Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(2):386-394. doi:10.7150/ijbs.28356 This issue Cite
Research Paper
Synergistic anti-tumor effects of arsenic trioxide and blue LED irradiation on human osteosarcoma
Chao Feng2*, Rui Gong2*, Qiuyan Zheng2*, Gege Yan2, Mingyu He2, Hong Lei2, Xingda Li2,3, Lai Zhang2, Zihang Xu2, Shenzhen Liu2, Meixi Yu2, Tianshuai Ma2, Manqi Gao2, Djibril Bamba2, Elina Idiiatullina5, Naufal Zagidullin5, Valentin Pavlov5, Chaoqian Xu2,4, Ye Yuan 2 ![]() , Lei Yang1
, Lei Yang1 ![]()
1. Department of Orthopedics, The First Affiliated Hospital of Harbin Medical University, Harbin 150001, China.
2. Department of Pharmacy, The Second Affiliated Hospital of Harbin Medical University, Harbin 150086, China; and Department of Pharmacology, College of Pharmacy, Harbin Medical University, Harbin 150081, China.
3. Department of Pharmacy, The First Affiliated Hospital of Harbin Medical University, Harbin 150086, China.
4. Department of Pharmacology, College of Pharmacy, Mudanjiang Medical University, Mudanjiang 157011, China.
5. Central Laboratory of Scientific Research, Bashkir State Medical University, Ufa 450008, Russia.
*Equal contributors to this work
Received 2018-7-7; Accepted 2018-11-11; Published 2019-1-1
Abstract

Arsenic trioxide (ATO) has been well recognized as an anti-tumor agent for various human cancers. Recently, the blue light emitting diodes (LEDs)-based therapy has also been demonstrated to be potential therapeutic strategies for several cancers. However, the combination effects of ATO and blue LED on tumor suppression are still unclear. In this study, we determined whether combination of ATO and blue LED irradiation at 470 nm in wavelength exhibited superior anti-tumor activity in human osteosarcoma (OS). We observed that combination treatments of ATO and blue LED much more significantly decreased the percentages of proliferative cells, and increased apoptotic rate compared with any single treatments in U-2 OS cells. Furthermore, we found suppression of cell migration and invasion were much more pronounced in ATO plus blue LED treated group than single treated groups. Moreover, reactive oxygen species (ROS) assay and immunostaining of γ-H2A.X and p53 indicated that the combined treatments resulted in further markedly increases in ROS accumulation, DNA damage and p53 activity. Taken together, our study demonstrated synergistical anti-tumor effects of combined treatments of ATO and blue LED on human OS cells, which were associated with an increased ROS accumulation, DNA damaged mediated p53 activation.
Keywords: Osteosarcoma (OS), Blue LED irradiation, Arsenic trioxide (ATO), DNA damage, p53
Introduction
Osteosarcoma (OS), the most common type of primary malignant tumor of bone, mainly occurs in teenagers with the international incidence of 3.4 per million globally [1, 2]. Although current treatments for OS have made significant progress in increasing survival rate of patients [3], some side effects still hurt patients' daily function [4] which is urgent to be solved. As we all known, arsenic trioxide (ATO), an ancient poison in China, is highly effective anti-leukemic agent in acute promyelocytic leukemia (APL) which has been proven firstly by Ting-Dong Zhang et al. in 1973. In the following decades, as an available chemotherapy, ATO has been applied for a wide variety of cancers including lung cancer [5], cervical cancer [6], rhabdomyosarcoma [7], sarcoma [8], and OS as well [9]. However, the clinical applications of ATO are still limited due to its inescapable side-effects in high doses. Thus, we were attempting to lower these side-effects through combination of ATO and other treatments. In recent years, light-emitting diodes (LEDs)-based therapies especially blue LEDs, at wavelengths ranging from 400-500 nm, have exhibited their beneficial effects in treating several cancers such as melanoma [10], lymphoid cells [11], and skin tumors [12]. Notably, Niu et al., showed that combination of curcumin with blue LED light united red-light irradiation can produced much higher efficiency in suppressing cell proliferation and inducing apoptosis [13]. In the present study, we determined whether the combination treatments of ATO and blue LED would exert anti-tumor effects in a synergistic manner in human OS cells.
Materials and methods
Cell culture
U-2 OS (ATCC® HTB-96™) cells were cultured in Dulbecco`s modified Eagle medium (DMEM) (Life Technologies Corporation, California, United States) containing 4,500 mg/L glucose supplemented with 10% fetal bovine serum (FBS) (Biological Industries, Israel) and at 37°C in an atmosphere containing 5% CO2.
