med Genet. 1995;55:500-4
16. Fonteles CSR, Finnell RH, George TM. et al. Craniosynostosis: current conceptions and misconceptions. Aims Genetics. 2016;3:99-129
17. Governale LS. Craniosynostosis. Pediatr Neurol. 2015;53:394-401
18. Cohen SR, Persing JA. Intracranial pressure in single-suture craniosynostosis. Cleft Palate Craniofac J. 1998;35:194-6
19. Thompson DN, Harkness W, Jones B. et al. Subdural intracranial pressure monitoring in craniosynostosis: its role in surgical management. Childs Nerv Syst. 1995;11:269-75
20. Will AJ, Cova G, Osterwalder M. et al. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog). Nat Genet. 2017;49:1539-45
21. Sharma VP, Fenwick AL, Brockop MS. et al. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet. 2013;45:304-7
22. Nur BG, Pehlivanoglu S, Mihci E. et al. Clinicogenetic study of Turkish patients with syndromic craniosynostosis and literature review. Pediatr Neurol. 2014;50:482-90
23. Johnson D, Wilkie AO. Craniosynostosis. Eur J Hum Genet. 2011;19:369-76
24. Ko JM. Genetic Syndromes Associated with Craniosynostosis. J Korean Neurosurg S. 2016;59:187
25. Merrill AE, Sarukhanov A, Krejci P. et al. Bent Bone Dysplasia-FGFR2 type, a Distinct Skeletal Disorder, Has Deficient Canonical FGF Signaling. Am J Hum Genet. 2012;90:550-7
26. Marie PJ, Huang Y, Meng T. et al. Twist1- and Twist2-Haploinsufficiency Results in Reduced Bone Formation. PLoS ONE. 2014;9:e99331
27. Xu Y, Sun S, Li N. et al. Identification and analysis of the genetic causes in nine unrelated probands with syndromic craniosynostosis. Gene. 2018;641:144-50
28. Chaudhry A, Sabatini P, Han L. et al. Heterozygous mutations in ERF cause syndromic craniosynostosis with multiple suture involvement. Am J Med Genet A. 2015;167A:2544-7
29. Azoury SC, Reddy S, Shukla V. et al. Fibroblast Growth Factor Receptor 2 (FGFR2) Mutation Related Syndromic Craniosynostosis. Int J Biol Sci. 2017;13:1479-88
30. Lattanzi W, Barba M, Di Pietro L. et al. Genetic advances in craniosynostosis. Am J Med Genet A. 2017;173:1406-29
31. Twigg SR, Wilkie AO. A Genetic-Pathophysiological Framework for Craniosynostosis. Am J Hum Genet. 2015;97:359-77
32. Lin Z, Fateh A, Salem DM. et al. Periosteum: biology and applications in craniofacial bone regeneration. J Dent Res. 2014;93:109-16
33. Ochareon P, Herring SW. Cell replication in craniofacial periosteum: appositional vs. resorptive sites. J Anat. 2011;218:285-97
34. Pagni G, Kaigler D, Rasperini G. et al. Bone repair cells for craniofacial regeneration. Adv Drug Deliv Rev. 2012;64:1310-9
35. Petrovic V, Zivkovic P, Petrovic D. et al. Craniofacial bone tissue engineering. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:e1-9
36. Zhao H, Feng J, Ho TV. et al. The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat Cell Biol. 2015;17:386-96
37. Maruyama T, Jeong J, Sheu TJ. et al. Stem cells of the suture mesenchyme in craniofacial bone development, repair and regeneration. Nat Commun. 2016;7:10526
38. Maruyama T, Mirando AJ, Deng CX. et al. The balance of WNT and FGF signaling influences mesenchymal stem cell fate during skeletal development. Sci Signal. 2010;3:ra40
39. Liu J, Nam HK, Campbell C. et al. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl-/- mouse model of infantile hypophosphatasia. Bone. 2014;67:81-94
40. Behr B, Longaker MT, Quarto N. Absence of endochondral ossification and craniosynostosis in posterior frontal cranial sutures of Axin2(-/-) mice. PLoS One. 2013;8:e70240
