Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(2):416-429. doi:10.7150/ijbs.26884 This issue Cite
Research Paper
Small RNA sequencing reveals dynamic microRNA expression of important nutrient metabolism during development of Camellia oleifera fruit
Xiao-Xia Liu, Xiao-Fang Luo, Ke-Xin Luo, Ya-Lin Liu, Ting Pan, Zhi-Zhang Li, Gregory J. Duns, Fu-Lin He ![]() , Zuo-Dong Qin
, Zuo-Dong Qin ![]()
Key Laboratory of Comprehensive Utilization of Advantage Plants Resources in Hunan South, Hunan Provincial Engineering Research Center for Ginkgo biloba, College of Chemistry and Bioengineering, Hunan University of Science and Engineering, Yongzhou 425100, China
Received 2018-4-25; Accepted 2018-11-10; Published 2019-1-1
Abstract

To obtain insight into the function of miRNAs in the synthesis and storage of important nutrients during the development of Camellia oleifera fruit, Illumina sequencing of flower and fruit small-RNA was conducted. The results revealed that 797 miRNAs were significantly differentially expressed between flower and fruit samples of Camellia oleifera. Through integrated GO and KEGG function annotations, it was determined that the miRNA target genes were mainly involved in metabolic pathways, plant hormone signal transduction, fruit development, mitosis and regulation of biosynthetic processes. Carbohydrate accumulation genes were differentially regulated by miR156, miR390 and miR395 in the fruit growth and development process. MiR477 is the key miRNA functioning in regulation of genes and involved in fatty acid synthesis. Additionally, miR156 also has the function of regulating glycolysis and nutrient transformation genes.
Keywords: Camellia oleifera, deeping sequencing, microRNA, development of fruit, nutrient metabolism regulation
Introduction
Camellia oleifera is a small evergreen arbor of the Camellia genus, Theaceae, which originated in China [1]. As an important woody oil plant, edible oil can be obtained (camellia oil) from Camellia oleifera seeds. Approximately 95% of the global production of camellia oil comes from China, about half of which comes from Hunan Province [2, 3]. Camellia oil has medicinal and food value [4, 5], and contains a high content of unsaturated fatty acids (including oleic acid, linoleic acid and palmitic acid) and the lowest level of saturated fatty acids. Its fatty acid composition is similar to that of olive oil, thus it is often referred to as “Oriental olive oil” [6, 7]. Xianglin No.210 is an excellent Camellia clonal plant widely planted in Hunan Province. It has the characteristics of drought resistance, barren resistance, and high resistance to various pest diseases and high yield performance, as the oil rate of its dry seeds can reach over 35% [1]. The flowering period is moderate in length, usually from October to late December. The fruit enlargement period is mainly from July to September, but the fully matured fruiting stage will be extended to October. The fat content accumulation generally starts from July until the fruit fully matures [8].
MicroRNAs (miRNAs) are a class of small (approximately 22 nt), non-coding single strand molecules that negatively regulate gene expressions at both the transcriptional and posttranscriptional levels [9-13]. Recent studies have shown that they play a key role in diverse biological processes such as organ development [14-17], cell proliferation [15], tumorigenesis [18-20], fat metabolism [21, 22] and embryogenesis [14, 23]. In plants, functional mature miRNAs are incorporated into Argonaute 1 (AGO1)-containing RNA-induced silencing complex (RISC) target endogenous transcripts for degradation or translational repression in a sequence-specific manner [24, 25]. The specific position of the target AGO1 protein is usually between the 10th and 11th nucleotides of the mRNA, which is complementary miRNA [25, 26]. Studies of Camellia oleifera have shown that some of the genes involved in lipid metabolism [3], drought resistance [5] and cold acclimation [27] and miRNAs involved in lipid metabolism [2] were defined. Omega-6 fatty acid desaturase 2 (FAD2) genes can encode microsomal oleate desaturase, which is regarded as a key factor determining the quality of the edible oil [3]. In drought stress conditions, 21 unigenes associated with superoxide dismutase (SOD), 52 unigenes related with catalase (CAT) and 100 unigenes associated with peroxidase (POD) were up or down regulated [5]. In cold acclimation, many of the genes encoding transmembrane sugar transporters (such as comp214280_c0, comp215715_c0, and comp220377_c0) were differentially expressed in Camellia oleifera leaves [27]. In natural dried Camellia oleifera and C. meiocarpa seeds, miR5067 was identified as encoding 3-ketoacyl-CoA synthase III in fatty acid synthesis, while miR2118 and miR482 regulated long-chain-alcohol oxidase FAO2 and miR1861 is involved in lysophospholipase for fatty acid catabolism [2].
In recent years, molecular methods have been used to study the growth and development, metabolism regulation and resistance of Camellia oleifera. However, the basic molecular research on Camellia oleifera is weak and poorly understood, especially for the molecular mechanisms which determine the yield and quality of camellia oil and growth traits of Camellia oleifera, which certainly hinders the sustainable development of the camellia oil industry. In order to explore the potential role of miRNAs in the synthesis and storage of important nutrients during the development of Camellia oleifera fruit, miRNA expression profiles of flower and fruit samples were investigated using high throughput next generation small RNA sequencing technology. This permits the unraveling of the differentially expressed miRNAs to help understand their involvement in the regulation of the synthesis and storage of important nutrients in fruit.
Materials and Methods
Plant Material Collection
The samples of Camellia oleifera flowers and fruits were collected in October and July in 2016, respectively. They were grown over six years, in a garden located in Yongzhou city, Hunan province, China. The young flowers and fruits were collected and then frozen in liquid nitrogen, to be used for subsequent total RNA extraction, with three replicates per group.
Scanning Electron Microscope Analysis
A Hitachi SU8010 cold field emission scanning electron microscope (SEM) was used for the internal ultrastructural analysis of the Camellia oleifera seed samples. The samples were ensured to be clean and dry prior to analysis. Secondary electron resolution was 1.3 nm.
Camellia Oil Extraction
The Soxhlet method was used to extract camellia oil [28]. The dry Camellia oleifera seeds were milled into a powder by a pulverizer, and then about 2 g of powder (m0) were weighed out, packed in a folded filter paper tube, sealed with absorbent cotton, bound and compacted. The sample was then sealed with cotton thread, put into a Soxhlet extraction thimble, and 180mL of petroleum ether added. The extraction took place at 88°C for 6 h, using vacuum distillation to remove the solvent. The residual Camellia oil was thus obtained and weighed (m1) to determine the oil content by the following:
Seed oil content=
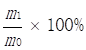
Fatty Acid Composition Analysis of Camellia Oil
In order to determine the fatty acid composition of the camellia oil, the fatty acids were first converted to methyl esters [28]. For this procedure, 200mg of camellia oil in a tube were dissolved by 20 ml sodium hydroxide-methanol solution (0.5 mol/L), shaken, and immersed in a 60°C water bath for 30 min. After this, 50ml of n-hexane were added, mixed well, and left to stand for a while. After the solution stratified, the upper layer of solution was removed and stored at 4°C until needed for further analysis.