Cell treatments and Cell counting assay
Cells were plated in 6-cm diameter dishes. The medium was subsequently exchanged with fresh medium containing different concentrations of ATO. The cells were counted using Easy cell analysis (Count star, Shang Hai, China) after 24 hrs.
To detect the combination effects of blue LED irradiation and ATO treatment, the U-2 OS cells were irradiated by a blue LED (470 nm) at a power density of 100 mW/cm2 for 180 J/cm2 after subjected to ATO for 24 hrs. The rates of dead cells were subsequently measured by Easy cell analysis (Count star). ATO was purchased from pharmaceuticals limited company of Harbin medical university (Harbin, China).
Ethynyl-2-deoxyuridine (EdU) cell proliferation assay
The assay was described previously [14] using EdU Apollo DNA in vitro kit (Ribobio, Guangzhou, China). Briefly, cells were fixed with 4% paraformaldehyde (m/v) for 30 min, and followed by incubation of 30 μM EdU at 37°C for 90 min. After permeabilized in 0.5% Triton X-100, cells were added in Apollo staining solution using a shaker for 30 min in the dark. Finally, the cells were incubated with 20 μg/mL 4',6-diamidino-2-phenylindole (DAPI) for 20 min. The EdU index (%) was the average ratio of the EdU-positive cells over total cells in five randomly selected areas under the confocal laser scanning microscope (FV10i, Olympus, Tokyo, Japan).
Cell apoptosis assay
The percentage of apoptotic cells was determined using In Situ Cell Death Detection Kit (Roche, Basel, Switzerland) according to the manufacturer's instructions. After cell fixation and permeabilization, the cells were then introduced to Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) reaction mixture (viaL1:viaL2=1:9) for 1 hr at 37°C under a humidified atmosphere in the dark. It was followed by incubation with 20 μg/mL DAPI for 20 min. The TUNEL index (%) was the average ratio of the number of TUNEL-positive cells over total cells in randomly selected areas under fluorescence microscopy (FV10i).
Cell migration assay
Cells were cultured into 6-well culture plates at the density of 2.5×105. When the confluence of cells reached to 70%, the cells were starved in DMEM (contained with 0.4% FBS) for 12 hrs. After starvation, a wound was created by scraping the cells with a 200 μL pipette tip. After washing twice with PBS, the cells were maintained in DMED (contained with 0.4% FBS) for 72 hrs. Images were captured at 0, 24, 48 and 72 hrs after wounding with a standard light microscopy (ECLIPSE TS100, Nikon, Japan), and the wound area was measured using ImageJ.
Cell invasion assay
Invasion assays were performed using Trans-well filter chambers coated with matrigel (Corning, USA) and put the chambers in 24-well culture plates. Cell suspensions (each with 5×104 cells in 200 μL of serum-free DMEM medium) were starved for 12 hrs. DMEM medium contained with 10% FBS were added into the lower chamber. After 24 hrs incubation, the cells migrated through the membrane were stained with 0.1% crystal violet (Beyotime Biotechnology, Shanghai, China) for 15 min and then visualized using a light microscopy.
γ-H2A.X Immunostaining
Cells were fixed using 4% paraformaldehyde (m/v) at 37°C for 15 min and permeabilized using a 0.3% Triton X100 solution for 15 min. The cells were then incubated in PBS supplemented with 0.1% Tween and 2% fetal bovine serum (FBS) (Embriolife®, Vitrocell, Brazil) for 1 hr. Cells were then detected using mouse anti γ-H2A.X phospho S139 (200× diluted, ab26350, Abcam, Cambridge, UK) and p53 (1000×, Cell Signaling Technology (CST), Danvers, Massachusetts, United State, cat. no. 2524). A secondary incubation step was performed using Alexa Fluor® 594 goat anti-mouse (200× diluted, ab150116, Abcam) and Alexa Flour® 488 Conjugate anti-rabbit (200× diluted, 5429, Cell Signaling). After a PBS wash, the cells were progressively dehydrated with alcohol, and then, nuclear staining was performed with DAPI (2.5 μg/mL). Fluorescence signals were visualized under a confocal laser scanning microscope (FV10i, Olympus Optical).
Measurement of reactive oxygen species (ROS) production
The intracellular ROS were measured using a Reactive Oxygen Species Assay Kit (S0033, Beyotime Biotechnology) according to the manufacturer's instructions. After washing three times with PBS, cells were incubated with 10 μM DCFH-DA probes for 30 min at 37°C. Following the removal of the superfluous DCFH-DA probes, the cells were fixed in 4% paraformaldehyde (m/v) for 30 min and stained with DAPI (20 μg/mL) for 20 min. The cells were imaged with a fluorescence microscope (Olympus Optical). ROS scavenger N-acetyl cysteine (NAC) (5mM) was purchased from Aladdin (A105420, Shanghai, China).