41. Opperman LA, Fernandez CR, So S. et al. Erk1/2 signaling is required for Tgf-beta 2-induced suture closure. Dev Dyn. 2006;235:1292-9
42. Opperman LA, Adab K, Gakunga PT. Transforming growth factor-beta 2 and TGF-beta 3 regulate fetal rat cranial suture morphogenesis by regulating rates of cell proliferation and apoptosis. Dev Dyn. 2000;219:237-47
43. Kramer K, Yang J, Swanson WB. et al. Rapamycin rescues BMP mediated midline craniosynostosis phenotype through reduction of mTOR signaling in a mouse model. Genesis. 2018;56:e23220
44. Hayano S, Komatsu Y, Pan H. et al. Augmented BMP signaling in the neural crest inhibits nasal cartilage morphogenesis by inducing p53-mediated apoptosis. Development. 2015;142:1357-67
45. Komatsu Y, Yu PB, Kamiya N. et al. Augmentation of Smad-dependent BMP signaling in neural crest cells causes craniosynostosis in mice. J Bone Miner Res. 2013;28:1422-33
46. Jin SW, Sim KB, Kim SD. Development and Growth of the Normal Cranial Vault: An Embryologic Review. J Korean Neurosurg Soc. 2016;59:192-6
47. Deckelbaum RA, Holmes G, Zhao Z. et al. Regulation of cranial morphogenesis and cell fate at the neural crest-mesoderm boundary by engrailed 1. Development. 2012;139:1346-58
48. Merrill AE, Bochukova EG, Brugger SM. et al. Cell mixing at a neural crest-mesoderm boundary and deficient ephrin-Eph signaling in the pathogenesis of craniosynostosis. Hum Mol Genet. 2006;15:1319-28
49. Ting MC, Wu NL, Roybal PG. et al. EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development. 2009;136:855-64
50. Yen HY, Ting MC, Maxson RE. Jagged1 functions downstream of Twist1 in the specification of the coronal suture and the formation of a boundary between osteogenic and non-osteogenic cells. Dev Biol. 2010;347:258-70
51. Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22:138-47
52. Carter DH, Sloan P, Aaron JE. Immunolocalization of collagen types I and III, tenascin, and fibronectin in intramembranous bone. J Histochem Cytochem. 1991;39:599-606
53. Neve A, Corrado A, Cantatore FP. Osteoblast physiology in normal and pathological conditions. Cell Tissue Res. 2011;343:289-302
54. Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446-65
55. Lenton K, James AW, Manu A. et al. Indian hedgehog positively regulates calvarial ossification and modulates bone morphogenetic protein signaling. Genesis. 2011;49:784-96
56. Connerney J, Andreeva V, Leshem Y. et al. Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev Biol. 2008;318:323-34
57. Sosic D, Richardson JA, Yu K. et al. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappa B activity. Cell. 2003;112:169-80
58. Bialek P, Kern B, Yang X. et al. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423-35
59. Connerney J, Andreeva V, Leshem Y. et al. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev Dynam. 2012;241(2):433-433
60. Goos JA, Fenwick AL, Swagemakers SM. et al. Identification of Intragenic Exon Deletions and Duplication of TCF12 by Whole Genome or Targeted Sequencing as a Cause of TCF12-Related Craniosynostosis. Hum Mutat. 2016;37:732-6
61. Luo XY, Yang Y, Sun HQ. Effect of FGF/FGFR Signal in Fluid Shear Stress and Estrogen Regulating Bone Metabolism. Mol Biol. 2017:7
62. Kress W, Petersen B, Collmann H. et al. An unusual FGFR1 mutation (fibroblast growth factor receptor 1 mutation) in a girl with non-syndromic trigonocephaly. Cytogenet Cell Genet. 2000;91:138-40
