Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(2):312-324. doi:10.7150/ijbs.29135 This issue Cite
Research Paper
Verteporfin blocks Clusterin which is required for survival of gastric cancer stem cell by modulating HSP90 function
Jixian Xiong1,2* ![]() , Shaoxiang Wang1*, Tie Chen1*, Xingsheng Shu1*, Xianming Mo3
, Shaoxiang Wang1*, Tie Chen1*, Xingsheng Shu1*, Xianming Mo3 ![]() , Gang Chang1, Jia-Jie Chen1, Chenyang Li1, Hui Luo1, Jiing-Dwan Lee1
, Gang Chang1, Jia-Jie Chen1, Chenyang Li1, Hui Luo1, Jiing-Dwan Lee1
1. School of Medicine, Shenzhen University, Shenzhen 518055, China
2. Shenzhen University International Cancer Center, Shenzhen University, Shenzhen 518055, China
3. Laboratory of Stem Cell Biology, State Key Laboratory of Biotherapy/Collaborative Innovation Center of Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, China
* These authors contribute equally to this work.
Received 2018-8-10; Accepted 2018-11-11; Published 2019-1-1
Abstract

Gastric cancer stem cell (GCSC) is implicated in gastric cancer relapse, metastasis and drug resistance. However, the key molecule(s) involved in GCSC survival and the targeting drugs are poorly understood. We discovered increased secreted clusterin (S-Clu) protein expression during the sphere-forming growth of GCSC via mass spectrometry. Overexpression of clusterin was detected in 69/90 (77%) of primary GC tissues and significantly associated with T stage, lymph node metastasis and TNM stage. Depletion of clusterin (Clu, the full-length intracellular clusterin) led to the declustering of GCSC tumorspheres and apoptosis of GCSC. Subsequently, we found clusterin was in complex with heat shock protein 90 beta (HSP90) and involved in regulating the cellular level of HSP90 client proteins. Furthermore, by screening a collection of drugs/inhibitors, we found that verteporfin (VP), a phototherapy drug, blocked clusterin gene expression, decreased the HSP90 client proteins and caused cell death of GCSC. VP treatment is more effective in eradicating GCSCs than in killing GC cells. Both clusterin silencing or VP treatment deterred tumor growth in human GCSC xenografts. These findings collectively suggest that GC patients can promptly benefit from clusterin-targeted therapy as well as VP treatment in combination with or subsequent to conventional chemotherapy for reducing mortality of GC.
Keywords: Gastric cancer stem cell, Clusterin, Verteporfin, HSP90
Introduction
Gastric cancer (GC) is one of the most frequently occurring and malignant types of cancer [1]. The current therapeutic approach is surgery followed by chemotherapy and/or radiotherapy [2]. However, the five-year survival rate is very low. Most post-treatment deaths of patients are due to the recurrence accompanied by metastasis [3].
Cancer stem cell (CSC) has been implicated in cancer relapse and metastasis which may be caused by CSC's capacity in uncontrolled growth, resistance to chemo- and radiotherapies as well as in Epithelial-Mesenchymal Transition [4-6]. Therefore, understanding the weak spot of CSC and targeting this vulnerability will provide more effective treatment and/or long-term cure for cancer patients [7].
Gastric cancer stem cell (GCSC) was first identified from human gastric cancer cell lines [8]. Our group identified CSC in human gastric adenocarcinoma (GAC) tumor tissues, and showed that these cells can be expanded in vitro [9]. Cancer stem cells from gastric cancer cell lines or GC tumor tissues could be isolated using several surface markers such as CD24, CD44, CD54, CD90, CD133, Lgr5, ALDH1 and EpCAM[10]. However, the biological function of these markers in GC is often unclear. GCSC is characterized by the properties of sustained self-renewal, high proliferative capacities, high expression of CSC markers, and can form sphere in non-adherent medium, shows high tumorigenic potential when xenografted into immunocompromised mice.
GCSC-targeting therapies are currently being investigated, including chemotherapeutic and biological agents that targeting GCSC surface markers, signaling pathways, the CSC microenvironment, and others [10]. However, few agents against these molecules can efficiently eliminate GCSC, which leads to relapse, chemoresistance, and treatment failure. Therefore, finding the key molecule(s) involved in GCSC survival, and developing the targeting drugs are crucial for GCSC-targeting therapies.
In the present study, we revealed that intracellular clusterin is required for sustaining GCSC survival via modulating HSP90 function. We also discovered that verteporfin (VP), a FDA approved drug, was able to effectively inhibit clusterin gene expression in GCSC, suppressed the viability of GCSC and hindered the growth of GCSC-derived tumor xenografts in nude mice, thus suggesting clusterin-targeted therapy and VP treatment can be effective in inhibiting GCSC-mediated tumor growth in cancer patients through modulating HSP90 function.
Materials and methods
GC cell lines, chemicals and antibodies
MGC-803 and BGC-823 GC cell lines were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China), where they were characterized by mycoplasma detection, DNA-Fingerprinting, isozyme detection and cell vitality detection. MGC-803 and BGC-823 cells were maintained in RPMI-1640 with 10% fetal bovine serum and 100 U/mL penicillin-streptomycin, at 37°C and 5% CO2. VP, cisplatin, 5-Fu, 17-AAG and SNX2112 were purchased from Selleck Chemicals (Houston, TX). Doxycycline (Dox) was from Sigma (St. Louis, MO). Anti-Clusterin-α (Santa Cruz, sc6419) was purchased from Santa Cruz Biotechnologies. Anti-HSP90 (Abcam, ab1429) was purchased from Abcam. Anti-Sox2 (CST, no.2748), anti-Cleaved PARP (CST, no.5625), anti-pSer807/Ser811-Rb (CST, no.8516), anti-AKT (CST, no.4691), anti-CDK4 (CST, no.12790), anti-HER2 (CST, no.4290), anti-c-Raf (CST, no.53745), anti-EGFR (CST, no.2646), anti-IGF-1R (CST, no.9750), anti-YAP (CST, no.14074), anti-flag (CST, no.2368) antibodies were from Cell Signaling Technology. Anti-β-actin (Sigma, A2228) antibody was from Sigma.
Gastric cancer stem cell culture
Fresh isolated, primary tumor-derived GCSC which from a GAC patient was obtained from Dr. Xianming Mo, grown and maintained as previously described [9].
Mass spectrometry analysis of conditioned medium from human GCSC tumorsphere
Human GCSC tumorspheres were grown for 5 days and the conditioned medium was harvested at the end of each day. Protein extracts of these conditioned medium (day 1 to day 5) and plain growth medium (day 0) were isolated and trypsin digested, then labeled with iTRAQ reagents according to the manufacturer's instructions (Applied Biosystems Inc., Foster City, CA, USA). These iTRAQ peptides of six samples were pooled and analyzed by data-dependent LC/MS/MS with an AB Sciex hybrid quadruple TOF mass spectrometer. All MS and MS/MS data were processed with the ProteinPilot software, which assigned peptide identity with a <1% false discovery rate with a target-decoy database search approach, and quantitated with extracted iTRAQ reporter ions.
Construction of clusterin, HSP90 expression plasmid
The full-length ORF of clusterin, HSP90 were amplified from total cDNA of GCSC using high-fidelity AccuPrime Taq polymerase (Life Technology), and cloned into the MluI and EcoRI sites of pLVX-TetOne-Puro inducible expression vector (Clontech) using In-Fusion HD Cloning Kit (Clontech) following the manufacturer's instructions. A FLAG tag (DYKDDDDK) was added to the C terminal of Clusterin protein. pLVX-TetOne-Puro inducible expression vector (Clontech) was used as control.