Western blot
The Western blot analysis was described previously [15]. Briefly, cells were lysed in RIPA buffer (Beyotime Biotechnology) on ice for 1 hr. Protein fractions were collected by centrifugation at 13,500 rpm for 15 min, and then supernatants were heated with SDS buffer at 95°C for 7 min. Proteins were subjected to SDS-PAGE and transferred onto nitrocellulose membranes. After blocked with 5% fat-free milk and incubated with specific antibodies overnight, the membranes subjected to secondary antibodies at room temperature for 1 hr. Odyssey v1.2 software (LI-COR Biosciences, Lincoln, NE, United States) was used to visualize the proteins. The following primary antibodies and concentrations were used: anti γ-H2A.X phospho S139 (1:500 diluted, ab26350, Abcam) and p53 (1:1000, CST) and β-actin (1:1000, cat. no. 8457S, CST). All antibodies were diluted in Phosphate buffered saline (PBS).
Statistical Analysis
All assays were repeated at least three times, and values were given as mean ± SEM. Statistical analysis was performed by one-way analysis of variance (ANOVA) (GraphPad Software Inc., San Diego, CA). P values < 0.05 were considered statistically significant.
Results
Blue LED irradiation enhanced ATO induced OS cell growth suppressive effect
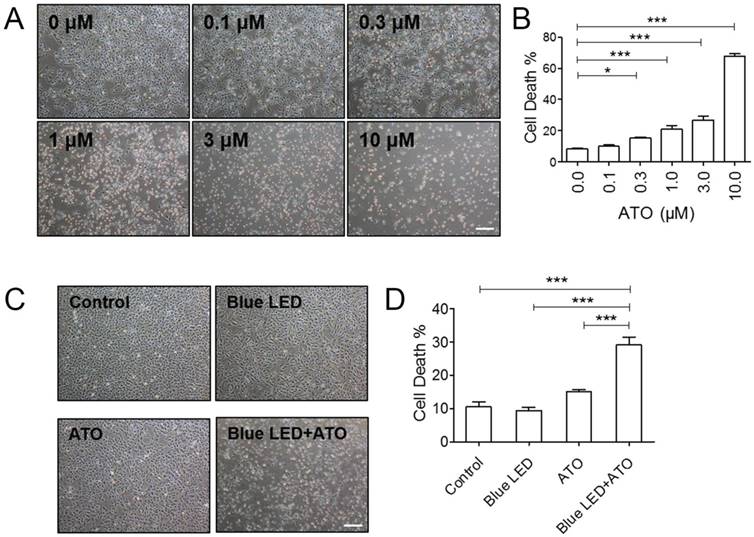
To detect the effects of ATO on OS cell growth, we firstly treated U-2 OS cells with 0 μM, 0.1 μM, 0.3 μM, 1.0 μM, 3.0 μM and 10.0 μM of ATO. As shown in Figure 1A and 1B, trypan blue staining revealed that ATO induced a dose-dependent increase in dead cells as compare with untreated control group. We selected relative low concentration of 0.3 μM ATO for our follow-up experiments. Next, to confirm whether blue LED irradiation combined with ATO treatment exerted synergistic anti-tumor effect on OS cell growth, we irradiated U-2 OS cells with blue LEDs for 180 J/cm2, and then treated cells with 0.3 μM ATO. We found that blue LED irradiation plus ATO treatments were more effective than single treatments of ATO or LED in inhibiting OS cell growth (Figure 1C and 1D).
Blue LED irradiation enhanced ATO induced cell growth suppressive effect in OS cells. (A) U-2 OS cells were treated with ATO at the concentration of 0 μM, 0.1 μM, 0.3 μM, 1.0 μM, 3.0 μM or 10.0 μM for 24 hrs respectively. Representative images were taken under a microscope (magnification ×4) after ATO treatment. (B) The histogram shows the changes of the cell mortality rates. N=3. (C) U-2 OS cells were irradiated with blue LED at the density of 100 mW/cm2 for 180 J/cm2 in the presence or absence of ATO at the concentration of 0.3 μM or treated with ATO alone. Representative images were taken under a microscope (magnification ×4). (D) The histogram shows the change of the cell mortality rates. N=5. Bar: 100 μm. Data are expressed as the mean ± SEM. ***P < 0.001.