63. Tartaglia M, Valeri S, Velardi F. et al. Trp290Cys mutation in exon IIIa of the fibroblast growth factor receptor 2 (FGFR2) gene is associated with Pfeiffer syndrome. Hum Genet. 1997;99:602-6
64. Kim BG, Kim HJ, Park HJ. et al. Runx2 phosphorylation induced by fibroblast growth factor-2/protein kinase C pathways. Proteomics. 2006;6:1166-74
65. Miraoui H, Oudina K, Petite H. et al. Fibroblast Growth Factor Receptor 2 Promotes Osteogenic Differentiation in Mesenchymal Cells via ERK1/2 and Protein Kinase C Signaling. J Biol Chem. 2009;284:4897-904
66. Jeon E, Yun YR, Kang W. et al. Investigating the Role of FGF18 in the Cultivation and Osteogenic Differentiation of Mesenchymal Stem Cells. Plos One. 2012:7
67. Raucci A, Bellosta P, Grassi R. et al. Osteoblast proliferation or differentiation is regulated by relative strengths of opposing signaling pathways. J Cell Physiol. 2008;215:442-51
68. Mefford HC, Shafer N, Antonacci F. et al. Copy Number Variation Analysis in Single-Suture Craniosynostosis: Multiple Rare Variants Including RUNX2 Duplication in Two Cousins With Metopic Craniosynostosis. Am J Med Genet A. 2010;152a:2203-10
69. Varvagiannis K, Stefanidou A, Gyftodimou Y. et al. Pure de novo partial trisomy 6p in a girl with craniosynostosis. Am J Med Genet A. 2013;161a:343-51
70. Fluck CE, Tajima T, Pandey AV. et al. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat Genet. 2004;36:228-30
71. Laue K, Pogoda HM, Daniel PB. et al. Craniosynostosis and Multiple Skeletal Anomalies in Humans and Zebrafish Result from a Defect in the Localized Degradation of Retinoic Acid. Am J Hum Genet. 2011;89:595-606
72. Ribes V, Otto DME, Dickmann L. et al. Rescue of cytochrome P450 oxidoreductase (Por) mouse mutants reveals functions in vasculogenesis, brain and limb patterning linked to retinoic acid homeostasis. Dev Biol. 2007;303:66-81
73. Schmidt K, Hughes C, Chudek JA. et al. Cholesterol Metabolism: the Main Pathway Acting Downstream of Cytochrome P450 Oxidoreductase in Skeletal Development of the Limb. Mol Cell Biol. 2009;29:2716-29
74. Nieminen P, Morgan NV, Fenwick AL. et al. Inactivation of IL11 Signaling Causes Craniosynostosis, Delayed Tooth Eruption, and Supernumerary Teeth. Am J Hum Genet. 2011;89:67-81
75. Kido S, Kuriwaka-Kido R, Imamura T. et al. Mechanical stress induces Interleukin-11 expression to stimulate osteoblast differentiation. Bone. 2009;45:1125-32
76. Suga K, Saitoh M, Fukushima S. et al. Interleukin-11 induces osteoblast differentiation and acts synergistically with bone morphogenetic protein-2 in C3H10T1/2 cells. J Interferon Cytokine Res. 2001;21:695-707
77. Suga K, Saitoh M, Kokubo S. et al. Interleukin-11 acts synergistically with bone morphogenetic protein-2 to accelerate bone formation in a rat ectopic model. J Interferon Cytokine Res. 2003;23:203-7
78. Sims NA, Jenkins BJ, Nakamura A. et al. Interleukin-11 receptor signaling is required for normal bone remodeling. J Bone Miner Res. 2005;20:1093-102
79. Alderman BW, Bradley CM, Greene C. et al. Increased Risk of Craniosynostosis with Maternal Cigarette-Smoking during Pregnancy. Teratology. 1994;50:13-8
80. Kallen K. Maternal smoking and craniosynostosis. Teratology. 1999;60:146-50
81. Gardner JS, Guyard-Boileau B, Alderman BW. et al. Maternal exposure to prescription and non prescription pharmaceuticals or drugs of abuse and risk of craniosynostosis. Int J Epidemiol. 1998;27:64-7