Generation of inducible knockdown (clusterin, YAP or non-targeting control) GCSC lines as well as inducible clusterin, HSP90 expressing and control GCSC lines
Lentiviruses carrying inducible shRNA targeting human clusterin, YAP or non-targeting control lentiviral vectors (GV307) were from GeneChem. The viruses were used to infect cells in the presence of Polybrene. Forty-eight hours later, cells were cultured in medium containing puromycin for the selection of the stable clones. The resulting clones were induced by Doxycycline (Dox) (2.5 μg/ml) for 48 hr. Lentiviruses carrying inducible expressing construct encoding clusterin, HSP90 or an empty inducible expressing vector were used to infect cells in the presence of Polybrene. The stable clones by selection on puromycin were induced by Doxycycline (Dox) (2.5 μg/ml) for 48 hr. Stable repression of clusterin or YAP in GCSC cells, or overexpression of clusterin or HSP90 in GCSC cells, respectively, by lentiviral transduction were confirmed by western blotting.
RNA extraction and quantitative reverse-transcription PCR (qRT-PCR)
RNA was extracted using the RNeasy Mini Kit (QIAGEN) and reverse-transcribed using M-MLV reverse transcriptase (TIANGEN). Quantitative PCR was carried out with Power Up SYBR Green Master Mix (Thermo Fisher Scientific). A total volume of 20 μl containing 10 μl SYBR Green-based PCR Master Mix, 0.2 μl forward primer (10 μM), 0.2 μl reverse primer (10 μM) and template cDNA were mixed and plated for gene expression analyses using the Applied Biosystems QuantStudio Real-Time PCR System (Thermo Fisher Scientific) in accordance with the manufacturer's instructions. GAPDH was used as an internal control. The sequences of primers used in this study were as follows:
Clusterin-F: 5'TGATGAAGACTCTGCTGCTG3'
Clusterin-R: 5'ACTTACTTCCCTGATTGGAC 3'
GAPDH-F: 5'CGAGATCCCTCCAAAATCAA 3'
GAPDH-R: 5'ATCCACAGTCTTCTGGGTGG 3'
Western Blotting
Cells were lysed in cold RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentration was determined by BCA assay (Thermo Fischer Scientific). Equal amounts of protein were resolved by 4-10% Bis-Tris/PAGE, transferred to PVDF membranes (BioRad) and probed overnight at 4°C with the following primary antibodies: anti-Clusterin-α (1:3000), anti-Sox2 (1:2000), anti-HSP90 (1:2000), anti-Cleaved PARP (1:2000), anti-pSer807/Ser811-Rb (1:2000), anti-AKT (1:2000), anti-CDK4 (1:2000), anti-HER2 (1:2000), anti-c-Raf (1:2000), anti-EGFR (1:2000), anti-IGF-1R (1:2000), anti-YAP (1:2000), anti-flag (1:2000), anti-β-actin (1:20000). Secondary antibodies were anti-goat-HRP (Santa Cruz sc2020; 1:5000), anti-mouse-HRP (Cell Signaling 7076; 1:5000) or anti-rabbit-HRP (Cell Signaling 7074; 1:5000). Blots were developed by using Immobilon Western Chemiluminescent HRP substrate (Millipore) or SuperSignal West Chemiluminescent substrate (Thermo Fisher Scientific), and imaged in ChemiDoc MP imaging system (BioRad).
Immunostainning of tissue arrays
Tissue arrays of gastric adenocarcinomas (HStm-Ade180Sur-05) were obtained from Shanghai Outdo Biotech (Shanghai Biochip Co.,Ltd, Shanghai, People's Republic of China) approved by National Human Genetic Resources sharing Service Platform (China, 2005DKA21300) for Medical Research ethical review panel. The goat polyclonal antibody anti-human clusterin (Santa Cruz, sc6419) was diluted 1:5000 in DAKO antibody diluent. The EnVision+ detection system (Dako) was used according to the manufacturer's instructions. Immunostained microarrays were scored by multiplying the intensity (0-3) and extent (0-100) of staining for each tissue point as previously described [11]. Ten independent microscopic fields (400x) were selected for each patient sample to ensure representativeness and homogeneity. The evaluation of clusterin staining was performed without knowledge of the clinicopathologic data by two independent investigators. Statistical analyses were carried out with SPSS 12.0 software (SPSS, Chicago,IL).
TUNEL assay
The DNA fragmentation indicative of apoptosis was examined using terminal deoxynucleotidyl transferase-mediated dUTP nickend labeling method (TUNEL). TUNEL assay was performed using Insitu Cell Death Detection Kit (Cat. NO. 11684817910, Roche Molecular Biochemicals, Germany) according to the instructions of the manufacturer. Briefly, cells were fixed in 4% paraformaldehyde at room temperature for 1h, and then rinsed with phosphate-buffered saline (PBS). The cells were incubated with 3% H2O2 (in methanol) at room temperature for 10 min, and then rinsed with PBS. The cells were permeated with 0.1% Triton X-100 for 2 min on ice. TUNEL enzyme and label solution were mixed and applied to the prepared cell climbing slices, which were incubated again in the humidified chamber for 1h at 37°C. Slices were thoroughly rinsed with PBS, counterstained with DAPI for nuclear staining and analyzed in a drop of PBS under the fluorescence microscope. The nuclei of apoptotic cells were with green fluorescence (stained with FITC fluorescein-dUTP). The TUNEL positive cells (apoptotic cells) were counted. Three fields in each section were measured. Percentage apoptotic cells were quantified by green cells over total cells times 100%.
Cell viability assay
The cell viability was analyzed using a CCK-8 kit (Dojindo Laboratories, Kumamoto, Japan). Exponentially growing cells were seeded into 96-well culture plates (1 × 105 cells/mL) in 100 μl medium for 24 hr. Cells were treated with 17-AAG (0.2 μM), and/or Dox (2.5 μg/ml) for 24 hr, along with an equal volume of DMSO as the control. After adding 10 μl CCK-8 solution per well, the plates were incubated at 37oC for 2 hr. The absorbance was measured at 450 nm using a microplate reader (Infinite M1000 Pro, Tecan US, Morrisville, NC). Cell viability was calculated as (optical density of experimental sample/optical density of control) × 100%.
Immunoprecipitation
For whole cell extracts, cells were lysed in buffer containing 50 mM Tris-HCl (pH7.5), 150 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 1% Triton X-100, protease inhibitors (Pierce), phosphatase inhibitors (Pierce) and cleared by centrifugation. Lysates were incubated with 5 μg of anti-clusterin or anti-HSP90 antibodies, followed by incubation with protein G beads (GE healthcare). The beads were washed four times by lysis buffer and immunoprecipitates were eluted with SDS sample buffer by boiling for 5 min followed by western blot analysis.
MS-dependent identification of clusterin-interacting proteins
Inducible clusterin (Flag-tagged) GCSC line was treated with Dox (2.5 μg/ml) for 48 hr. Lysate from these cells was immunoprecipitated with beads conjugated with anti-Flag antibody. After that, proteins captured were eluted, digested with trypsin and analyzed by data-dependent LC/MS/MS with an AB Sciex hybrid quadruple TOF mass spectrometer. All MS and MS/MS data were processed with the ProteinPilot software, which assigned peptide identity with a <1% false discovery rate with a target-decoy database search approach.
