Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(2):493-506. doi:10.7150/ijbs.29404 This issue Cite
Research Paper
CD9 regulates keratinocyte migration by negatively modulating the sheddase activity of ADAM17
Jie Liu1, Guoqin Zhu1, Naixin Jia1, Weiyi Wang1, Yuan Wang1, Meifang Yin1, Xuping Jiang1, Yuesheng Huang1, Jiaping Zhang1,2 ![]()
1. Institute of Burn Research, State Key Laboratory of Trauma, Burns and Combined Injury, Southwest Hospital, Third Military Medical University(Army Medical University), Chongqing, China.
2. Department of plastic Surgery, Southwest Hospital, Third Military Medical University(Army Medical University), Chongqing, China.
Received 2018-8-22; Accepted 2018-12-13; Published 2019-1-1
Abstract

CD9 is a trans-membrane protein, and has recently been implicated in different physiological and cellular processes, such as cell migration and adhesion. According to previous study, down-regulation of CD9 contributes to keratinocyte migration, critical for wound re-epithelialization. Nevertheless, it is widely believed that tetraspanin CD9 does not have ligands or function as the cell surface receptor, rather it is thought to associate with other transmembrane molecules, thereby mediate keratinocyte migration. Little is known about how CD9 associates with transmembrane molecules in migratory keratinocytes. Here, using confocal microscopy, we observed that tetraspanin CD9 and ADAM17 co-localized on the surface of keratinocytes in the course of wound repair in vivo and in vitro. Co-immunoprecipitation experiments demonstrated a direct association between CD9 and ADAM17 in HaCaT cells and C57-MKs. Functional studies revealed that down-regulation or over-expression of CD9 exerted negative regulatory effects on ADAM17 sheddase activity. This activity is involved in CD9-regulated cell motility and migration. Further studies found that ADAM17 inhibitor-TAPI-2 or siADAM17 significantly abolished the enhanced effect of keratinocyte migration induced by CD9 down-regulation. Meanwhile, the sheddase activity of ADAM17 was inhibited by TAPI-2, which decreased this release of AREG and HB-EGF in CD9-silenced HaCat cells and C57-MKs. Importantly, neutralizing antibody against HB-EGF significant weakened keratinocyte migration and motility in CD9-silenced keratinocytes, and the inhibition of CD9-regulated keratinocyte migration by siADAM17 was rescued by addition of recombinant HB-EGF, activating EGFR/ERK pathway. Collectively, our results suggest that ADAM17 sheddase activity is activated by down-regulation of CD9, thereby mediating shedding of HB-EGF and activation of EGFR/ERK signaling, which crucially affects the keratinocyte migration and wound healing.
Keywords: CD9, ADAM17, HB-EGF, EGFR, keratinocyte migration, wound healing
Introduction
Re-epithelialization of wounds involves several keratinocyte functions: proliferation, migration, and differentiation. The key step is keratinocyte migration, an essential aspect for understanding chronic non-healing wounds [1, 2]. According to our previous study, wound repair was delayed in CD9-knockout mice, indicating that CD9 played a key role in wound healing [3]. CD9 down-regulation is beneficial to keratinocyte migration [3, 4]. CD9 is a highly conserved and intact transmembrane protein composed by two extracellular loops and short intracellular ends as well as four transmembrane domains; it plays an important role in cell migration, motility and adhesion [5, 6]. By contrast with many other cell surface proteins, CD9 does not have an obvious receptor function, CD9 may participates in the organization of surface multiprotein complexes through association with other transmembrane molecules, including integrins, ADAMs and signaling receptors, thereby mediating various cellular and physiological processes [7-9]. However, the regulation mechanisms of interaction between CD9 and other transmembrane molecules in keratinocyte migration are poorly understood. Our previous study found that CD9 down-regulation triggered the switch from αvβ5 to αvβ6 integrin in keratinocytes, a critical step for cell migration; however, the functional regions mediating CD9 and integrin αvβ5 interaction in keratinocyte migration remains unclear [10]. Moreover, CD9 complexes with integrins including α2β1 or α3β1 do not participate in keratinocyte migration [5, 10-12]. Therefore, CD9 may regulate keratinocyte migration by activating other non-integrins transmembrane molecules.