82. Higashino T, Hirabayashi S. A secondary craniosynostosis associated with juvenile hyperthyroidism. J Plast Reconstr Aesthet Surg. 2013;66:e284-6
83. Chawla R, Alden TD, Bizhanova A. et al. Squamosal Suture Craniosynostosis Due to Hyperthyroidism Caused by an Activating Thyrotropin Receptor Mutation (T632I). Thyroid. 2015;25:1167-72
84. Zeiger JS, Beaty TH, Hetmanski JB. et al. Genetic and environmental risk factors for sagittal craniosynostosis. J Craniofac Surg. 2002;13:602-6
85. Bradley CM, Alderman BW, Williams MA. et al. Parental Occupations as Risk-Factors for Craniosynostosis in Offspring. Epidemiology. 1995;6:306-10
86. Sanchez-Lara PA, Carmichael SL, Graham JM. et al. Fetal constraint as a potential risk factor for craniosynostosis. Am J Med Genet A. 2010;152A:394-400
87. Jacob S, Wu C, Freeman TA. et al. Expression of Indian Hedgehog, BMP-4 and Noggin in craniosynostosis induced by fetal constraint. Ann Plast Surg. 2007;58:215-21
88. Hunenko O, Karmacharya J, Ong G. et al. Toward an understanding of nonsyndromic craniosynostosis: Altered patterns of TGF-beta receptor and FGF receptor expression induced by intrauterine head constraint. Ann Plas Surg. 2001;46:546-53
89. Cinalli G, Sainte-Rose C, Kollar EM. et al. Hydrocephalus and craniosynostosis. J Neurosurg. 1998;88:209-14
90. Albright AL, Tyler-Kabara E. Slit-ventricle syndrome secondary to shunt-induced suture ossification. Neurosurgery. 2001;48:764-70
91. Weinzweig J, Bartlett SP, Chen JC. et al. Cranial vault expansion in the management of postshunt craniosynostosis and slit ventricle syndrome. Plast Reconstr Surg. 2008;122:1171-80
92. Heller JB, Gabbay JS, Wasson K. et al. Cranial suture response to stress: expression patterns of Noggin and Runx2. Plast Reconstr Surg. 2007;119:2037-45
93. Green PJ, Walsh FS, Doherty P. Promiscuity of fibroblast growth factor receptors. Bioessays. 1996;18:639-46
94. Teven CM, Farina EM, Rivas J. et al. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis. 2014;1:199-213
95. Hunter T. Signaling-2000 and beyond. Cell. 2000;100:113-27
96. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211-25
97. Wang F, Kan M, Yan GC. et al. Alternately Spliced Nh2-Terminal Immunoglobulin-Like Loop-I in the Ectodomain of the Fibroblast Growth-Factor (Fgf) Receptor-1 Lowers Affinity for Both Heparin and Fgf-1. J Biol Chem. 1995;270:10231-5
98. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235-53
99. Mckeehan WL, Kan M. Heparan-Sulfate Fibroblast Growth Factor Receptor Complex: Structure-Function Relationships. Mol Reprod Dev. 1994;39:69-82
100. Wang F, Lu WQ, McKeehan K. et al. Common and specific determinants for fibroblast: Growth factors in the ectodomain of the receptor kinase complex. Biochemistry-Us. 1999;38:160-71
101. Zhang XQ, Ibrahimi OA, Olsen SK. et al. Receptor specificity of the fibroblast growth factor family - The complete mammalian FGF family. J Biol Chem. 2006;281:15694-700
102. Wiedemann M, Trueb B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics. 2000;69:275-9
103. Trueb B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell Mol Life Sci. 2011;68:951-64
104. Sleeman M, Fraser J, McDonald M. et al. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. 2001;271:171-82
105. Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth F R. 2005;16:139-49
106. Yun YR, Won JE, Jeon E. et al. Fibroblast growth factors: biology, function, and application for tissue regeneration. J Tissue Eng. 2010;2010:218142