Immunofluorescence staining
Living cells were washed with PBS, then fixed with 4% formaldehyde and permeabilized with PBS containing 0.5% Triton X-100. Cells were blocked, and incubated with anti-clusterin, anti-HSP90 antibodies at 4oC overnight, washed with PBS, visualized with secondary antibodies conjugated with Alexa Fluor 488, 594 (Thermo Fisher Scientific), and counterstained with DAPI for nuclear staining.
In Vivo Animal Studies
The following animal handling and procedures were approved by the Shenzhen University Animal Care and Use Committee and followed the ARRIVE guidelines.
GCSC xenograft model for testing the effect of clusterin knockdown
BALB/c nude mice (6 weeks old, 18.0 ± 2.0 g) were subcutaneously injected with the single-cell GCSC lines carrying inducible lentiviral shRNA vector targeting clusterin (right flank, shClu) or with inducible lentiviral control shRNA vector (left flank, shCtrl) (4x105 cells in 100 μl PBS, day 0). On the second day (day 1) after injection, mice were randomized into two groups (5 animals for each group). The Dox+ group was fed with Dox at the dose of 1 mg/ml in water with 0.5% glucose. The Dox- group was fed with water with 0.5% glucose as control. The water was changed every three days. Tumor size was measured using caliper, and tumor volume was determined by using the formula: L×W2×0.52, where L is the longest diameter and W is the shortest diameter. After 12 days on Dox treatment, all mice were euthanized.
GCSC xenograft model for examining the effect of VP treatment
Single-cell GCSC cells were subcutaneously injected with 3x106 cells in BALB/c nude mice (6 weeks old, 18.0 ± 2.0 g), n=9 per group. 10 days after inoculation, VP was diluted in 10% DMSO (in PBS) and administered by intraperitoneal injection (100 mg/kg/mouse) every three days for 16 days. Control group was injected with same volume of vehicle.
Statistical Analysis
Statistical analyses were performed by GraphPad Prism 6 (La Jolla, CA, USA). Results were expressed as mean ± SEM, unless indicated otherwise. Groups were compared with either a 2-tailed Student's test for analysis of 2 groups or using 2-way ANOVA to compare multiple groups. Significance was accepted when p was less than 0.05.
Data Availibility
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Clusterin is expressed during the growth of GCSC tumorsphere, and overexpression of Clu correlates with poor survival outcome in GC
Sphere-formation is one of the characteristic of cancer stem cells [12]. We analyzed the secretomics of the tumorsphere generated by GCSC using mass spectrometry (MS). We found that 15 proteins in the supernatant of GCSC tumorsphere culture (Table S1. partial data in Figure 1a). Among them, clusterin is known to enhance aggregation of a variety of cell types and has cytoprotective effect during various situations [13-19]. We confirmed clusterin secretion in the supernatant of GCSC tumorsphere using western blot (Figure 1b). Moreover, intracellular clusterin (together with Sox2 protein, a stem cell marker) in GCSC also steadily increased during the growth of GCSC sphere (Figure 1c).
Clusterin expresses in GCSC during tumorsphere growth, and overexpression of Clu correlates with poor survival outcome in GC. (a) Relative concentrations of proteins from supernatant of GCSC tumorspheres at day 0 (noted as ↓), 1, 2, 3, 4 and 5 (noted as *) (full list is shown in Table S1). The concentration of each protein at day 1 (second bar of each protein) was taken as 1. (b) The conditioned medium of GCSC tumorsphere at indicated date was concentrated, and the levels of secreted clusterin protein expression were detected by western blot and subsequently quantified by densitometry. The amount of secreted clusterin of day 1 was taken as 1. n = 3, ± standard error of mean (SEM). (c) The cell lysates from GCSC tumorsphere at indicated time were analyzed by western blot using anti-clusterin, anti-Sox2 and anti-β-actin antibodies as noted (left panel). The relative expression levels of clusterin and Sox2 proteins were quantified by densitometry and normalized against β-actin. The levels of clusterin and Sox2 at day 0 were taken as 1. n = 3, ± standard error of mean (SEM) (right panel). (d) Clusterin immunostaining images in cross sections of different stage (T1-T4) gastric tumor and normal gastric tissue (left panel). Scale bar, 50μM. Quantification of clusterin expression according to IHC scores in T1(8), T2(7), T3(59) and T4(14) (right panel). Significance was determined using the One-Way ANOVA test.
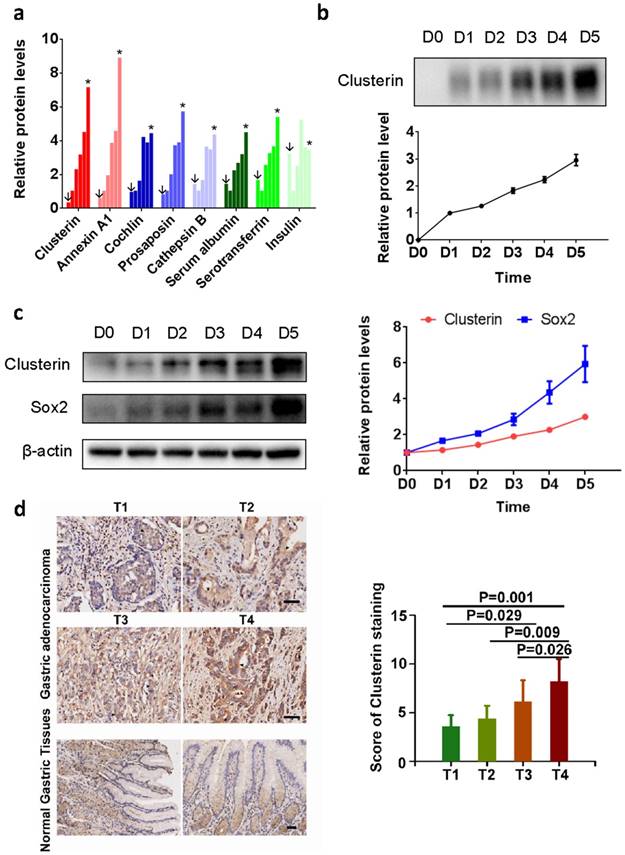
To investigate the clinical significance of clusterin in gastric cancer, clusterin expression in protein level was also studied using GC tissue microarray (TMA) containing 90 primary GC cases. The results showed that overexpression of clusterin was significantly associated with T stage (P=0.0004, Figure 1d), lymph node metastasis (P=0.026) and TNM stage (P=0.013) (Table S2). Thus, clusterin may be involved in providing GCSC and/or cancer cell with higher invasive and metastatic potential leading to cancer spread and higher mortality rate of patient.