A disintegrin and metalloproteinases(ADAMs) are a family of transmembrane proteinases which take charge of proteolytic cleavage as well as release of various substrates from the cell surface, which greatly impact the wound healing, and tumorigenesis [7, 13]. The ADAM family consists of 22 known members; however, only ADAM17 is thought to be a key regulator in repair of skin [14]. ADAM17-/- mice are short of various kinds of EGFR ligands, and, due to the decreases of HB-EGF and TGF-α, suffer many serious failures with regard to the epithelial morphogenesis and maturation [15, 16]. In ADAM17 active epidermis, EGF family molecules such as HB-EGF, TGF-α and amphiregulin (AREG) are released, and EGFR activates enhancement [17]. It is known that EGFR ligands promote the migration of keratinocyte, thereby significantly improve wound healing, especially in the early reaction to wounding [18]. In particular, HB-EGF and TGF-α are responsible for keratinocyte migration during wound healing, meanwhile the HB-EGF contributes to the re-epithelialization and accelerates wound healing [19-21].The data showed that ADAM17 played a key role in keratinocyte migration and wound re-epithelialization. Nevertheless, the mechanisms of ADAM17 maturation or activation have not been elucidated in keratinocytes. According to a study conducted recently using the super-resolution microscopy, it seemed that a majority of tetraspanin nanoclusters contain ADAMs [22]. This is beneficial for explaining the specific function of tetraspanin CD9 with transmembrane metalloproteinases. Here, we described investigations of the interactions between CD9 and ADAM17 in keratinocyte migration. We demonstrated that the sheddase activity of ADAM17 was activated by CD9 down-regulation, increasing cleavage and release of HB-EGF, as well as activation of the EGFR/ERK signaling pathway, promoting keratinocyte migration and wound healing.
Materials and methods
Ethics Statement
C57 mice (male and female, about 1.5 g) used in the experiment were provided from the Experimental Animal Department of the Army Medical University (Third Military Medical University). All the animal experimental procedures had obtained the approval from the Animal Experiment Ethics Committee of the Army Medical University. Methods employed here were carried out following the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Pub. No. 85-23, revised 1996).
Cell culture
Primary keratinocytes culture
Keratinocytes were taken from the skin of newborn C57 mice one to three days after birth, as previously stated [23]. 0.25% trypsin/0.04% EDTA solution (Invitrogen, USA) was used to isolate keratinocytes from mice's skin at 4 °C overnight. Then, the obtained keratinocytes were plated into dishes and then cultured in RPMI 1640 medium (Hyclone SH30809.01, USA) which contained 100 U/ml penicillin (Invitrogen, USA), 100 mg/ml streptomycin (Invitrogen, USA), and 10% fetal bovine serum (Hyclone SV30087.02, USA). The cells then accepted 24h incubation at 37 °C in 5% CO2 and 95% humidity, followed by a gentle washing by using warm phosphate-buffered saline (PBS) aiming at eliminating non-adherent cells. Subsequently, the media were refreshed. The study used the keratinocytes at passage 2 or passage 3.
HaCaT cells culture
HaCaT cells used in the experiment were provided by the Cell Bank of the Chinese Academy of Sciences (CAS), China. Cells were cultured in RPMI 1640 medium which contained 100mg/ml streptomycin, 100U/ml penicillin, as well as 10% FBS. The HaCaT cells were incubated at 37°C, in the environment with 5% CO2, and 95% humidity.
Immunohistochemistry and immunocytochemistry
A biopsy punch was used to punch a full-thickness wound of which the diameter was 3 mm on the dorsal midline of 8-week-old C57 male mice. Wound specimen sections firstly accepted dewaxing and rehydration, and microwave treatment was adopted to carry out 6 min antigen retrieval at 650 W in citrate buffer (pH 6.0). HaCaT cell line were then cultured on glass cover slips (16-mm), followed by being fixed with a solution of PBS containing 4% paraformaldehyde. For double-staining, rabbit anti-CD9 primary antibody (1:100 dilution; Abcam ab92726, UK) was used to incubate the fixed keratinocytes at 4°C overnight, then it was properly washed, followed by another 1h incubation at 37°C using FITC goat anti-rabbit (1:100 dilution; Proteintech SA00003-11, USA). Next, goat anti-ADAM17 primary antibody (1:100 dilution; Abcam ab13535, UK) was used to incubate the fixed keratinocytes at 4°C overnight, then it was properly washed, followed by another 1h incubation at 37°C using CY3 donkey anti-goat (1:100 dilution; Proteintech SA00009-3, USA). DAPI (Sigma F6057, USA) was adopted to stain Nuclei for 3min (Sigma F6057, USA). A Leica confocal microscope (Leica Microsystems, Wetzlar, Germany) was used to observe the fluorescence.