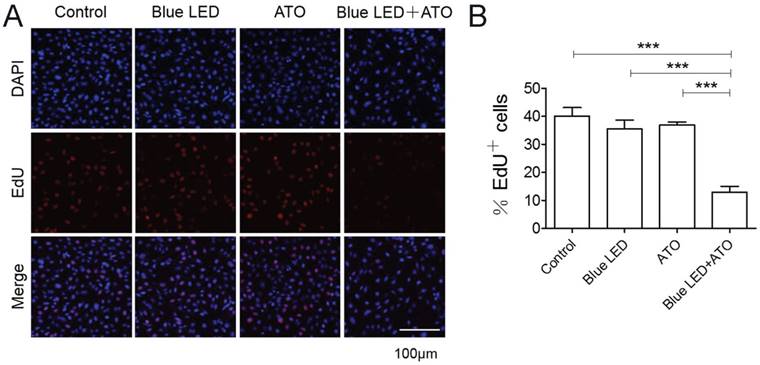
Furthermore, we measured OS cell proliferation after blue LED irradiation and/or ATO treatments. As shown in Figure 2A and 2B, the single treatments of blue LED irradiation or ATO had no effects on U-2 OS cell proliferation. However, the combination treatments caused a dramatical reduction in proliferating cells compared with single-treated or non-treated control groups. Overall, these results suggested that combination treatments of blue LED irradiation and ATO synergistically suppressed cell growth and proliferation in OS.
Combination treatments of Blue LED and ATO inhibit OS cell proliferation. U-2 OS cell proliferation was detected 24 hrs after treatment with blue-LED and/or ATO by EdU staining. (A) Representative images of EdU staining were shown in blue (DAPI) and red (EdU). (B) The histogram shows percentages of proliferative cells (EdU positive). Bar: 100 μm. Data are expressed as the mean ± SEM. N=6. ***P < 0.001.
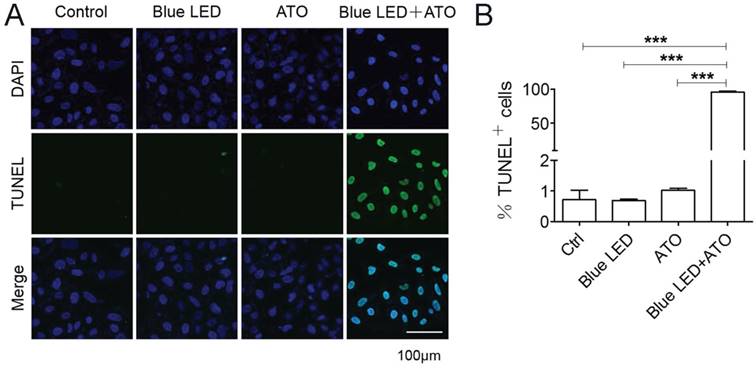
Synergistic effect of blue LED irradiation and ATO on promoting OS apoptosis
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was further performed to elucidate the effects of blue LED irradiation plus ATO treatment on OS cell apoptosis. No obvious difference was observed after single treatments of blue LED irradiation or ATO in U-2 OS cells. Surprisingly, we observed that the percentages of apoptotic cells were markedly increased about to 95% following combination treatments with blue LED irradiation and ATO in U-2 OS cells (Figure 3A and 3B).
Blue LED irradiations combined with ATO accelerate OS cell apoptosis. U-2 OS cell apoptosis was detected 24 hrs the treatment of blue LED and/or ATO by TUNEL staining. (A) Representative images of TUNEL staining were shown in blue (DAPI) and green (TUNEL). (B) The histogram shows the percentages of the apoptotic cells (TUNEL positive). Bar: 100 μm. Data are expressed as the mean ± SEM. N=4. ***P < 0.001.
Synergistic effect of blue LED irradiation and ATO on inhibition of OS migration and invasion
To evaluate the combination effects of blue LED irradiation and ATO on cell invasion and migration, Wound healing and Trans-well assays were then employed in U-2 OS cells. As shown in Figure 4A and 4B, combination treatments of blue LED and ATO significantly suppressed the migration of U-2 OS cells compared with single-treated or untreated groups.
Blue LED irradiations plus ATO treatment inhibit OS cell migration and invasion. U-2 OS cells were irradiated with blue-LED for 180 J/cm2 in the presence or absence of ATO at the concentration of 0.3μM or treated with ATO alone. (A) Representative images of Wound-healing assay at 0, 24, 48 72 hrs after treatments. (B) The relative mean distance of migrated cells. N=5 (C) Representative images of Trans-well assay. (D) The histogram shows the absorbance value of crystal violet at 570 nm reflecting the cell invasion capacity. Bar: 100 μm. N=6. Data are expressed as the mean ± SEM. ***P < 0.001.