The fatty acid composition analysis of camellia oil was determined by GC-MS (gas chromatography-mass spectrometry) using the above methylated samples. An HP-5 capillary GC column (30m×0.25mm×0.25µm) was used in a GC equipped with an MS detector (Shimadzu, QP2010S), with an inlet temperature of 250°C. The injection volume was set at 1 µL, and the split ratio was 20:1. High purity helium was used as the carrier gas and the flow rate was set at 1.0 mL·min-1, the oven temperature of the instrument was raised from 180°C (held for 5 min) to 230°C (held 15min) at 3°C·min-1. For GC-MS detection, an electron ionization energy system with ionization energy of 70 eV was used. The electron ionization energy system temperature was 230°C, the temperature of the quadrupole rod was 150°C, and the transmission line temperature was 280°C. The mass range of the quadrupole mass spectrometer was scanned over the range 30-450 amu, with a solvent delay of 3min.
Small RNA Library Preparation and Sequencing
Total RNAs were isolated from flower and fruit tissues of Camellia oleifera using TRlzol Reagent (Invitrogen) according to the manufacturer's protocol. The purity of the RNA was checked using the Nanodrop 2000 (Thermo, MA, USA) system. The concentration of the RNA was measured using a Qubit® RNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). The integrity of the RNA samples was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). RNA isolation was carried out individually for each sample with three biological replicates, and then the purified RNAs were used to construct small RNA libraries according to Illumina recommendation and high-throughput sequencing using the Hiseq2500 platform.
Identification of Camellia oleifera miRNAs Using Deep Sequencing
After sequencing, the raw reads (fastq format) were firstly processed through custom Perl and Python scripts to obtain clean reads, and then a certain range of lengths was chosen from the clean reads to perform all the downstream analyses. The small RNA tags were mapped to the reference sequence by Bowtie [29] without mismatch to analyze their expression and distribution on the reference. The mapped small RNA tags were aligned to Repeat Masker, Rfam database or those types of data from the specified species itself to remove tags originating from protein-coding genes, repeat sequences, rRNA, tRNA, snRNA, and snoRNA. The remaining tags were then aligned with known miRNA and mapped to miRBase21.0 [30] and modified by mirdeep2 software [31]. Novel miRNA were predicted using the characteristics of the hairpin structure of the miRNA precursor, and the miREvo [32] and mirdeep2 software [31] were integrated to predict novel miRNAs through exploring the secondary structure, the Dicer cleavage site and the minimum free energy of the small RNA tags unannotated in the former steps. The known families were identified using miFam.dat (http://www.mirbase.org/ftp.shtml) and the novel miRNA precursors were submitted to Rfam (http://rfam.sanger.ac.uk/search/) to search for Rfam families.
Differential Expression Analysis of MiRNAs
MiRNA expression levels were estimated by TPM (transcript per million) through the following criteria [33] using a normalization formula:
Normalized expression = Mapped read count/Total reads*1000000
Differential expression analysis of two samples was performed using the DESeq R package (1.8.3). The P-values were adjusted using the Benjamini & Hochberg method. The corrected P-value of 0.05 was set as the threshold for significantly different differential expression by default.
Target Prediction, GO Enrichment and KEGG Pathway Analysis
Significantly expressed conserved and novel miRNAs were used for target prediction using the psRNA Target server by psRobot_tar in psRobot[34]. Gene Ontology (GO) enrichment analysis was used on the target gene candidates of differentially expressed miRNAs (“target gene candidates” in the following). GOseq based Wallenius non-central hyper-geometric distribution [35], which could adjust for gene length bias, was implemented for GO enrichment analysis. KEGG [36] is a database resource for understanding high-level functions and utilities of biological systems, such as cells, organisms and ecosystems, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies (http://www.genome.jp/kegg/). We used KOBAS [37] software to test the statistical enrichment of the target gene candidates in KEGG pathways.
Validation of miRNA Expressions with qRT-PCR Analysis
Total RNA was isolated from CBMCs using TRIzol reagent (Invitrogen) as per the manufacturer's instructions. The RNAs were reverse transcribed to cDNA with Reverse Transcriptase M-MLV (RNase H-, Takara Bio Inc.) and then quantified on a Step OnePlus Real-Time PCR system (Applied Biosystems, Foster City, USA) using SYBR Green qPCR Super-Mix-UDG with Rox (Invitrogen). The PCR reaction was conducted at 95˚C for 5 min, followed by incubations at 95˚C for 15 sec, and 60˚C for 31 sec repeated for 40 cycles. Each PCR analysis was repeated at least three times. The relative expression level of each miRNA was normalized against EF1a2 expression levels. The fold-change was calculated according to the 2-ΔΔCT method. The qRT-PCR primers are listed in the file (Table S1).
Results
SEM results of Camellia oleifera fruit
SEM was used to investigate the morphological features of the internal cellular structure of Camellia oleifera seeds. The results of the SEM observations confirmed that oil grains existed in Camellia oleifera seeds (Figure 1C), while the fresh Camellia oleifera fruit and seeds are shown in Figure 1A and Figure 1B, respectively.
The features of Camellia oleifera fruit. (A) Fresh fruit. (B) Fresh seed. (C) SEM view of seed.
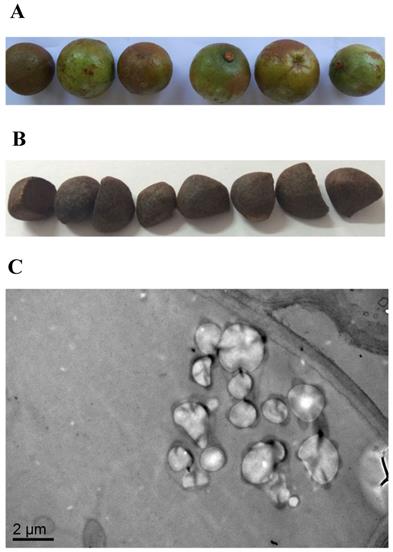
Fatty acid composition of camellia oil
Using the Soxhlet extraction method, the oil content of Camellia oleifera fruit, which was a light yellow liquid at room temperature, was determined to be 21.54%. The results of the analysis of the fatty acid composition of camellia oil by GC-MS (Figure 2) show that the highest fatty acid proportion consisted of oleic acid (76.91±0.33%), followed by palmitic acid (10.33±0.08%), linoleic acid (9.08±0.34%), and finally stearic acid (0.51±0.08%).
The fatty acid composition of camellia oil by GC-MS.