Immunoprecipitation
Cells underwent cytolysis in 1 ml RIPA buffer, followed by 10 min incubation on ice. Total cell lysates accepted 10 min centrifugation at 10000 xg, at 4°C. Then the supernatants received 30 min incubation using 20ul protein A/G PLUS- agarose (Santa Cruz sc-2003) at 4°C. Pellet beads received 5 min centrifugation at 2,500 rpm, at 4°C. 10ul of primary antibody was used to incubate the supernatants for 60min. The complexes were then precipitated through the addition of 20ul protein A/G PLUS- agarose into the lysate, followed by being incubated at 4°C for overnight. The 30 s centrifugation on beads was performed at 2,500 rpm, at 4°C, then ice-cold RIPA buffer was employed to wash the beads four times. The samples were then suspended and denatured in SDS sample buffer (which contained 100 mM dithiothreitol, 10% glycerol, 50 mM Tris pH 6.8, 2% SDS, and 0.01% bromophenol blue).
Recombinant adenovirus vector to silence CD9 expression
The recombinant adenovirus vector to silence CD9 expression (CD9-shRNA-GFP) and the negative-control adenovirus vector which contained non-specific shRNA (Vector) were provided by Shanghai GeneChem, Co. Ltd. Before the further experiments, keratinocytes received infection with CD9-shRNA or mock vector at a multiplicity of infection (MOI, known as infection index) of 10 for 48 hours.
Recombinant adenovirus vector for CD9 overexpression
Ad-CD9-GFP and CD9 mock vector were purchased by Shanghai GeneChem, Co. Ltd. The GFP sequences were encoded by vectors as a marker gene. Western blotting was employed to test the transgene expression of all recombinant adenoviruses in keratinocytes. Before the further experiments, keratinocytes received infection with Ad-CD9 or a negative vector at a multiplicity of infection (MOI, known as infection index) of 10 for 48 hours.
siRNA knockdown of ADAM17 and pharmacological reagents
On-target siRNA specific to ADAM17 (GenePharma, China) was used to carry out siRNA knockdown on cells under culture. Briefly, penicillin- and streptomycin-free medium, which contained 0.1 μM ADAM17 or non-targeting pool (NTP) siRNA, were used to incubate the keratinocytes for 24 h. Western blot was employed to evaluate ADAM17 expression. Keratinocytes were treated with ADAM17 inhibitor TAPI-2(Sigma CAS 187034-31-7, USA) at final concentration 40 μM and recombinant human heparin-binding EGF (Abcam ab167830, UK) at final concentration 50 mg.
ADAM17 Activity Assay
As above mentioned, the fluorimetric SensoLyte 520 TACE/ ADAM17 (α-Secretase) Aactivity Aassay Kkit (AnaSpec AS-72085, USA) was adopted to determine the ADAM17 shedding activity according to the protocol of manufacturer [24], which applies a FRET peptide substrate to continuously measuring the ADAM17 enzyme activity. After using active enzyme for FRET peptide cleavage, the complete FRET peptide 5-carboxyfluorescein normally quenched via the QXL 520 saw fluorescence recovery, followed by being monitored at the excitation/emission of 490/520 nm. During assay, trypan blue as well as a hemocytometer were used to count the number of viable cells as above mentioned.
Scratch wounding assay of keratinocyte
Scratch wounding assay, is an in vitro incisional model and was carried out, as previously described [25]. Six-well plates (BD Biosciences, USA) were incubated 24 hours in RPMI 1640 medium containing 40μg/ml human fibronectin (PROSPEC, Tany TechnoGene Ltd. ,USA). Cells gradually covered in the plates coated with fibronectin in medium RPMI 1640 (serum conditioned). A sterile p10 pipette tip was used to create scratch wounds were created in confluent monolayers. For standardizing quantitative analysis, three perpendicular marks were made on each scratch on the outside surface of the well. We first washed the suspended cells for three times, and then put the wounded monolayers in RPMI 1640 medium for culture. Following 24h incubation, phase-contrast microscopy (OLYMPUS, Japan) was employed to observe repopulation of the wounded areas. The NIH ImageJ image processing program was used to confirm the scale of the denuded area at each time point from the digital images. Keratinocytes were treated with ADAM17 inhibitor TAPI-2 or siRNA knockdown of ADAM17 to clear the dependency of migration on ADAM17. Anti-AREG and anti-HB-EGF as neutralizing antibodies, were used to remove the EGF family growth factors from the supernatant.