107. Dorey K, Amaya E. FGF signalling: diverse roles during early vertebrate embryogenesis. Development. 2010;137:3731-42
108. Dailey L, Ambrosetti D, Mansukhani A. et al. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth F R. 2005;16:233-47
109. Lee SW, Choi KY, Cho JY. et al. TGF-beta 2 stimulates cranial suture closure through activation of the Erk-MAPK pathway. J Cell Biochem. 2006;98:981-91
110. Shukla V, Coumoul X, Wang RH. et al. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet. 2007;39:1145-50
111. Morita J, Nakamura M, Kobayashi Y. et al. Soluble form of FGFR2 with S252W partially prevents craniosynostosis of the apert mouse model. Dev Dynam. 2014;243:560-7
112. Yin L, Du X, Li CL. et al. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone. 2008;42:631-43
113. Connerney J, Andreeva V, Leshem Y. et al. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev Dynam. 2006;235:1345-57
114. Twigg SRF, Kan R, Babbs C. et al. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. P Natl Acad Sci USA. 2004;101:8652-7
115. Jabs EW, Muller U, Li X. et al. A Mutation in the Homeodomain of the Human Msx2 Gene in a Family Affected with Autosomal-Dominant Craniosynostosis. Cell. 1993;75:443-50
116. Loeys BL, Chen J, Neptune ER. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275-81
117. Roth DA, Gold LI, Han VK. et al. Immunolocalization of transforming growth factor beta 1, beta 2, and beta 3 and insulin-like growth factor I in premature cranial suture fusion. Plast Reconstr Surg. 1997;99:300-9 discussion 10-6
118. Opperman LA, Galanis V, Williams AR. et al. Transforming growth factor-beta3 (Tgf-beta3) down-regulates Tgf-beta3 receptor type I (Tbetar-I) during rescue of cranial sutures from osseous obliteration. Orthod Craniofac Res. 2002;5:5-16
119. Zhou H, Zou S, Lan Y. et al. Smad7 Modulates TGFβ Signaling During Cranial Suture Development to Maintain Suture Patency. J Bone Miner Res. 2014;29:716-24
120. Ishii M, Han J, Yen HY. et al. Combined deficiencies of Msx1 and Msx2 cause impaired patterning and survival of the cranial neural crest. Development. 2005;132:4937-50
121. Katsianou MA, Adamopoulos C, Vastardis H. et al. Signaling mechanisms implicated in cranial sutures pathophysiology: Craniosynostosis. BBA Clin. 2016;6:165-76
122. Timberlake AT, Choi J, Zaidi S. et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife. 2016:5
123. Timberlake AT, Wu R, Nelson-Williams C. et al. Co-occurrence of frameshift mutations in SMAD6 and TCF12 in a child with complex craniosynostosis. Hum Genome Var. 2018;5:14
124. Warren SM, Brunet LJ, Harland RM. et al. The BMP antagonist noggin regulates cranial suture fusion. Nature. 2003;422:625-9
125. Roybal PG, Wu NL, Sun JJ. et al. Inactivation of Msx1 and Msx2 in neural crest reveals an unexpected role in suppressing heterotopic bone formation in the head. Dev Biol. 2010;343:28-39
126. Satokata I, Maas R. Msx1 Deficient Mice Exhibit Cleft-Palate and Abnormalities of Craniofacial and Tooth Development. Nat Genet. 1994;6:348-56
127. Bariana M, Dwivedi P, Ranjitkar S. et al. Glypican-based drug releasing titania implants to regulate BMP2 bioactivity as a potential approach for craniosynostosis therapy. Nanomedicine. 2017
128. Hermann CD, Wilson DS, Lawrence KA. et al. Rapidly polymerizing injectable click hydrogel therapy to delay bone growth in a murine re-synostosis model. Biomaterials. 2014;35:9698-708