Clusterin is critical for the survival of GCSC
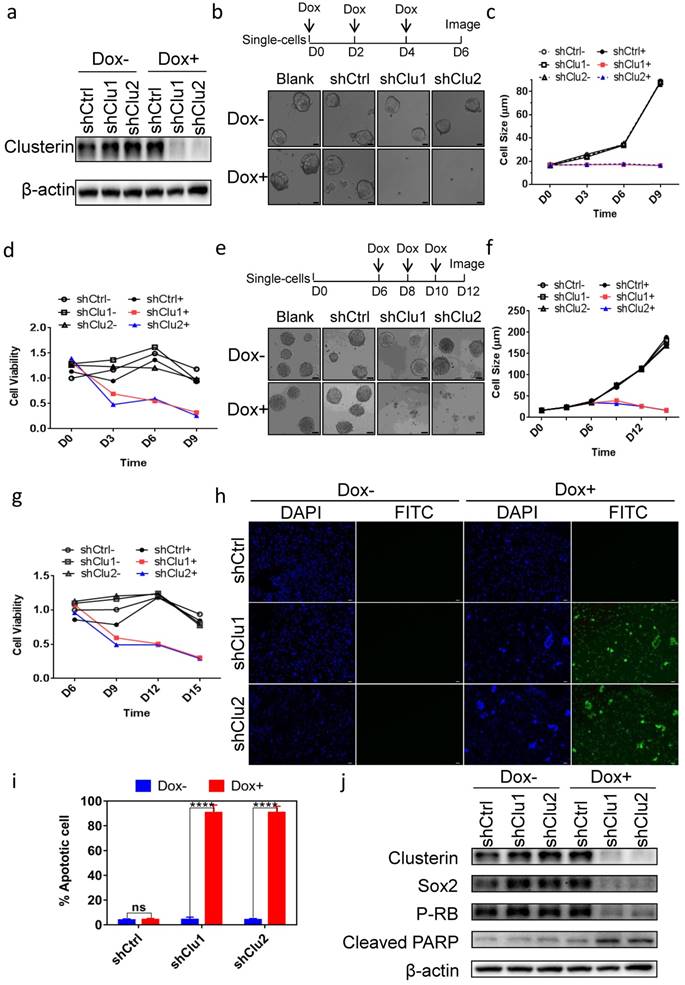
To assess the role of clusterin in GCSC, we tested several constructs encoding inducible shRNA targeting clusterin message RNA and found two shRNA sequences effectively depleted intracellular clusterin in GCSC after doxycycline (Dox) induction (Figure 2a). Clusterin reduction in GCSC inhibited tumorsphere formation (Figure 2b & 2c) as well as lowered the viability of GCSC (Figure 2d). Interestingly, when we introduced Dox to medium after GCSC sphere was formed, the tumor sphere started to “declustering” (Figure 2e), the size of sphere and the viability of GCSC both progressively decreased (Figure 2f & 2g). Further analysis showed that these clusterin knockdown GCSC were mostly positive for TUNEL assay (Figure 2h & 2i) indicating programmed cell death in these cells. We screened for alteration (after clusterin depletion) of an array of critical proteins involved in cell growth, survival and stemness by western blot (partial data shown in Figure 2j, and Figure S1). And found that stem cell-related markers such as Sox2, Nanog, Oct4A and the phosphorylation level of Rb protein (P-RB) were reduced whereas cleaved PARP was moderately increased (Figure 2j and Figure S1). These results indicate that after clusterin silencing, the decrease of Sox2, Nanog, Oct4A proteins may be involved in the reduction of GCSC stemness, the decrease of the phosphorylation level of Rb may connect to the slowdown of GCSC growth and the increase of cleaved PARP may contribute to the apoptosis of GCSC.
Clusterin expression is required for the survival of GCSC. (a) GCSC lines were established by infecting lentiviral inducible shRNA control (shCtrl) or inducible shRNA targeting clusterin (shClu1, shClu2) as described in Materials and methods. These cell lines were treated with or without Dox (2.5 μg/ml) for 48 hr as noted. The expression levels of clusterin and β-actin (loading control) were examined by western blot. (b) Single cell of GCSC, GCSC-shCtrl, -shClu1 and -shClu2 lines were incubated and treated with Dox (Blank+, shCtrl+, shClu1+, shClu2+) as indicated in scheme (top panel) or without Dox (Blank-, shCtrl-, shClu1-, shClu2-) for 6 days. Phase-contrast microscopic images of these cells/spheres were taken at D6 as indicated. Scale bar, 20 μm. (c) The cell/sphere sizes of GCSCs in (b) were measured at the indicated day. n >50, ± standard error of mean (SEM). (d) The cell viability of GCSCs in (b) was analyzed using Cell Counting Kit-8 (CCK8) assay at indicated day. The cell viability of shCtrl GCSC of day 0 was taken as 1. n = 3, ± standard error of mean (SEM). (e) Single cell of GCSC, GCSC-shCtrl, -shClu1 and -shClu2 were incubated for 6 days. After that, these cells were treated with Dox (Blank+, shCtrl+, shClu1+, shClu2+) as indicated in scheme (top panel) or without Dox (Blank-, shCtrl-, shClu1-, shClu2-) for six more days. Phase-contrast microscopic images of these cells/spheres were taken at D12. Scale bar, 50 μm. (f) The sphere sizes of GCSCs in (e) were measured at the indicated day. n >50, ± standard error of mean (SEM). (g) The cell viability of GCSCs in (e) was analyzed as in (d) at indicated day. The cell viability of GCSC-shCtrl of day 6 was taken as 1. n = 3, ± standard error of mean (SEM). (h) TUNEL analysis of the GCSCs from (e) at D12. DAPI staning (blue) of these cells were shown in column 1 and column 3. Immunofluorescent analysis of TUNEL (green) of these cells were shown in column 2 and column 4. Scale bar, 50 μm. (i) Percentage of apoptotic cells were calculated by the following formula (the number of TUNEL positive cell / the total cell number of GCSC-shCtrl, -shClu1 or -shClu2 in (h) X 100%). n = 3, ± SEM, *p < 0.0001. (j) The cell lysates of GCSC-shCtrl, -shClu1 and -shClu2 in (e) were prepared at D12. The expression levels of clusterin, Sox2, phosphorylated RB (P-RB), cleaved PARP, and β-actin in these cells were examined by western blot.