Cell motility assay
Cell motility was analyzed as previously reported [26]. Cells were seeded at a density of 0.5 × 104/ml. Twenty-four-well plates (BD Biosciences, USA) received overnight incubation in 0.6 mL RPMI 1640 media which contained 10% FBS. A Zeiss imaging system (Carl Zeiss Meditec, Jena, Germany) equipped with a CO2- controlled and temperature-controlled chamber was used to carry out the time-lapse imaging and acquired the image each five minutes. NIH ImageJ software was used to analyze the time-lapse image. Anti-AREG (Proteintech 66433-1-lg, USA) and anti-HB-EGF (Abcam ab89241, UK) as neutralizing antibodies, were used to remove the EGF family growth factors from the supernatant.
ELISA
For evaluating the production of ADAM17 substrates, the keratinocytes of scratch wound assay received 24h incubation in medium which did not contain any exogenous EGFR ligands, and were then collected following the process above mentioned [17]. Relevant DuoSet ELISA kits (Cloud-clone Corp, AREG SEA006Hu, HB-EGF SEB479Hu, TGF-α SEA123Hu, China) were used to analyze the collected medium specimens regarding the proteins. Supernatants were collected in triplicate for each cell line, and results from the vector and CD9-silenced keratinocytes treated with ADAM17 inhibitor TAPI-2 (Sigma, USA) at final concentration 40 μM.
Western blot analysis
Ice-cold PBS was used to wash the cells which were then harvested in 100-200 μL lysis buffer, followed by being homogenized. 15 minutes centrifugation at 14000 rpm at 4°C was performed to separate lysates.BCA protein assay kit (Sigma, USA) was used to determine the concentration of protein. The lysates which contained 10 or 20 μg proteins were separated on 10% SDS-PAGE gel, followed by being transferred to polyvinylidene difluoride (PVDF) membranes in an electrophoretic manner. The primary antibodies were used at 1:1000 dilution, the loading control anti-GAPDH was used at 1:5000 dilution, and the secondary antibody was used at 1:4000 dilution. The blots were probed using primary antibodies: anti-GAPDH (Proteintech HRP-60004, USA), anti-EGFR (Abcam ab52894, UK) and anti-p-EGFR (Abcam ab24928, UK), anti-CD9, anti-ADAM17, anti-JNK (Abcam ab176662, UK), anti-p-JNK (Abcam ab207477, UK), anti-ERK(Abcam ab54230, UK) and anti-p-ERK (Abcam ab192591, UK), anti-p-P38 (Cell Signaling #4511, USA) and anti-P38 (Cell Signaling #8690, USA). Horseradish peroxidase-conjugated GAPDH were used as loading control. An improved chemiluminescence detection kit (Amersham Pharmacia) was used to image the immunocomplexes on ChemiDoc imaging system (Bio-Rad, USA). Quantity One 4.1 software (Bio-Rad, USA) was adopted to quantify the images.
Statistical analysis
Data obtained in the study were represented as the mean ± standard error of mean (SEM).The differences between various groups was measured by two-tailed Student's t-tests. Comparison in different groups was performed by carrying out the Tukey's HSD test or Dunnett's test. P < 0.05 was considered statistically significant.
Results
1. CD9 and ADAM17 were associated on epidermal cells
For evaluating the possible connection between ADAM17 and CD9 on HaCaT cell lines and C57-MKs, double immunofluorescence staining was performed on these molecules before the confocal microscopy analysis. Although CD9 and ADAM17 were expressed both on the cell surface and in the cytoplasm, co-localization of ADAM17 and CD9 was particularly evident on the cell surface (Fig. 1A).
Co-localization and association of ADAM17 and CD9 on keratinocytes. (A) Immunofluorescence staining of ADAM17 and CD9 on keratinocytes in vitro and in vivo; (B) Immunoprecipitation analysis of the association between ADAM17 and CD9 in HaCaT cells and C57-MKs. Bar = 50μm.