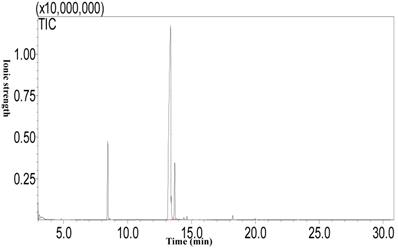
Sequence analysis of Small RNAs
In order to obtain the comprehensive profiles of miRNAs in Camellia oleifera flower and fruit, two small RNA libraries were constructed and sequenced. Average totals of 13,461,290 and 12,824,110 raw reads were generated from Camellia oleifera flower and fruit, respectively (Table S2). After removing low quality reads and adaptors, totals of 13, 459,418 and 12,823,613 clean reads were obtained from flower and fruit groups, respectively (Table S2). The clean reads were used for analysing the distribution of 18 to 40 nt length small RNAs and the majority of small RNAs were 21 to 24 nt in length, producing similar length distributions in both sample types. The most abundant sRNAs were 21 (27.35%) and 24nt (22.95%) lengths in flower and 22 (13.03%) and 24nt (40.32%) lengths in fruit, respectively (Figure 3). These clean reads were further annotated by Bowtie; 7,948,865 and 3, 854,767 mapped small RNA tags were obtained in Camellia oleifera flower and fruit, respectively, and were classified into miRNAs, tRNAs, rRNAs and others by Blasting with Rfam and miRBase (Table 1).
Small RNA categorization in Camellia oleifera flower and fruit samples.
| RNA Type | flower | Fruit |
|---|---|---|
| Mapped | 7948865 | 3854767 |
| rRNA | 3338103 | 762599 |
| tRNA | 404718 | 543946 |
| snRNA | 13407 | 12460 |
| snoRNA | 210 | 353 |
| ta_siRNA | 1534 | 1329 |
| Other | 3999134 | 2417738 |
| Known_miRNA | 135670 | 89524 |
| Novel_miRNA | 56087 | 26818 |
| Total | 12105443 | 10898416 |
Length distribution of clean small RNAs in Camellia oleifera flower and fruit samples.
Identification of miRNAs from Deep Sequencing
To identify miRNAs, the clean sequence tags were aligned to all plant miRNA mature sequences deposited in miRBase 21.0. The averages of 135,670 and 89,524 redundant reads of Camellia oleifera flower and fruit were matched to the currently known miRNA sequences, respectively (Table 1). In total, 1,231 miRNAs, which belong to 42 families, were obtained from sequencing in two libraries (Table S3). Among the 42 families, miR156 was the largest represented with 284 members, followed by miR167 and miR160 with 156 and 108 members, respectively (Table S3). Five miRNA families (miR168, miR159, miR166, miR396, miR858) were represented with the top read abundance above 10, 000 in all libraries (Table S3). In addition, the miRNA sequences which met the threshold of the miRDeep2 analysis but did not have any known homologous miRNA gene families in miRBase were further analyzed to predict novel miRNAs in the flower and fruit of Camellia oleifera. The miRNAs which met the total score of > 0 were considered to be true novel miRNAs. A total of 147 novel mature miRNA sequences belonging to 23 miRNA families were identified in flower and fruit (Table S4).
Identification of Differentially Expressed miRNAs
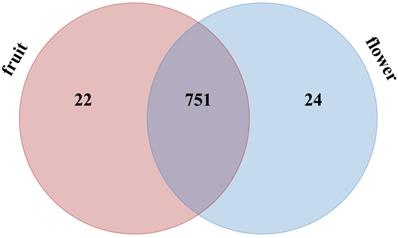