Clusterin is in complex with HSP90 and modulates cellular level of HSP90 client proteins
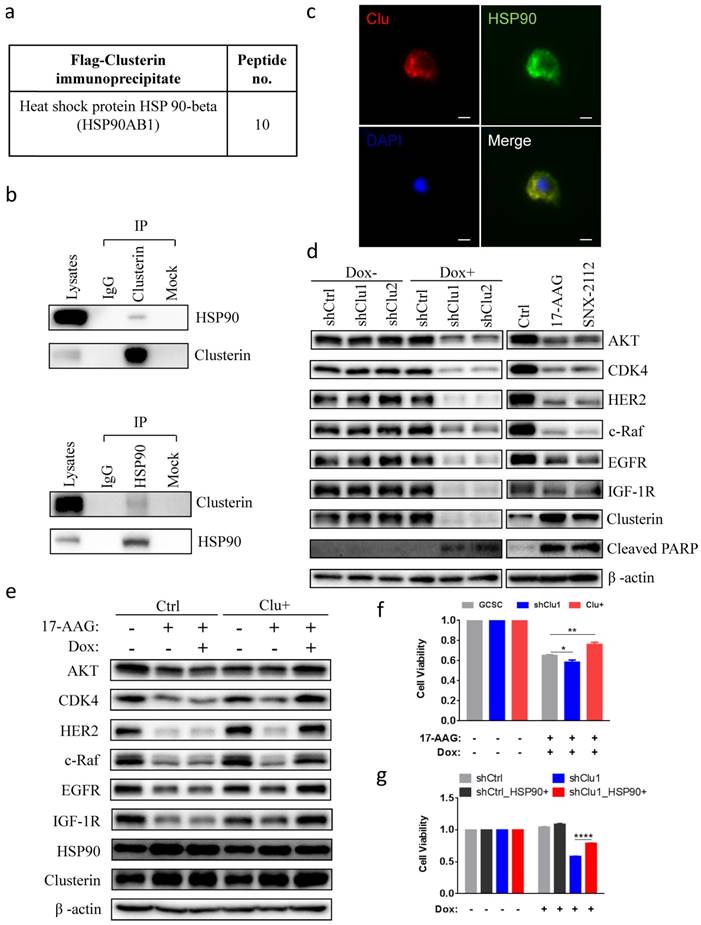
Next, we tried to elucidate the molecular mechanism of clusterin in sustaining GCSC survival. We immunoprecipitated flag-tagged clusterin in GCSC and analyzed those proteins in complex with clusterin using MS (Table S3). We discovered that heat shock protein 90 beta (HSP90), a protein which is important for cancer cell survival[20-22], was co-precipitated with clusterin (Figure 3a). Immunoprecipitation of cellular clusterin or HSP90 brought pulled down cellular HSP90 or clusterin, respectively (Figure 3b). Immuno-stainning of cells with overexpressed clusterin and HSP90 further indicated that these two proteins mainly co-localize in the cytosol (Figure 3c). Immunofluorescence imaging revealed co-localization of clusterin and HSP90 in the cell displayed as a yellow color in the merged image. Silencing clusterin in GCSC as well as treatment with HSP90 inhibitor 17-AAG and SNX2112, lowered the cellular levels of HSP90 client protein Akt, CDK4, Her2, c-Raf, EGFR, IGF1R, and increased expression of cleaved PARP (Figure 3d). Increased expression of clusterin in GCSC partially recovered the cellular level of HSP90 client proteins reduced by HSP90 inhibitor 17-AAG (Figure 3e). The reduction of GCSC cell viability by 17-AAG was worsen by silencing clusterin in GCSC (Figure 3f) or was partially rescued by increased clusterin expression in GCSC (Figure 3f). On the other hand, the viability of clusterin-depleted GCSC was significantly recovered by increased expression of HSP90 in these GCSC (Figure 3g). These results indicate that clusterin is in complex with HSP90 and is involved in modulating HSP90 client proteins.
Clusterin is in complex with HSP90 and modulates HSP90 function. (a) HSP90 in complex with clusterin was identified by MS as described in Materials and methods (a list of top clusterin-associated proteins is shown in Table S3). (b) Lysates from GCSCs were immunoprecipitated (IP) with IgG or anti-clusterin (top panel), anti-HSP90 (bottom panel), then immunoblotted with indicated antibodies. (c) Fluorescent microscopy images of GCSCs transfected with expression vectors encoding clusterin (Clu) and HSP90 (HSP90). These cells were stained with anti-clusterin (red, upper left panel) or anti-HSP90 antibody (green, upper right panel). Nuclei staining by DAPI (blue, lower left panel). Merged image (yellow, lower right panel). Scale bar, 10 μm. (d) Lysates from GCSC-shCtrl, -shClu1, and -shClu2 treated with or without Dox (2.5 μg/ml, 48 hr) (Left panel) and from GCSC treated with HSP90 inhibitors 17-AAG, SNX2112 (0.2 μM, 24 hr) (Right panel) were subjected to immunoblotting with antibodies as noted. One of three independent experiments (with similar result) was shown. (e) Inducible clusterin expressing (Clu+) and empty vector control (Ctrl) GCSC lines were treated with 17-AAG (0.2 μM) and/or Dox (2.5 μg/ml) for 24 hr. Lysates from these cell lines were subjected to immunoblotting using antibodies as indicated. One of three independent experiments (with similar result) was shown. (f) GCSC, shClu1, Clu+ were treated with 17-AAG (0.2 μM) and Dox (2.5 μg/ml) for 24 hr. The cell viability of these GCSC cells was analyzed using Cell Counting Kit-8 (CCK8) assay. The value of cell viability of untreated GCSC, shClu1 or Clu+ cells was taken as 1 when compared to those treated GCSC, shClu1, Clu+ cells, respectively. Data showed mean ± SEM generated from three technical replicates. Data were analyzed by Student's t-test. *P < 0.05, **P < 0.01. (g) shCtrl, shClu1 GCSC infected with lentiviral inducible expressing construct encoding HSP90 (HSP90+) were treated with or without Dox (2.5 μg/ml) for 48 hr. The cell viability of these GCSC cells was analyzed using Cell Counting Kit-8 (CCK8) assay. The cell viability of shCtrl, shClu1, shCtrl_HSP90+, shClu1_HSP90+ without Dox treatment was taken as 1. Data showed mean ± SEM generated from three technical replicates. Data were analyzed by Student's t-test. ****P < 0.0001.
VP blocks clusterin gene transcription in GCSC
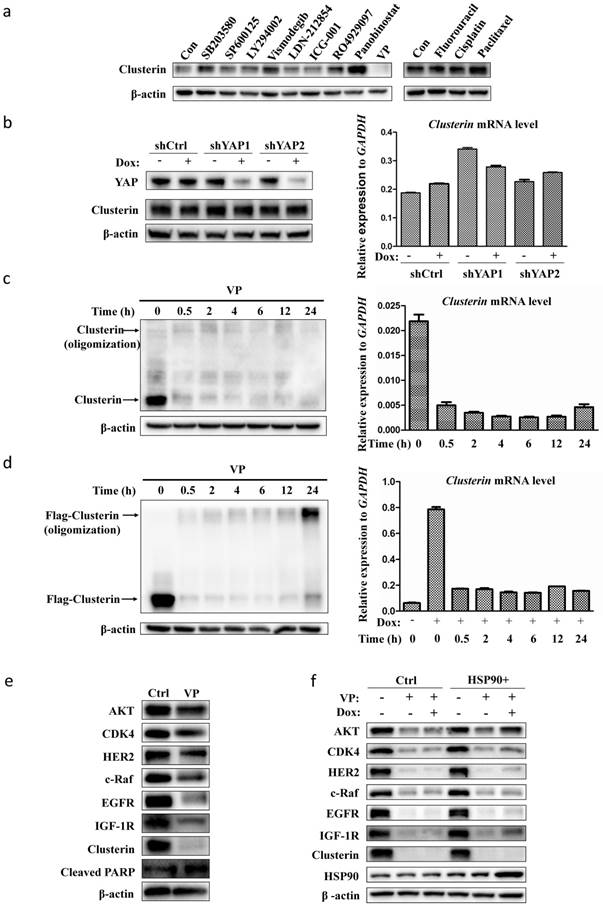
As our data indicated that clusterin is critical for GCSC survival, we scanned a collection of drugs/inhibitors for their influence on clusterin expression (Figure 4a). The inhibitors we used herein are known to block signaling pathways or to suppress molecules involved in CSC functions. SB203580 and SP600125 are inhibitors for p38 and JNK MAP kinase pathways, respectively. LY294002, Vismodegib, LDN212854, ICG-001, RO4929097 are inhibitors for PI3K, hedgehog, TGFβ, Wnt, Notch pathways, respectively. Panobinostat and VP are inhibitors for histone deacetylase and Yap transcription factor, in that order. We also tested common agents for cancer chemotherapy such as fluorouracil (5-Fu), cisplatin and paclitaxel. Most of these drugs/inhibitors had no or moderate effect on clusterin expression. Yet, VP drastically shuted down the clusterin expression while panobinostat stimulated the expression of clusterin in GCSC (Figure 4a). VP is known to interfere with protein expression/function either by blocking Yap-mediated gene transcription [23, 24] or by protein oligomerization [25]. We first induced Yap gene silencing in GCSC and found no obvious difference in both clusterin mRNA and protein levels compared to control (Figure 4b). We next checked whether VP treatment induced clusterin oligomerization (Figure 4c, left panel). There was no obvious oligomerization of clusterin within 24 hr of VP treatment, while clusterin protein was dramatically reduced after 0.5 hr VP introduction concomitant with the considerably reduction of clusterin mRNA (Figure 4c).