129. Geetha-Loganathan P, Nimmagadda S, Antoni L. et al. Expression of WNT signalling pathway genes during chicken craniofacial development. Dev Dyn. 2009;238:1150-65
130. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179-92
131. Potter AB, Rhodes JL, Vega RA. et al. Gene expression changes between patent and fused cranial sutures in a nonsyndromic craniosynostosis population. Eplasty. 2015;15:e12
132. Timberlake AT, Furey CG, Choi J. et al. De novo mutations in inhibitors of Wnt, BMP, and Ras/ERK signaling pathways in non-syndromic midline craniosynostosis. Proc Natl Acad Sci USA. 2017;114:E7341-E7
133. Cetinkaya A, Taskiran E, Soyer T. et al. Dermal fibroblast transcriptome indicates contribution of WNT signaling pathways in the pathogenesis of Apert syndrome. Turk J Pediatr. 2017;59:619-24
134. Yu HMI, Jerchow B, Sheu TJ. et al. The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development. 2005;132:1995-2005
135. Liu B, Yu HMI, Hsu W. Craniosynostosis caused by Axin2 deficiency is mediated through distinct functions of beta-catenin in proliferation and differentiation. Dev Biol. 2007;301:298-308
136. Liu B, Chen S, Johnson C. et al. A ciliopathy with hydrocephalus, isolated craniosynostosis, hypertelorism, and clefting caused by deletion of Kif3a. Reprod Toxicol. 2014;48:88-97
137. Jenkins D, Seelow D, Jehee FS. et al. RAB23 mutations in carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am J Hum Genet. 2007;80:1162-70
138. McDonald-McGinn DM, Feret H, Nah HD. et al. Metopic Craniosynostosis Due To Mutations in GLI3: A Novel Association. Am J Med Genet A. 2010;152a:1654-60
139. Ocbina PJ, Eggenschwiler JT, Moskowitz I. et al. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet. 2011;43:547-53
140. Takahara M, Katoh Y, Nakamura K. et al. Ciliopathy-associated mutations of IFT122 impair ciliary protein trafficking but not ciliogenesis. Hum Mol Genet. 2018;27:516-28
141. Smith C, Lamont RE, Wade A. et al. A relatively mild skeletal ciliopathy phenotype consistent with cranioectodermal dysplasia is associated with a homozygous nonsynonymous mutation in WDR35. Am J Med Genet A. 2016;170:760-5
142. Walczak-Sztulpa J, Wawrocka A, Swiader-Lesniak A. et al. Clinical and molecular genetic characterization of a male patient with Sensenbrenner syndrome (cranioectodermal dysplasia) and biallelic WDR35 mutations. Birth Defects Res. 2018;110:376-81
143. Alazami AM, Seidahmed MZ, Alzahrani F. et al. Novel IFT122 mutation associated with impaired ciliogenesis and cranioectodermal dysplasia. Mol Genet Genomic Med. 2014;2:103-6
144. Kunova Bosakova M, Varecha M, Hampl M. et al. Regulation of ciliary function by fibroblast growth factor signaling identifies FGFR3-related disorders achondroplasia and thanatophoric dysplasia as ciliopathies. Hum Mol Genet. 2018;27:1093-105
145. Flaherty K, Singh N, Richtsmeier JT. Understanding craniosynostosis as a growth disorder. Wiley Interdiscip Rev Dev Biol. 2016;5:429-59
Author contact
![]() Corresponding author: Yan Gu, guyan96com; Department of Orthodontics, Peking University School and Hospital of Stomatology, No. 22 Zhongguancun Avenue South, Haidian District, Beijing, 100081, PR. China; Tel: +86 010 82195739; Fax: +86 01082195336
Corresponding author: Yan Gu, guyan96com; Department of Orthodontics, Peking University School and Hospital of Stomatology, No. 22 Zhongguancun Avenue South, Haidian District, Beijing, 100081, PR. China; Tel: +86 010 82195739; Fax: +86 01082195336