A total of 797 miRNAs belonging to two libraries were significantly differently expressed based on fold change (≥1 or ≤-1) and P-value (<0.05) criteria (Table S5); among them, 388 miRNAs were up-regulated and 409 miRNAs were down-regulated. We evaluated the miRNA distribution among two libraries and determined that 751 miRNAs were commonly expressed in flower and fruit libraries (Figure 4). The results of comparing the specifically expressed miRNAs showed that 24 miRNAs were expressed only in flower and 22 miRNAs were expressed only in fruit, respectively (Table S5 and Figure 4).
Distribution of significantly different expressed Camellia oleifera miRNAs in flower and fruit samples.
Target Prediction, GO Enrichment and KEGG Pathway Analyses
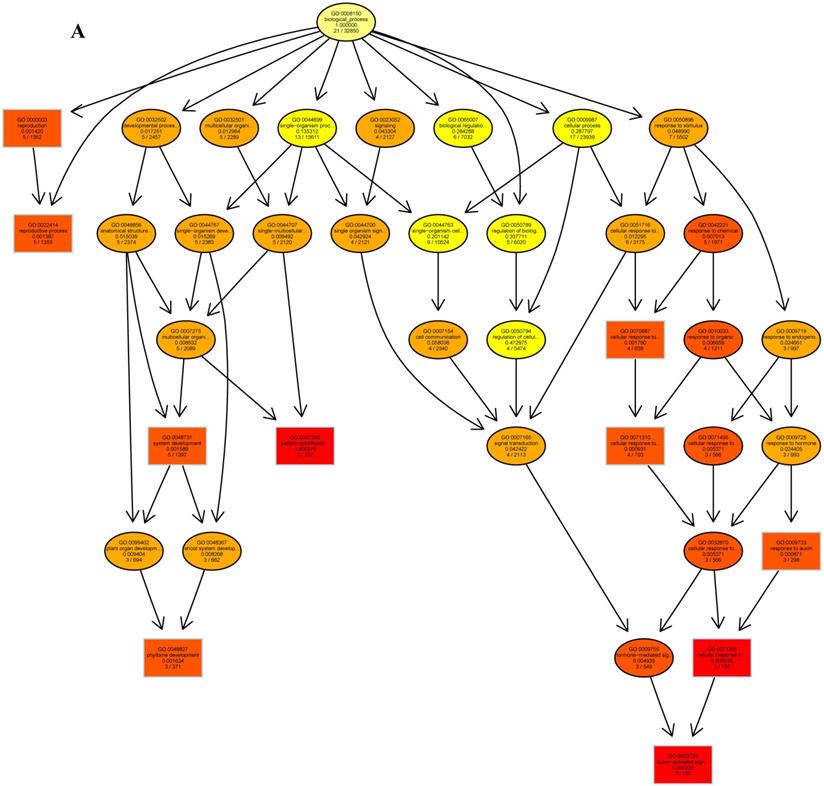
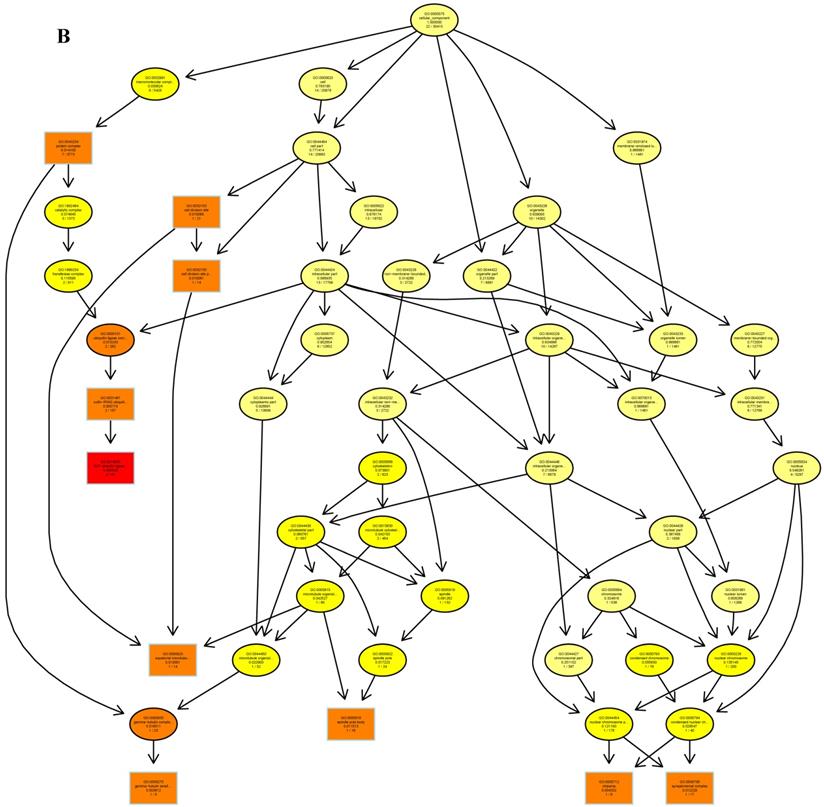
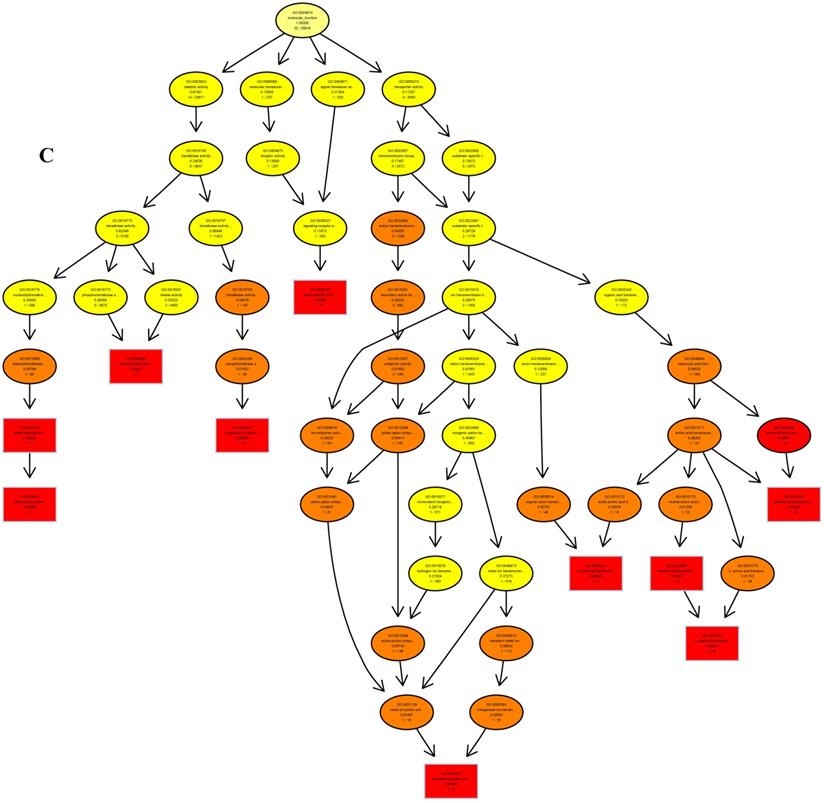
The psRNATarget server (http://plantgrn.noble.org/psRNATarget/) was used to predict miRNA target genes, and a total of 2041 miRNA target genes were obtained (Table S6). To further investigate the potential functions of these miRNA target genes, we applied gene ontology (GO) and KEGG pathway analyses to categorize the miRNA targets. The miRNA target genes were categorized according to biological process, cellular component and molecular function by GO-based enrichment analysis. A total of 358 potential miRNA targets were classified into 143 biological processes, 29 cellular components and 41 molecular functions (Table S7). Some of the significant biological processes with the highest annotation numbers were: cellular response to stimulus (GO:0051716), developmental process (GO:0032502), single-organism developmental process (GO:00447670), signaling (GO:0023052), system development (GO:0048731), response to organic substances (GO:0010033), and developmental processes involved in reproduction (GO:0003006) (Table S7). The common cellular component terms were: macromolecular complex (GO:0032991) and protein complex (GO:0043234) (Table S7). Active transmembrane transporter activity (GO:0022804), lipid binding (GO:0008289), hormone binding (GO:0042562), and mismatched DNA binding (GO:0030983) were among the most abundant classes in the molecular function category (Table S7). GO terms were submitted to enrichment analysis by the agriGO tool [38]. Among the GO terms related to biological processes, the results showed that several target genes were involved in organism development and signaling pathways (Figure 5A). In the cellular component category, the target genes were mainly related to chromosomal behavior in the nucleus (Figure 5B). In molecular function, the majority of the target gene functions were related to transmembrane transporter activity (Figure 5C).
The plots of GO ontology enrichment in Camellia oleifera flower and fruit. (A) Biological process. (B) Cellular component. (C) Molecular function. Red to yellow colours represent decreasing significance levels (Red is most, yellow is least significant).