VP suppresses the expression of clusterin in GCSC. (a) GCSCs were treated with indicated drugs and inhibitors for 24 hr at concentrations (SB203580, 10 μM; SP600125, 5 μM; LY294002, 10 μM; Vismodegib (GDC-0449), 20μM; LDN-212854, 2 μM; ICG-001, 5 μM; R04929097, 20 μM; Panobinostat, 0.03 μM; VP, 0.5 μM; Fluorouracil, 10 μM; Cisplatin, 2 μM; Paclitaxel, 0.005 μM). Clusterin expression was detected by anti-clusterin antibody using western blotting analysis. The level of β-actin was use as a loading control. One of three independent experiments (with similar result) was shown here. (b) GCSCs were infected with lentiviral inducible shRNA control (shCtrl) or lentiviral inducible shRNAs targeting two different sites of YAP gene, as designated by shYAP1 and shYAP2. These GCSC lines were incubated with or without Dox (2.5 μg/ml) for two days as noted. The expression levels of clusterin were analyzed by western blot (left panel) and quantitative RT-PCR (right panel). GAPDH was used as an internal control for qRT-PCR. Error bars represent the standard deviation (s.d.) of experiments (n = 3). qRT-PCR experiments were performed in three technical replicates and at least two biological replicates. (c) GCSCs were treated with VP (0.5 μM) for indicated time, and the expression levels of clusterin were analyzed by western blot (left panel) and qRT-PCR (right panel). Endogenous clusterin as well as oligomerized endogenous clusterin were indicated by arrows. Error bars represent the s.d. of experiments (n = 3). qRT-PCR experiments were performed in three technical replicates and at least two biological replicates. (d) GCSC line was established by infecting lentiviral inducible expressing construct encoding flag-tagged clusterin (f-clusterin) described in Materials and methods. The expression of f-clusterin in this GCSC line was induced for 48 hr by Dox (2.5 μg/ml) and then treated with VP (0.5 μM) for indicated time. The expression levels of f-clusterin were evaluated by western blot (left panel) and qRT-PCR (right panel). Oligomerized f-clusterin and f-clusterin were indicated by arrows. Error bars represent the s.d. of experiments (n = 3). qRT-PCR experiments were performed in three technical replicates and at least two biological replicates. (e) Lysates from GCSC treated with VP (0.5 μM) for 24 hr were subjected to immunoblotting using antibodies as indicated. One of three independent experiments (with similar result) was shown here. (f) GCSC infected lentiviral inducible expressing construct encoding HSP90 (HSP90+) or empty vector control (Ctrl) were treated with VP (0.5 μM) and/or Dox (2.5 μg/ml) as indicated for 24 hr. Lysates from these cell lines were subjected to immunoblotting using indicated antibodies. One of three independent experiments (with similar result) was shown.
Additionally, we checked the effect of VP on ectopically expressed clusterin driven by an inducible minimal CMV promoter (Figure 4d). Interestingly, the expression of this type of clusterin was also blocked by VP, which was most likely due to suppression of this minimal CMV promoter within 0.5 hr treatment of VP (Figure 4d). Clusterin oligomerization was only observed in ectopically expressed clusterin after 24 hr of VP treatment (Figure 4d, left panel) and not in endogenous clusterin (Figure 4c, left panel). We analyzed the promoter region of clusterin gene (-2 kb to +1kb) as well as the minimal CMV promoter that drive the transcription of cloned clusterin cDNA in inducible system by TFScan program (http://www.bioinformatics.nl/cgi-bin/emboss/tfscan) and found no predictive binding site of YAP, TAZ, or TEAD proteins. Further, we also searched the genome-wide loci binding by these three transcription factors (YAP, TAZ, or TEAD) which were identified by ChIP-seq experiments, and did not find any binding enrichment of these transcription factors in clusterin gene promoter and related regions in these datasets[26]. These results suggest that VP has a strong inhibitory effect on common element(s) in transcriptional machinery shared by both clusterin promoter and minimal CMV promoter. As VP blocked clusterin expression and clusterin had an effect on the cellular level of HSP90 client proteins, we tested whether VP also affects those proteins regulated by HSP90. Indeed, we found that VP also reduced the levels of various HSP90 client proteins as predicted (Figure 4e). And, increased expression of HSP90 can partially recover the VP-induced reduction of HSP90 client proteins in GCSC (Figure 4f). These results implicate that VP blocks clusterin expression via inhibiting clusterin gene transcription.
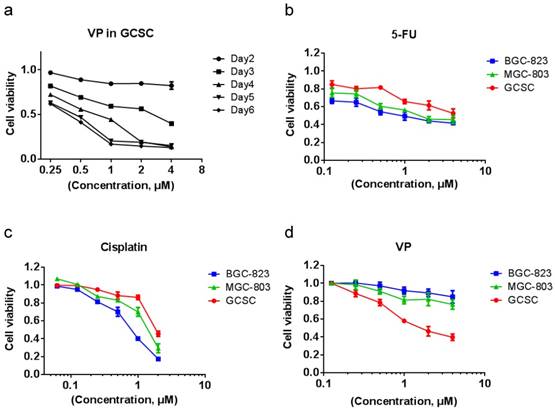
VP treatment is more effective in eradicating GCSC than in killing GC cells
We tested the effect of VP on GCSC viability and found that resembling to clusterin depletion, VP efficiently caused the death of GCSC (Figure 5a). Cancer stem cells are well-known for resistant to chemotherapy. Indeed, in comparison with MGC-803 and BGC-823 GC cells, GCSC showed more resilient to fluorouracil (5-FU) and cisplatin which are common chemotherapy drugs for GC (Figure 5b & 5c). In contrast, GCSC was more sensitive to VP treatment compared to that of GC cells (Figure 5d). The reason for GCSC was more susceptible to VP treatment may be in part because of the dependence of clusterin for GCSC survival and the capacity of VP in shutting down clusterin expression, and the consequent reduction of HSP90 client protein levels in GCSC. This discovery may provide important knowledge for designing novel approach in targeting GCSC during chemotherapy of GC.
VP treatment is more effective in eradicating GCSCs than in eliminating GC cells. (a) GCSCs were treated with VP at a serial of concentrations as indicated, and the viability of cells was measured as in Figure 2d at indicated time points. The viability of cells treated with vehicle at each day was set as 1 for calculating cell viability of GCSCs under various VP concentration of each day. Error bars represent the s.d. of experiments (n = 3). (b) GCSCs and two GC cells (MGC-803 and BGC-823) were treated with 5-Fu at a serial of concentrations as indicated for 72 hr, and the viability of cells was measured by CCK-8 assay. The viability of cells treated with vehicle at 72 hr was set as 1. Error bars represent the s.d. of experiments (n = 3). (c) GCSCs, MGC-803 and BGC-823 GC cells were treated with cisplatin at a serial of concentrations as indicated for 72 hr, and the viability of cells was measured by CCK-8 assay. The viability of cells treated with vehicle at 72 hr was set as 1. Error bars represent the s.d. of experiments (n = 3). (d) GCSCs, MGC-803 and BGC-823 GC cells were treated with VP at a serial of concentrations as indicated for 72 hr, and the viability of cells was measured by CCK-8 assay. The viability of cells treated with vehicle at 72 hr was set as 1. Error bars represent the s.d. of experiments (n = 3).