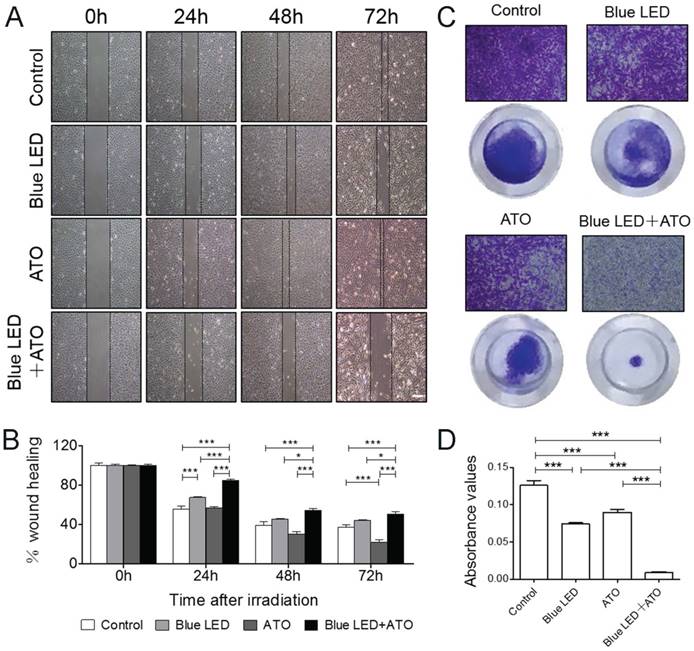
Additionally, the cell invasion was inhibited by the single treatments of blue LED irradiation or ATO compared with non-treated control group. Furthermore, this suppressive effect was further dramatically increased after the blue LED plus ATO combination treatments in U-2 OS cells (Figure 4C and 4D). Taken together, these findings demonstrated that the synergistic inhibition effects of blue LED irradiation and ATO on human OS cell migration and invasion.
Blue LED irradiation in combination with ATO induced ROS accumulation in OS cells
Reactive oxygen species (ROS) has been usually reported to facilitate tumorigenesis in the initial steps of malignant transformation, but in some contexts ROS are responsible for triggering cell death in tumors [16]. We then performed DCF-DA staining to evaluate combination effects of ATO and blue LED irradiation on ROS production in U-2 OS cells. As shown in Figure 5A, we found that there was no significant difference between single treatments of blue LED irradiation and ATO. However, an obvious increase in ROS production was induced in U-2 OS cells following combination treatments. This effect was significantly abrogated by ROS scavenger N-acetyl cysteine (NAC).
Combination treatments of Blue LED and ATO stimulate ROS accumulation and induce DNA damage in OS cells. (A) ROS production were measured after treatments of U-2 OS cells with blue LED for 180 J/cm2 in the presence or absence of ATO at the concentration of 0.3μM, or ATO alone. Representative images were shown in blue (DAPI) and green (ROS). Bar: 100 μm. (B) γ-H2A.X immunostaining followed by confocal microscopy was performed to determine the status of DNA damage after combination treatments in U-2 OS cells. Representative images were shown in blue (DAPI) and red (γ-H2A.X). (C) The histogram shows percentages of γ-H2A.X positive cells. Bar: 100 μm. Data are expressed as the mean ± SEM. N=5. ***P < 0.001. (D) Protein levels of γ-H2A.X were determined by Western blot. They were normalized to GAPDH.