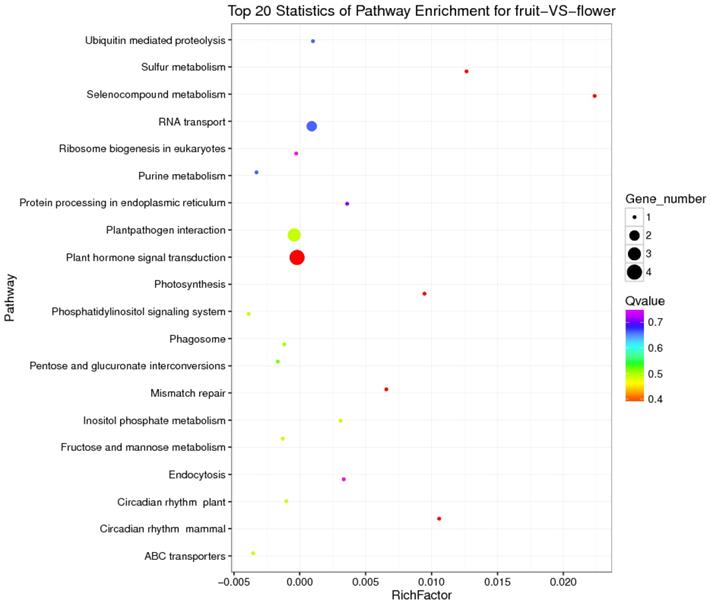
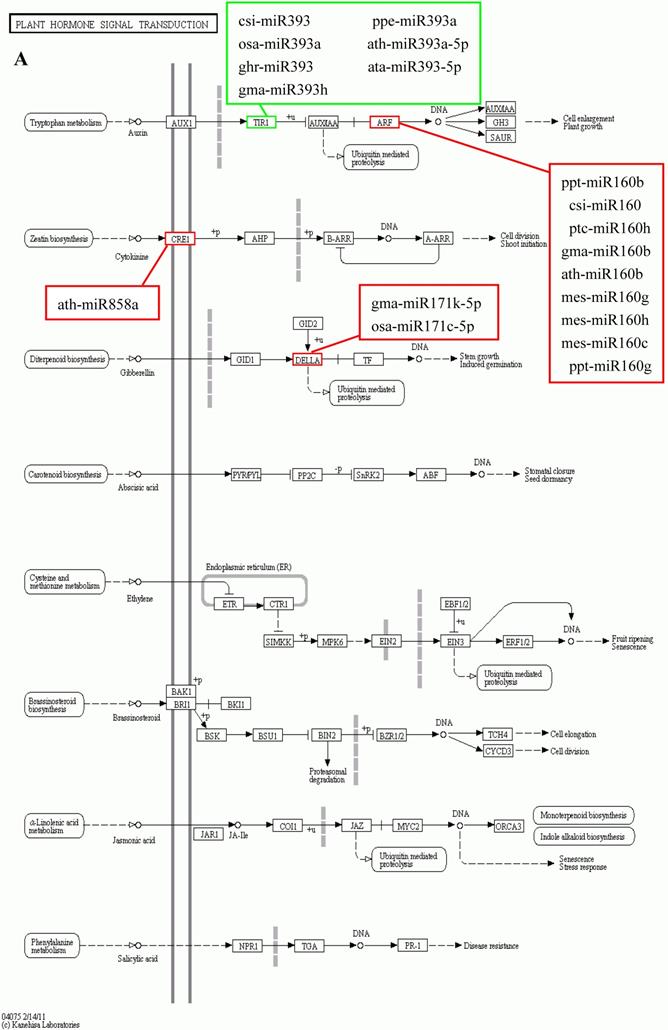
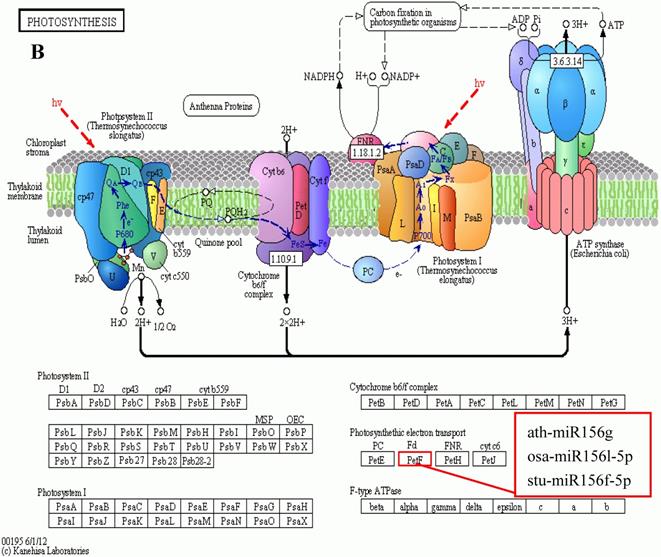
KEGG is a major public database for Pathway Mapping; the most important biochemical metabolic and signal transduction pathways of differentially expressed genes participation were determined by Pathway significant enrichment. The KEGG pathway analysis revealed that 112 pathways including 109 genes were obtained involving metabolic pathways, plant hormone signal transduction, RNA transport, starch and sucrose metabolism, RNA degradation, pentose and glucuronate interconversions, lipid metabolism and fructose and mannose metabolism (Table S8). The top 20 statistics of pathway enrichment for fruit-VS-flower are shown in Figure 6. The plant hormone signal transduction pathway was represented by 4 genes and contained auxin-, cytokinine- and gibberellin- inducible via flower developed to fruit (Figure 7). For example, tryptophan metabolism was involved in the pathway, contributes to auxin synthesis, enriched transport inhibitor response 1 (TIR1) and auxin response factor (ARF). These genes were regulated by the miR393 and miR160 families, respectively (Figure 7). Arabidopsis histidine kinase 2/3/4 (CRE1) was enriched to zeatin biosynthesis associated with cytokine synthesis, and the gene was targeted by ath-miR858a (Figure 7A). Similarly, DELLA protein (DELLA) was enriched to gibberellin biosynthesis associated with cysteine and methionine metabolism, and the gene was targeted by gma-miR171k-5p and osa-miR171c-5p. The photosynthesis pathway was represented by 1 gene and involved in photosynthetic electron transport inducible via flower developed to fruit, and contained ferredoxin (PetF) (Figure 7B).
The top 20 statistics of pathway enrichment for fruit-VS-flower by Q value from small to large.
The KEGG pathways and related miRNAs as determined for Camellia oleifera. (A) Plant hormone signal transduction. (B) Photosynthesis.
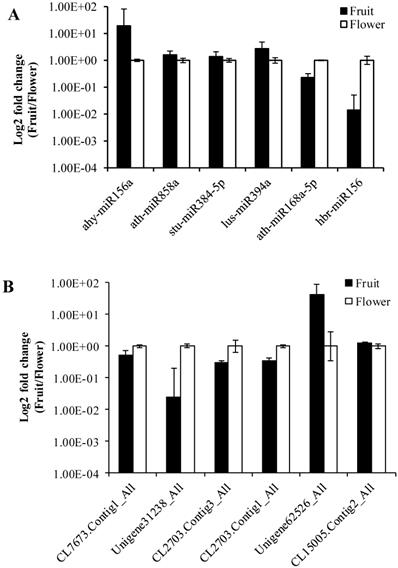
Validation of differentially expressed miRNAs by qRT-PCR
To validate the data obtained from the high-throughput sequencing, we randomly selected six miRNAs (ahy-miR156a, ath-miR858a, stu-miR384-5p, lus-miR394a, ath-miR168a-5p, hbr-miR156) and their corresponding target genes (CL7673.Contig1_All, Unigene31238_All, CL2703.Contig3_All, CL2703.Contig1_All, Unigene62526_All, CL15005.Contig2_All). For this purpose, we determined the expression levels between Camellia oleifera flower and fruit using log2 fold change (2-∆∆Ct) values with three technical and three biological replicates. The expression results showed that miRNAs were both up-regulated (ahy-miR156a, ath-miR858a, stu-miR384-5p, lus-miR394a) and down-regulated (ath-miR168a-5p, hbr-miR156) in the qRT-PCR analysis, which were similar to the deep sequencing data results (Figure 8A). Similarly, the RT-PCR up-regulated and down-regulated results of the miRNAs corresponding target genes were consistent with the prediction (Figure 8B).