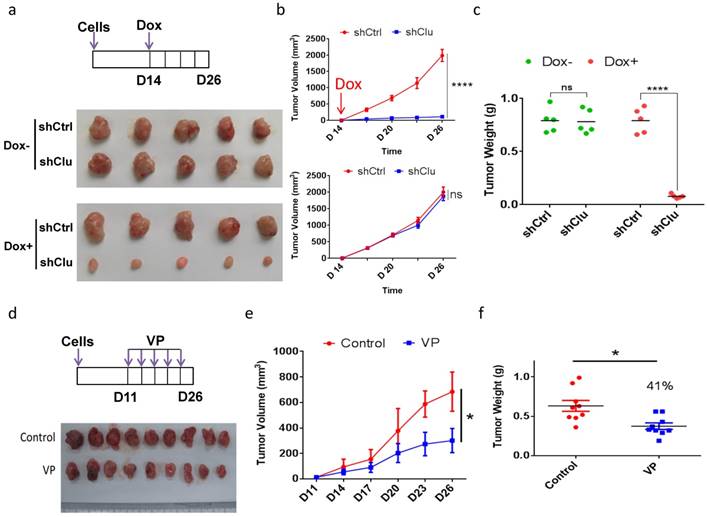
Induced clusterin depletion as well as VP treatment reduces the growth of GCSC-derived tumor xenografts in animal
As clusterin depletion and VP treatment both substantially reduced the viability of GCSC in cell culture, we next examined the effect of these two types of treatment for tumor growth in animal. We injected single-cell GCSC carrying vector encoding inducible shRNA targeting clusterin (shClu) and single-cell GCSC carrying vector encoding inducible control shRNA (shCtrl) into the right and left flank of the same mouse, respectively. Under Dox induction (Figure 6a top panel), we found that the growth of the shClu tumor was significantly slowdown compared to that of shCtrl tumors treated with Dox (Figure 6a bottom panel and Figure 6b top panel & Figure 6c). On the other hand, without Dox induction, the growth rate between shCtrl and shClu tumor had no noticeable difference (Figure 6a middle panel, Figure 6b bottom panel & Figure 6c). Next, we checked whether VP had similar effect as clusterin knockdown in suppressing GCSC-derived nude mice xenografts (Figure 6d, 6e & 6f). As predicted, the growth of GCSC tumors in VP treated mice was noticeably suppressed in comparison with that in vehicle treated mice (Figure 6e & 6f). These results suggest that both clusterin reduction and VP treatment can effectively suppress the growth of GCSC-derived tumor xenografts in animals.
Induced clusterin depletion as well as VP treatment reduces the growth of GCSC-derived tumor xenografts in animal. (a) (Top panel) Schematic representation of the induced clusterin knockdown in GCSC-derived nude mice xenograft model. The shCtrl and shClu GCSC lines were injected subcutaneously into the left and right flanks of the same nude mice, respectively. After 2 weeks, these tumor-bearing mice were fed with Dox (1 mg/ml, in 0.5% glucose solution, noted as Dox+), or without Dox (0.5% glucose solution, noted as Dox-). The water was changed every three days, and the tumor volume was measured every 2 or 3 days. On day 26, mice were sacrificed. (Bottom panel) Images of nude mice xenografts at the end of experiment from Dox- and Dox+ groups, respectively. (b) The tumor volume of Dox+ (top panel) and Dox- (bottom panel) groups of mice in (a) was measured at indicated time. n=5, ± standard error of mean (SEM), data were analyzed using Student's t-test. *p<0.0001. ns, no significance. (c) The tumor weights of Dox+ and Dox- (as indicated) groups of mice in (a) were measured. n=5, ± standard error of mean (SEM), data were analyzed using Student's t-test. *p<0.0001. ns, no significance. (d) (Top panel) Schematic representation of VP treatment to GCSC-derived nude mice xenograft model. GCSC were injected subcutaneously into the flanks of nude mice. 10 days later, these mice were intraperitoneally injected every 3 days for 16 days with VP (in PBS with 10% DMSO, noted as VP) or vehicle (PBS with 10% DMSO, noted as Control) as indicated. (Bottom panel) Images of nude mice xenografts at the end of experiment from VP and control groups, respectively. (e) The tumor volume of mice in (d) was measured at the day indicated. n=9, ± standard error of mean (SEM), Data were analyzed using Student's t-test. *p<0.01. (f) The tumor weights of mice in (d) was measured. n=9 mice, ± standard error of mean (SEM), Data were analyzed using Student's t-test. *p<0.01.
Discussion
GC ranks third in cause of worldwide cancer-related death [1]. The current therapeutic approach is surgery followed by chemotherapy and/or radiotherapy [2]. However, the five-year survival rate is very low (<20%). Most post-treatment deaths are due to the recurrence accompanied by metastasis [3]. For that reason, indentifying key molecule(s) involved in gastric cancer spreading, and thereby finding effective therapeutic approach for gastric cancer patients are crucial for reducing mortality of GC. CSC is implicated in cancer relapse, drug resistance and metastasis [4-6]. Herein, we found that not only was clusterin expressed in GCSC, but was also critical for GCSC survival. Additionally, clusterin depletion suppressed the growth of GCSC xenografted tumor in mice. Clusterin is implicated in cytoprotective effect in ischemia [27, 28] and providing antiapoptotic activity for cancer cell in chemotherapy [19, 29, 30]. From there, it seems that GCSC also makes use of the anti-apoptotic function of clusterin to sustain their survival. Based on our data in cell culture and in animal, it appears clusterin is a promising drug target in suppressing GCSC population in gastric cancer and consequently attenuating malignancy exerted by GCSC in patients. Moreover, Clusterin is overexpressed in many other human cancers such as hepatocellular carcinoma, pancreatic cancer, and squamous cell carcinoma. Therefore, it might be the general weak spot of CSC, and targeting this vulnerability will provide more effective treatment and/or long-term cure for cancer patients.
HSP90 is a chaperone that can interact with many cellular proteins in helping their folding and/stabilization [20-22]. HSP90 was found to stabilize many proteins that are important for cancer cell function and/or survival, thus HSP90 becomes an important drug target for cancer therapy [20-22]. Our data suggest that clusterin regulates HSP90 function and consequently affecting the level of HSP90 client proteins in GCSC. This information implies that clusterin-targeted cancer treatment may have similar therapeutic effect as HSP90 inhibitors in cancer patients. However, as clusterin also regulates other cellular proteins critical for cell survival such as activated Bax [19], targeting clusterin likely has additional effect on GCSC compared to that of HSP90 targeted treatment.
Importantly, we discovered a drug used in photodynamic therapy, VP, was able to effectively inhibit clusterin expression in GCSC, suppressed the viability of GCSC and hindered the growth of GCSC xenografted tumor in mice. As VP is already being approved for treating neovascular macular degeneration [31], it should be much quicker and cheaper, compared to traditional drug development, to repurpose VP for preventing recurrence and/or metastasis mediated by GCSC in GC patients. Notably, we demonstrated that VP also had an effect on HSP90 client proteins most likely via its function on suppressing clusterin expression. Moreover, VP is also a well-known inhibitor for Yap [24], a protein important for the stemness and cancerous properties of cancer cell [32], therefore VP treatment should have additional therapeutic effect on GCSC compared to that of HSP90 inhibitors which may increase its efficacy on treating cancer patients. Herein we also showed that VP was more effective in eradicating GCSC than in eliminating GC cells in contrast to other common GC chemotherapeutic agents which were less potent in killing GCSC than in exterminating GC cells. Together, it is logical to deduce that GC patients can promptly benefit from VP treatment in combination with or subsequent to conventional chemotherapy for slowing down or stopping the spreading of GC to other vital organs which is the major cause of mortality in GC.