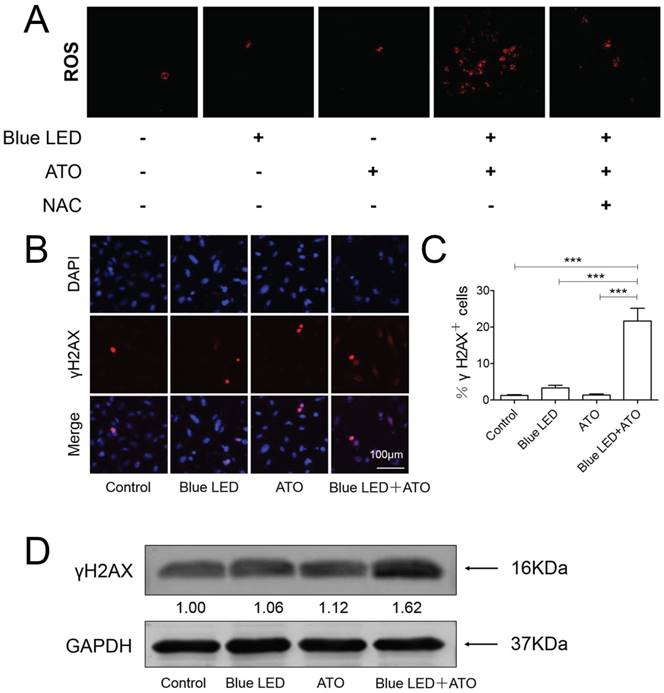
Blue LED plus ATO treatments induced DNA damage and p53 activation in OS
As previously reported, ATO treatment has been proven to promote apoptotic cell death caused by accumulated DNA damage in OS [9]. To determine whether DNA damage was the mechanism underlying the synergistic tumor suppressive effects of blue LED irradiation and ATO, we performed γ-H2A.X immunostaining in U-2 OS cells. As compared to non-treated control groups, no obvious change was observed in single treatments of blue LED irradiation or ATO. However, the percentages of γ-H2A.X positive cells were significantly increased in the combination treatment group of blue LED irradiation and ATO (Figure 5B and 5C). Additionally, blue LED plus ATO treatments resulted in increased γ-H2A.X protein expression levels as revealed by Western blot analysis (Figure 5D).
It has been reported that DNA damage response pathways might mediate tumor suppression by activating tumor suppressor p53 [17]. We then evaluated whether combination treatments of blue LED and ATO influences the expression levels and subcellular localization of p53 in U-2 OS cells. As shown in Figure 6A and 6B, combination treatments of blue LED and ATO led to an increase in p53 protein levels in comparison with non-treated or single treated groups in U-2 OS cells.
Elevated levels of p53 was induced by combination treatments with blue LED and ATO in OS cells. (A) p53 immunostaining followed by confocal microscopy was performed to assess the grade malignancy of OS after combination treatments with blue LED in the presence or absence of ATO, or with ATO alone in U-2 OS cells. Representative images were shown in blue (DAPI) and green (p53). (B) Protein levels of p53 were determined by Western blot. They were normalized to GAPDH.
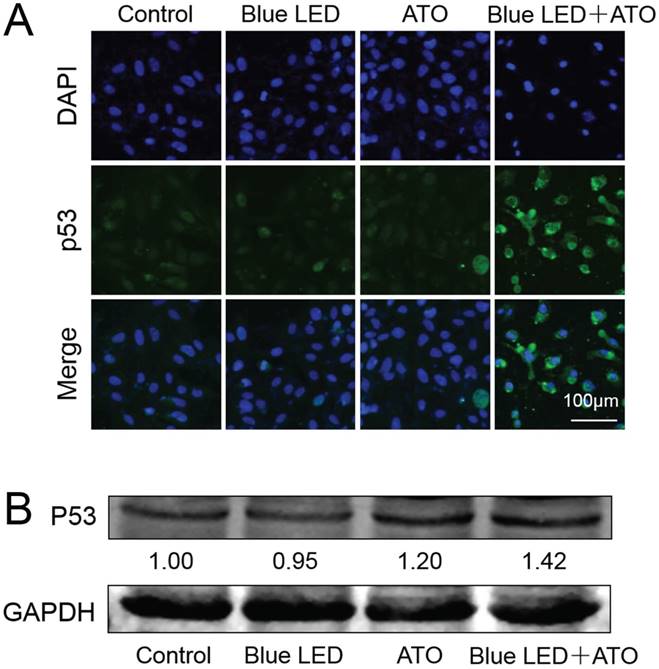
Collectively, these results suggested that the synergistic effects of blue LED irradiation and ATO treatment on cell growth, proliferation, apoptosis, migration and invasion are mainly via increased ROS accumulation and induction of DNA damage mediated p53 activation in human OS.
Discussion
In this study, we demonstrated that combination treatments of blue LED and ATO inhibited cell proliferation, migration and invasion, as well as induced apoptosis in human OS cells. The underlying mechanism involves an increase of ROS accumulation, DNA damage and p53 activation.