Validation of sequencing results by RT-qPCR. (A) MiRNAs. (B) Target genes. Error bars indicate mean ± SE (n = 3 per breed).
Discussion
Camellia oleifera is one of the most important woody oil species in China and has very high economic value [1]. The fruit is the main source of the economic benefits of Camellia oleifera, and how to achieve high yield, quality and efficiency of the species and its fruit has become an important research area [39]. The fruit is a plant reproductive organ that develops from the ovary or other parts of the flower (such as receptacle, calyx, etc.) by pistils after pollination fertilization [40]. The growth and development of fruit include the whole process from fertilization to fruit growth, maturation and senescence, and it is also the process of continuously accumulating and transforming nutrients and fruit morphogenesis construction [41]. During the fruit growth and development process, the accumulation of nutrients mainly includes carbohydrates, proteins and lipids, most of which are transported by other organs of the plant, such as roots and leaves, while carbohydrate metabolism is the basis of regulation in its process [42, 43]. During the early stages of Camellia oleifera fruit development, carbohydrates come from the photosynthesis of leaves and fruits [44].
In this study, we observed differentially expressed miRNA in the fruit and flower of Camellia oleifera by using high-throughput RNA sequencing. We found 42 families of known miRNAs and 23 families of novel miRNAs, respectively. Among the 65 families, the most highly expressing miRNAs were miR168, miR166, miR396, miR157, miR156, miR390 and so on, which were similar to the high expression miRNA of flowers and fruits in Camelina sativa[45]. The results of comparing the differentially expressed miRNAs showed that 751 miRNAs were overlapped between fruit and flower miRNAs, such as miR168, miR160, miR396, miR164, miR156, miR393, miR858, and those were similar to the overlaps of Camelina sativa [45].
Chloroplasts are important sites for plant photosynthesis, which converts light energy into chemical energy (through electron transfer reactions), and supplies NADPH and ATP for CO2 fixation into carbohydrates and for the synthesis of several essential compounds of plant growth [46-49]. In photosynthetic electron transfer reactions, electrons are generated by splitting water by light energy, and are transferred via photosystem II (PSII), cytochrome (Cyt) b6f complex and photosystem I (PSI) to ferredoxin (Fd). Fd is the first soluble electron acceptor of the electron transport chain, and determines the efficiency of the electron transfer between the thylakoid membrane and the soluble enzymes dependent on these electrons [50]. In this study, we found that miR156 targets Fd (Table S8 and Table S9) were expressed up-regulated in fruit, and the photosynthesis in fruit can be improved by increasing the efficiency of the electron transfer, which is similar to the results of reducing levels of miR156 promoting the rate of photosynthesis in plant leaves[51-54]. Ferredoxin-NADP+ oxidoreductase (FNR) is the key enzyme linking the light reactions of photosynthesis to carbon assimilation, which functions in the last step of the linear electron transfer (LET), mediating electron flow from reduced Fd to NADPH for fixation of CO2 in the Calvin cycle[46, 47, 50].
Studies have shown that miR156 and miR390 have the same developmental process oppositely regulating developmental timing in plants [55]. In the development of Camellia oleifera fruit, miR390 targets FNR expression up-regulation in fruit which is conducive to the generation of NADPH (Table S8 and Table S9), and it is possibly related to the miR156 and miR390 regulated tasiRNA network. NAD-dependent malate dehydrogenase (NADP-MDH) is a key enzyme controlling the "malate valve", which converts oxaloacetate to malate using NADPH from photosynthetic electron transport, facilitating the regeneration of the electron acceptor NADP+ in the chloroplasts, thus being indirectly involved in CO2 fixation [56-58]. Compared with flower, miR395 targets NADP-MDH were expressed up-regulation (Table S8 and Table S9) in fruit and consistent with the expression of Fd and FNR, and also similar to the over-expression of NADP-MDH resulting in an increased synthesis of tobacco biomass [56]. These results suggest that the concurrently low-level expressions of miR156, miR390 and miR395 in Camellia oleifera fruit may be the main reason for the accumulation of carbohydrates in the fruit growth and development process.
As an important oil crop, oil is one of the most important accumulated nutrients in the growth and development process of the fruit of Camellia oleifera. The main component of vegetable oil is triacylglycerol (TAG), which results from glycerol-3-phosphate (G3P) and fatty acid CoA condensation [59]. The glycolytic metabolic pathway is the upstream metabolic pathway of Camellia oleifera fruit oil synthesis; the intermediate product dihydroxyacetone phosphate (DHAP) of the glycolytic pathway is reduced to G3P by glycerol-3-phosphate dehydrogenase (GPD1) catalyst [60]. The pyruvate dehydrogenase complex converts pyruvate, which is the final product of glycolysis, into acetyl-CoA, then acetyl-CoA is carboxylated by acetyl CoA carboxylase (ACCase) to malonyl-CoA which is the substrate for biological fatty acid synthesis [61]. β-D-fructose-6-phosphate is one of the important intermediates in glycolysis and it has a very wide range of sources. One major source of it is the conversion of L-sorbose-1-phosphate to D-sorbitol-6-phosphate by the reversible catalysis of sorbitol-6-phosphate 2-dehydrogenase, then the reverse catalysis of the sorbitol-6-phosphate 2-dehydrogenase to β-D-fructose-6-phosphate. In this study, we found that miR156 target butanol dehydrogenase was up-regulated (Table S8 and Table S9), which may be involved in increasing the β-D-fructose-6-phosphate to promote glycolysis. In nature, acetyl-CoA carboxylase has two functional forms and each contains the four different components required for activity: biotin carboxylase, biotin carboxyl carrier protein, α- carboxyl transferases and β-carboxyl transferases. One form is heteromeric, in which the all four enzymatic components as individual subunits are combined into a multienzyme complex. The other form is homomeric, whereby all four enzymatic components are concatenated into a single polypeptide, and is the predominant form for de novo fatty acid synthesis [62]. In fruit, miR477 induced the target acetyl-CoA carboxylase carboxyl transferase subunit alpha up-regulated (Table S8 and Table S9), and it may be related to increasing the efficiency of fatty acid synthesis. There are two major biochemical pathways of linoleic acid (LA) to oxidized LA metabolites (OXLAMs) in LA metabolism; they begin with formation of the conjugated hydroperoxydienes 9- hydroperoxyoctadecadienoic acid (9-HPODE) and 13- hydroperoxyoctadecadienoic acid (13-HPODE), which are then rapidly reduced by glutathione-dependent peroxidase to the alcohols 9-hydroxy-octadecadienoic acid (9-HODE) and 13-hydroxy-octadecadienoic acid (13-HODE), respectively. Finally, the 9-HODE and 13-HODE can be converted to the ketones 9-oxo-octadecadienoic acid (9-oxoODE) and 13-oxo-octadecadienoic acid (13-oxoODE) via hydroxy-fatty acid dehydrogenase, respectively. MiR156 targets butanol dehydrogenase up-regulated expression (Table S8 and Table S9) which will be beneficial to the 13-HODE transformed to 13-oxoODE, which may be related to the relative content of LA being decreased in Camellia oleifera fruit during the ripening process [63].