Abbreviations
5-FU: fluorouracil
Clu: the full-length intracellular clusterin
CSC: cancer stem cell
Dox: doxycycline
GAC: gastric adenocarcinoma
GC: gastric cancer
GCSC: gastric cancer stem cell
HSP90: heat shock protein 90 beta
IHC: Immunohistochemistry
IP: immunoprecipitate
iTRAQ: isobaric tags for relative and absolute quantification
MS: mass spectrometry
P-RB: phosphorylation level of Rb protein
S-Clu: secreted clusterin
SEM: standard error of mean
TMA: tissue microarray
TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nickend labeling
VP: verteporfin
Supplementary Material
Supplementary figure and tables.
Acknowledgements
We thank Mr. Shuiming Li (Shenzhen University, Shenzhen, China) for mass spectrometry analysis. This work was supported by the Shenzhen Basic Research Program (JCYJ20150324141711574 and JCYJ20160520175200003), the Nature Science Foundation of Guangdong Province (2018A030310586).
Ethics approval and consent to participate
This study was approved by the institutional review board of Shenzhen University. Informed consents were obtained for the original human work that produced the tissue samples.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v1.0, cancer incidence and mortality worldwide: IARC CancerBase No. 11. Accessed October 11 2017. http://globocan.iarc.fr
2. House MG, Brennan MF. Neoadjuvant therapy for gastric cancer. Adv Surg. 2008;42:151-168
3. Anderson WF, Camargo MC, Fraumeni JF Jr, Correa P, Rosenberg PS, Rabkin CS. Age-specific trends in incidence of noncardia gastric cancer in US adults. JAMA. 2010;303(17):1723-1728
4. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13(10):727-738
5. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7(11):834-846
6. Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6(8):637-645
7. Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120(1):41-50
8. Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, Gordon SA, Shimada Y, Wang TC. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27(5):1006-1020
9. Chen T, Yang K, Yu J, Meng W, Yuan D, Bi F, Liu F, Liu J, Dai B, Chen X, Wang F, Zeng F, Xu H, Hu J, Mo X. Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell Res. 2012;22(1):248-258
10. Brungs D, Aghmesheh M, Vine KL, Becker TM, Carolan MG, Ranson M. Gastric cancer stem cells: evidence, potential markers, and clinical implications. J Gastroenterol. 2016;51(4):313-326
11. Detre S, Saclani Jotti G, Dowsett M. A "quickscore" method for immunohistochemical semiquantitation: validation for oestrogen receptor in breast carcinomas. J Clin Pathol. 1995;48(9):876-878
12. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755-768
13. Jones SE, Jomary C. Clusterin. Int J Biochem Cell Biol. 2002;34(5):427-431
14. Shannan B, Seifert M, Leskov K, Willis J, Boothman D, Tilgen W, Reichrath J. Challenge and promise: roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. 2006;13(1):12-19
15. Trougakos IP, Djeu JY, Gonos ES, Boothman DA. Advances and Challenges in Basic and Translational Research on Clusterin. Cancer Res. 2009;69(2):403-406
16. Zoubeidi A, Chi K, Gleave M. Targeting the Cytoprotective Chaperone, Clusterin, for Treatment of Advanced Cancer. Clin Cancer Res. 2010;16(4):1088-1093
17. Wilson MR, Easterbrook-Smith SB. Clusterin is a secreted mammalian chaperone. Trends Biochem Sci. 2000;25(3):95-98
18. Han BH, DeMattos RB, Dugan LL, Kim-Han JS, Brendza RP, Fryer JD, Kierson M, Cirrito J, Quick K, Harmony JA, Aronow BJ, Holtzman DM. Clusterin contributes to caspase-3-independent brain injury following neonatal hypoxia-ischemia. Nat Med. 2001;7(3):338-343
19. Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R, Wang CY. Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol. 2005;7(9):909-915
20. Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10(8):537-549
21. Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S. Heat Shock Proteins and Cancer. Trends Pharmacol Sci. 2017;38(3):226-256
22. Neckers L, Workman P. HSP90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64-76
23. Gibault F, Corvaisier M, Bailly F, Huet G, Melnyk P, Cotelle P. Non-Photoinduced Biological Properties of Verteporfin. Curr Med Chem. 2016;23(11):1171-1184
24. Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26(12):1300-1305
25. Zhang H, Ramakrishnan SK, Triner D, Centofanti B, Maitra D, Győrffy B, Sebolt-Leopold JS, Dame MK, Varani J, Brenner DE, Fearon ER, Omary MB, Shah YM. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal. 2015;8(397):ra98
26. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17(9):1218-1227
27. Zhou W, Guan Q, Kwan CC, Chen H, Gleave ME, Nguan CY, Du C. Loss of clusterin expression worsens renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2010;298(3):F568-F578
28. Dairi G, Guan Q, Roshan-Moniri M, Collins CC, Ong CJ, Gleave ME, Nguan CY, Du C. Transcriptome-Based Analysis of Molecular Pathways for Clusterin Functions in Kidney Cells. J Cell Physiol. 2016;231(12):2628-2638
29. Al Nakouzi N, Wang CK, Beraldi E, Jager W, Ettinger S, Fazli L, Nappi L, Bishop J, Zhang F, Chauchereau A, Loriot Y, Gleave M. Clusterin knockdown sensitizes prostate cancer cells to taxane by modulating mitosis. EMBO Mol Med. 2016;8(7):761-778
30. Miyake H, Nelson C, Rennie PS, Gleave ME. Acquisition of chemoresistant phenotype by overexpression of the antiapoptotic gene testosterone-repressed prostate message-2 in prostate cancer xenograft models. Cancer Res. 2000;60(9):2547-2554
31. Kaiser PK. Verteporfin photodynamic therapy and anti-angiogenic drugs: potential for combination therapy in exudative age-related macular degeneration. Curr Med Res Opin. 2007;23(3):477-487
32. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29(6):783-803
Author contact
![]() Corresponding authors: Jixian Xiong, PhD, School of Medicine, Shenzhen University, Xueyuan Ave 1066, Shenzhen, Guangdong 518055, China. E-mail: xiongjixianedu.cn; Telephone/Fax: +86-755-86670623. Xianming Mo, PhD, Laboratory of Stem Cell Biology, State Key Laboratory of Biotherapy/Collaborative Innovation Center of Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, China. E-mail: xmingmoedu.cn; Telephone/Fax: +86-028-85164017
Corresponding authors: Jixian Xiong, PhD, School of Medicine, Shenzhen University, Xueyuan Ave 1066, Shenzhen, Guangdong 518055, China. E-mail: xiongjixianedu.cn; Telephone/Fax: +86-755-86670623. Xianming Mo, PhD, Laboratory of Stem Cell Biology, State Key Laboratory of Biotherapy/Collaborative Innovation Center of Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, China. E-mail: xmingmoedu.cn; Telephone/Fax: +86-028-85164017