It has been known that blue LED irradiation or ATO treatment alone resulted in cell death in multiple cell types by inducing DNA damage [9, 18-20]. The therapeutic application of ATO was limited by adverse side effects at a high dosage. In recent years, several studies have shown that combination therapy of ATO and other treatments caused cell death in multiple types of cancer cells. For example, survivinT34A and melatonin promotes ATO-induced apoptosis in Hepal-6 hepatocellular carcinoma [21] and MCF-7 breast cancer cells [22] respectively. Synergistic combination effects of ATO and fibroblast growth factor receptor inhibitor has been shown in squamous cell lung carcinoma [23]. And combination with lithium chloride (LiCl) showed reduction in cell viability reduction colony formation, and cell death in rhabdomyosarcomas (RMS) [24]. ATO plus PX-478 (hypoxia-inducible factor-1 inhibitor) achieves effective treatment in pancreatic ductal adenocarcinoma [25]. In addition, combination treatments of different LED colors have also been proven to exert synergistic effects in some cases [13]. In the present study, we found that synergistic effects of blue LED irradiation and ATO treatment in human OS cells. Therefore, based on our findings, this combination therapy is more effective and could provide new treatment option for OS patients. Besides, we revealed that the mechanism was an increase of ROS accumulation, DNA damage and p53 activation. Consistently, ATO has been shown to induce apoptosis in p53-dependent manner in cancer cells [26, 27]. P53 can be activated by DNA damage pathway which leads to tumor suppression [17, 28].
The limitations of this study involve: (1) We did not compare combined treatments with different concentrations of ATO and irradiation energy of blue LEDs. (2) We did not confirm whether these synergistic anti-cancer effects are also applicable to tumor growth in animal models. (3) If it works well in animal models, it will be needed to test effectiveness and safeness of combination treatments in preclinical studies.
In conclusion, we demonstrated the synergistic anti-tumor effects of ATO and blue LED irradiation was attributed to induction of ROS accumulation, DNA damage and p53 activation on human OS cells. It will provide a novel potential therapeutic strategy for OS patients in the future.
Acknowledgements
This work was supported by grants from National Key Research and Development Program of China (2017YFB0403802), the National Natural Science Fund of China (81603146), and China Postdoctoral Science Foundation (2018M630365).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. International journal of cancer. 2009;125:229-34
2. Kager L, Zoubek A, Dominkus M, Lang S, Bodmer N, Jundt G. et al. Osteosarcoma in very young children: experience of the Cooperative Osteosarcoma Study Group. Cancer. 2010;116:5316-24
3. Hong AM, Millington S, Ahern V, McCowage G, Boyle R, Tattersall M. et al. Limb preservation surgery with extracorporeal irradiation in the management of malignant bone tumor: the oncological outcomes of 101 patients. Ann Oncol. 2013;24:2676-80
4. Han G, Bi WZ, Xu M, Jia JP, Wang Y. Amputation Versus Limb-Salvage Surgery in Patients with Osteosarcoma: A Meta-analysis. World J Surg. 2016;40:2016-27
5. Chang KJ, Yang MH, Zheng JC, Li B, Nie W. Arsenic trioxide inhibits cancer stem-like cells via down-regulation of Gli1 in lung cancer. American journal of translational research. 2016;8:1133-43
6. Chang YW, Chen MW, Chiu CF, Hong CC, Cheng CC, Hsiao M. et al. Arsenic trioxide inhibits CXCR4-mediated metastasis by interfering miR-520h/PP2A/NF-kappaB signaling in cervical cancer. Ann Surg Oncol. 2014;21(Suppl 4):S687-95
7. Meister MT, Boedicker C, Graab U, Hugle M, Hahn H, Klingebiel T. et al. Arsenic trioxide induces Noxa-dependent apoptosis in rhabdomyosarcoma cells and synergizes with antimicrotubule drugs. Cancer Lett. 2016;381:287-95
8. Chiu HW, Tseng YC, Hsu YH, Lin YF, Foo NP, Guo HR. et al. Arsenic trioxide induces programmed cell death through stimulation of ER stress and inhibition of the ubiquitin-proteasome system in human sarcoma cells. Cancer Lett. 2015;356:762-72
9. Nakamura S, Nagano S, Nagao H, Ishidou Y, Yokouchi M, Abematsu M. et al. Arsenic trioxide prevents osteosarcoma growth by inhibition of GLI transcription via DNA damage accumulation. PloS one. 2013;8:e69466