These results illustrate that the down-regulation of miR156 and miR477 contributed to the fat accumulation and nutrients transformation in Camellia oleifera fruit during ripening, and previous studies have shown that the down-regulation of miR156 expression plays an important role in the synthesis of fatty acids [64]. This work accordingly increases understanding of the underlying processes and the role of miRNA in the development and accumulation of nutrients in Camellia oleifera.
Supplementary Material
Tables S1-S2.
Tables S3.
Tables S4.
Tables S5.
Tables S6.
Tables S7.
Tables S8.
Tables S9.
Acknowledgements
This work was supported by the Natural Science Foundation of Hunan (NO. 2016JJ3064 and NO. 2018JJ3199), University Industrialization Cultivation Project of Hunan Province (NO. 13CY029) and Hunan Provincial Department of Education key projects (NO. 15A072).
Author contributions
X. L., Z. Q. and F. H. designed the experiments and wrote the manuscript. K. L., Y. L. and T. P. collected plant materials, and X. L., F. H., X. L., Z. L. and G. D. performed the experiments and data analysis. Z. Q. improved the English of the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhuang R. Camellia oleifera in China. Beijing: China Forestry Press. 2008
2. Feng JL, Yang ZJ, Chen SP. et al. High throughput sequencing of small RNAs reveals dynamic microRNAs expression of lipid metabolism during Camellia oleifera and C. meiocarpa seed natural drying. BMC Genomics. 2017;18:546
3. Xia EH, Jiang J, Huang H. et al. Transcriptome analysis of the oil-rich tea plant, Camellia oleifera, reveals candidate genes related to lipid metabolism. PLoS One. 2014;9:e104150
4. Cheng YT, Lu C, Yen GC. Beneficial effects of camellia oil (Camellia oleifera Abel.) on hepatoprotective and gastroprotective activities. J Nutr Sci Vitaminol. 2015;61:S100-S2
5. Dong B, Wu B, Hong W. et al. Transcriptome analysis of the tea oil camellia (Camellia oleifera) reveals candidate drought stress genes. PLoS One. 2017;12:e0181835
6. Zeng QL, Chen RF, Zhao XQ. et al. Aluminum could be transported via phloem in Camellia oleifera Abel. Tree Physiology. 2012;33:96-105
7. Chaikul P, Sripisut T, Chanpirom S. et al. Melanogenesis inhibitory and antioxidant effects of Camellia oleifera seed oil. Adv Pharm Bull. 2017;7:473-7
8. Jiang ZN, Tan X, Yuan J. et al. Content variation of main nutrients in leaves and fruits of Camellia oleifera. Journal of Central South University of Forestry and Technology. 2012;32:42-5
9. Wang XL, Li XD, Zhang S. et al. Physiological and transcriptional responses of two contrasting Populus clones to nitrogen stress. Tree Physiology. 2016;36:628-42
10. Rane JK, Erb H, Nappo G. et al. Inhibition of the glucocorticoid receptor results in an enhanced miR-99a100-mediated radiation response in stem-like cells from human prostate cancers. Oncotarget. 2016;7:51965-80
11. Cui JW, Zhao J, Zhao JY. et al. Cytological and miRNA expression changes during the vascular cambial transition from the dormant stage to the active stage in Ginkgo biloba L. Trees. 2016;30:1-12
12. Ding Y, Wang Y, Jiang Z. et al. MicroRNA268 overexpression affects rice seedling growth under cadmium stress. J Agric Food Chem. 2017;65:5860-7
13. Wang M, Yu F, Wu W. et al. Epstein-Barr virus-encoded microRNAs as regulators in host immune responses. International Journal of Biological Sciences. 2018;14:565-76
14. Zhong T, Hu J, Xiao P. et al. Identification and characterization of microRNAs in the goat (Capra hircus) rumen during embryonic development. Front Genet. 2017;8:163
15. Rodriguez RE, Schommer C, Palatnik JF. Control of cell proliferation by microRNAs in plants. Curr Opin Plant Biol. 2016;34:68-76
16. Shulga OA, Nedoluzhko A, Shchennikova AV. et al. Profiling of microRNAs in wild type and early flowering transgenic Chrysanthemum morifolium by deep sequencing. Plant Cell, Tissue and Organ Culture. 2016;128:283-301
17. Wang Y, Zhang J, Cui W. et al. Improvement in fruit quality by overexpressing miR399a in woodland strawberry. J Agric Food Chem. 2017;65:7361-70
18. Xin R, Bai F, Feng Y. et al. MicroRNA-214 promotes peritoneal metastasis through regulating PTEN negatively in gastric cancer. Clin Res Hepatol Gastroenterol. 2016;40:748-54
19. Sulas P, Di T, Novello C. et al. A large set of miRNAs is dysregulated since the earliest steps of human hepatocellular carcinoma development. Am J Pathol. 2017;188:785-94
20. Li YW, Sarkar FH. MicroRNA targeted therapeutic approach for pancreatic cancer. International Journal of Biological Sciences. 2016;12:326-37
21. Liang R, Han B, Li Q. et al. Using RNA sequencing to identify putative competing endogenous RNAs (ceRNAs) potentially regulating fat metabolism in bovine liver. Scientific Reports. 2017;7:6396
22. Yang WC, Guo W, Zan LS. et al. Bta-miR-130a regulates the biosynthesis of bovine milk fat by targeting peroxisome proliferator-activated receptor gamma. J Anim Sci. 2017;95:2898-906
23. Wójcik AM, Gaj M. MiR393 contributes to the embryogenic transition induced in vitro in Arabidopsis via the modification of the tissue sensitivity to auxin treatment. Planta. 2016;244:231-43
24. Meng Y, Shao C, Ma X. et al. Expression-based functional investigation of the organ-specific microRNAs in Arabidopsis. PLoS One. 2017;7:e50870
25. Meyers BC, Axtell M, Bartel B. et al. Criteria for annotation of plant MicroRNAs. The Plant Cell. 2008;20:3186-90
26. Pan Y, Niu M, Liang J. et al. Identification of heat-responsive miRNAs to reveal the miRNA-mediated regulatory network of heat stress response in Betula luminifera. Trees. 2017;31:1635-52
27. Chen J, Yang X, Huang X. et al. Leaf transcriptome analysis of a subtropical evergreen broadleaf plant, wild oil-tea camellia (Camellia oleifera), revealing candidate genes for cold acclimation. BMC Genomics. 2017;18:211
28. Dangarembizi R, Chivandi E, Dawood S. et al. The fatty acid composition and physicochemical properties of the underutilised Cassia abbreviata seed oil. Pak J Pharm Sci. 2015;28:1005-8
29. Langmead B, Trapnell C, Pop M. et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25
30. Kozomara A, Sam G-J. MiRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68-73
31. Friedländer MR, Mackowiak S, Li N. et al. MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37-52
32. Wen M, Shen Y, Shi S. et al. MiREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics. 2012;13:140
33. Zhou L, Chen J, Li Z. et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS One. 2010;5:e15224
34. Wu HJ, Ma Y, Chen T. et al. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012;40:W22-8
35. Young MD, Wakeeld M, Smyth GK. et al. Goseq: Gene Ontology testing for RNA-seq datasets. Alicia Oshlack. 2010
36. Kanehisa M, Araki M, Goto S. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480-4
37. Mao X, Cai T, Olyarchuk JG. et al. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787-93
38. Candar-Cakir B, Arican E, Zhang B. Small RNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol J. 2016;14:1727-46
39. Tang J, Feng J, Yang ZJ. et al. Changes of endogenous hormones in fruit and their effects on the fruit development of Camellia oleifera. Journal of Forest and Environment. 2015:35
40. Pan RC. Beijing: Higher Education Press; 2001.
41. Losada JM, Bachelier J, Friedman WE. Prolonged embryogenesis in Austrobaileya scandens (Austrobaileyaceae): its ecological and evolutionary significance. New Phytol. 2017;215:851-64
42. Zhu S, Liang Y, An X. et al. Changes in sugar content and related enzyme activities in table grape (Vitis vinifera L.) in response to foliar selenium fertilizer. J Sci Food Agric. 2017;97:4094-102
43. Sano N, Ono H, Murata K. et al. Accumulation of long-lived mRNAs associated with germination in embryos during seed development of rice. J Exp Bot. 2015;66:4035-46
44. Yuan MXF, Wang SD, Zhang DW. et al. A single leaf of Camellia oleifera has two types of carbon assimilation pathway, C3 and crassulacean acid metabolism. Tree Physiology. 2012;32:188-99
45. Saroj PNA, Lu CF. Identification of MicroRNAs and Transcript Targets in Camelina sativa by Deep Sequencing and Computational Methods. PLoS ONE. 2015;10:e0121542
46. Checchetto V, Teardo E, Carraretto L. et al. Regulation of photosynthesis by ion channels in cyanobacteria and higher plants. Biophys Chem. 2013;182:51-7
47. Rokka A, Battchikova N, Salminen TA. et al. Posttranslational modifications of FERREDOXIN-NADP+ OXIDOREDUCTASE in Arabidopsis chloroplasts. Plant Physiol. 2014;166:1764-76
48. Sun C, Chen S, Jin Y. et al. Effects of the herbicide Imazethapyr on photosynthesis in PGR5- and NDH-deficient Arabidopsis thaliana at the biochemical, transcriptomic, and proteomic levels. J Agric Food Chem. 2016;64:4497-504
49. Liu YF, Qi HY, Bai CM. et al. Grafting helps improve photosynthesis and carbohydrate metabolism in leaves of muskmelon. International Journal of Biological Sciences. 2011;7:1161-70
50. Hanke G, Mulo P. Plant type ferredoxins and ferredoxin-dependent metabolism. Plant Cell Environ. 2013;36:1071-84
51. Yu S, Cao L, Zhou CM. et al. Sugar is an endogenous cue for juvenile-to-adult phase transition in plants. Elife. 2013;2:e00269
52. Feng S, Xu Y, Guo C. et al. Modulation of miR156 to identify traits associated with vegetative phase change in tobacco (Nicotiana tabacum). J Exp Bot. 2016;67:1493-504
53. Yoshikawa T, Ozawa S, Sentoku N. et al. Change of shoot architecture during juvenile-to-adult phase transition in soybean. Planta. 2013;238:229-37
54. Yu S, Lian H, Wang JW. Plant developmental transitions: the role of microRNAs and sugars. Curr Opin Plant Biol. 2015;27:1-7
55. Cho SH, Coruh C, Axtell MJ. MiR156 and miR390 regulate tasiRNA accumulation and developmental timing in Physcomitrella patens. Plant Cell. 2012;24:4837-49
56. Hebbelmann I, Selinski J, Wehmeyer C. et al. C. Multiple strategies to prevent oxidative stress in Arabidopsis plants lacking the malate valve enzyme NADP-malate dehydrogenase. J Exp Bot. 2012;63:1445-59
57. Scheibe R. NADP+-malate dehydrogenase in C3-plants: regulation and role of a light-activated enzyme. Physiologia Plantarum. 1987;71:393-400
58. Beeler S, Liu H, Stadler M. et al. Plastidial NAD-dependent malate dehydrogenase is critical for embryo development and heterotrophic metabolism in Arabidopsis. Plant Physiol. 2014;164:1175-90
59. Xue LL, Chen HH, Jiang JG. Implications of glycerol metabolism for lipid production. Prog Lipid Res. 2017;68:12-25
60. Morales-Sánchez D, Kim Y, Terng EL. et al. A multidomain enzyme, with glycerol-3-phosphate dehydrogenase and phosphatase activities, is involved in a chloroplastic pathway for glycerol synthesis in Chlamydomonas reinhardtii. Plant J. 2017;90:1079-92
61. Chhikara S, Abdullah H, Akbari P. et al. Engineering Camelina sativa (L.) Crantz for enhanced oil and seed yields by combining diacylglycerol acyltransferase1 and glycerol-3-phosphate dehydrogenase expression. Plant Biotechnol J. 2017:1-12
62. Salie MJ, Thelen J. Regulation and structure of the heteromeric acetyl-CoA carboxylase. Biochim Biophys Acta. 2016;1861:1207-13
63. Li H, Fang XZ, Zhong HY. et al. Variation of physicochemical properties and nutritional components of oil-tea camellia seeds during ripening. Forest Research. 2014;27:86-91
64. Wang J, Jian HJ, Wang TY. et al. Identification of microRNAs actively involved in fatty acid biosynthesis in developing Brassicanapus seeds using high-throughput sequencing. Front Plant Sci. 2016;7:1570
Author contact
![]() Corresponding authors: Zuo-Dong Qin, E-mail: dong6758068com and Fu-Lin He, E-mail: 2339695475com
Corresponding authors: Zuo-Dong Qin, E-mail: dong6758068com and Fu-Lin He, E-mail: 2339695475com