10. Oh PS, Na KS, Hwang H, Jeong HS, Lim S, Sohn MH. et al. Effect of blue light emitting diodes on melanoma cells: involvement of apoptotic signaling. Journal of photochemistry and photobiology B, Biology. 2015;142:197-203
11. Oh PS, Hwang H, Jeong HS, Kwon J, Kim HS, Kim M. et al. Blue light emitting diode induces apoptosis in lymphoid cells by stimulating autophagy. The international journal of biochemistry & cell biology. 2016;70:13-22
12. Ohara M, Kawashima Y, Kitajima S, Mitsuoka C, Watanabe H. Blue light inhibits the growth of skin tumors in the v-Ha-ras transgenic mouse. Cancer Sci. 2003;94:205-9
13. Niu T, Tian Y, Cai Q, Ren Q, Wei L. Red Light Combined with Blue Light Irradiation Regulates Proliferation and Apoptosis in Skin Keratinocytes in Combination with Low Concentrations of Curcumin. PloS one. 2015;10:e0138754
14. Cai B, Ma W, Bi C, Yang F, Zhang L, Han Z. et al. Long noncoding RNA H19 mediates melatonin inhibition of premature senescence of c-kit(+) cardiac progenitor cells by promoting miR-675. J Pineal Res. 2016;61:82-95
15. Yang F, Yang L, Li Y, Yan G, Feng C, Liu T. et al. Melatonin protects bone marrow mesenchymal stem cells against iron overload-induced aberrant differentiation and senescence. J Pineal Res. 2017:63
16. Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther. 2008;7:1875-84
17. Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9:714-23
18. Yuan Y, Yan G, Gong R, Zhang L, Liu T, Feng C. et al. Effects of Blue Light Emitting Diode Irradiation On the Proliferation, Apoptosis and Differentiation of Bone Marrow-Derived Mesenchymal Stem Cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;43:237-46
19. Kumar S, Yedjou CG, Tchounwou PB. Arsenic trioxide induces oxidative stress, DNA damage, and mitochondrial pathway of apoptosis in human leukemia (HL-60) cells. J Exp Clin Cancer Res. 2014;33:42
20. Godley BF, Shamsi FA, Liang FQ, Jarrett SG, Davies S, Boulton M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. The Journal of biological chemistry. 2005;280:21061-6
21. Huang A, Yue D, Liao D, Cheng L, Ma J, Wei Y. et al. SurvivinT34A increases the therapeutic efficacy of arsenic trioxide in mouse hepatocellular carcinoma models. Oncology reports. 2016;36:3283-90
22. Nooshinfar E, Bashash D, Safaroghli-Azar A, Bayati S, Rezaei-Tavirani M, Ghaffari SH. et al. Melatonin promotes ATO-induced apoptosis in MCF-7 cells: Proposing novel therapeutic potential for breast cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2016;83:456-65
23. Lam SK, Leung LL, Li YY, Zheng CY, Ho JC. Combination effects of arsenic trioxide and fibroblast growth factor receptor inhibitor in squamous cell lung carcinoma. Lung Cancer. 2016;101:111-9
24. Schleicher SB, Zaborski JJ, Riester R, Zenkner N, Handgretinger R, Kluba T. et al. Combined application of arsenic trioxide and lithium chloride augments viability reduction and apoptosis induction in human rhabdomyosarcoma cell lines. PloS one. 2017;12:e0178857
25. Lang M, Wang X, Wang H, Dong J, Lan C, Hao J. et al. Arsenic trioxide plus PX-478 achieves effective treatment in pancreatic ductal adenocarcinoma. Cancer Lett. 2016;378:87-96
26. Jiang XH, Wong BC, Yuen ST, Jiang SH, Cho CH, Lai KC. et al. Arsenic trioxide induces apoptosis in human gastric cancer cells through up-regulation of p53 and activation of caspase-3. International journal of cancer. 2001;91:173-9
27. Kircelli F, Akay C, Gazitt Y. Arsenic trioxide induces p53-dependent apoptotic signals in myeloma cells with SiRNA-silenced p53: MAP kinase pathway is preferentially activated in cells expressing inactivated p53. International journal of oncology. 2007;30:993-1001
28. Mellert H, Espinosa JM. Tumor suppression by p53: is apoptosis important or not? Cell Rep. 2013;3:1335-6
Author contact
![]() Corresponding authors: Prof. Lei Yang, Department of Orthopedics, The First Affiliated Hospital of Harbin Medical University, Harbin 150001, China, or Ye Yuan, Department of Pharmacy, The Second Affiliated Hospital of Harbin Medical University, Harbin 150086, China. E-mail: yangray83qq.com; yuanye_hmucom
Corresponding authors: Prof. Lei Yang, Department of Orthopedics, The First Affiliated Hospital of Harbin Medical University, Harbin 150001, China, or Ye Yuan, Department of Pharmacy, The Second Affiliated Hospital of Harbin Medical University, Harbin 150086, China. E-mail: yangray83qq.com; yuanye_hmucom