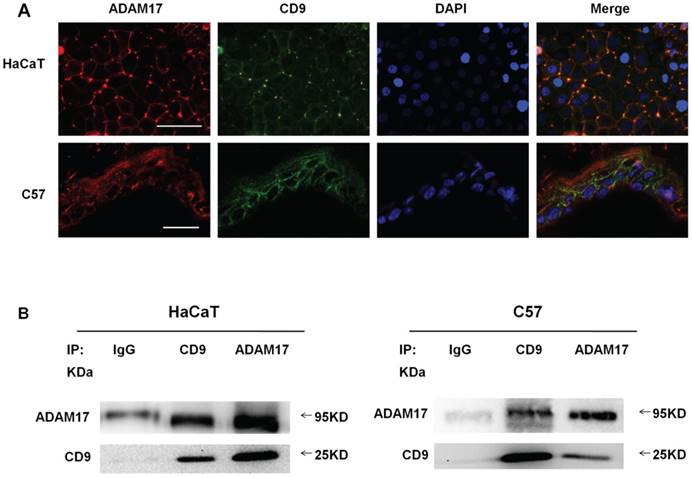
The co-localization in optical microscopy indicated that these molecules were associated with each other on the cell surface, IP approach was applied to further proving the certain interaction between CD9 and ADAM17. As shown in Fig.1B, CD9 immunoprecipitates of both cell types saw bands which correspond to the mature form of ADAM17, suggesting that these two proteins exhibited a cross-linking on the cell surface. These results suggest that CD9 and ADAM17 physical association and direct functional linkage on the cell membrane.
2. CD9 negatively regulated the shedding activity of ADAM17 in keratinocytes
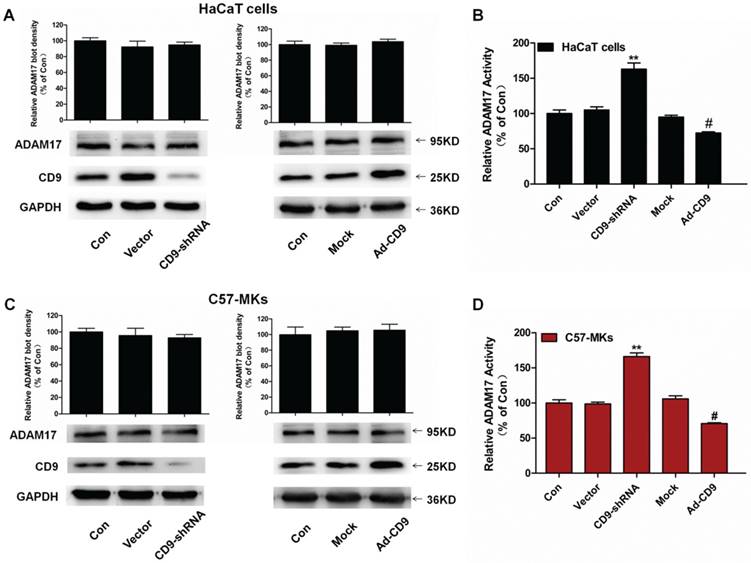
To assess the role of CD9 in ADAM17 maturation and activation in keratinocytes, we constructed recombinant lentivirus vectors to silence CD9 (CD9-shRNA) and overexpress CD9 (Ad-CD9) by infecting HaCaT cells and C57-MKs, and then carried out the Western blotting on CD9 and ADAM17. The enzyme activity of ADAM17 was assessed using a TACE protease activity kit. As shown in Fig. 2A and 2C, the mature form of ADAM17 was not influenced by down-regulation of CD9 or over-expression of CD9 compared with the vector or mock group (p>0.05). However, compared with the vector group, CD9-silenced keratinocytes caused significant increases of ADAM17 sheddase activity, 58% in HaCaT cells, and 67% in C57-MKs (Fig. 2B and 2D). Compared with the mock group, CD9-overexpression in keratinocytes caused a significant decrease of ADAM17 sheddase activity, decreasing 22.6% in HaCaT cells, and 35% in C57-MKs (Fig. 2B and 2D). These date suggest that CD9 associates with ADAM17 via negatively regulating the shedding activity of ADAM17 in keratinocytes.
The expression or enzyme activity of ADAM17 in CD9 down-regulation or over-expression keratinocytes. (A) Western blots showing expression of ADAM17 in CD9 silence or over-expression HaCaT cells; (B) TACE activity assay kit measurement of the effect of CD9 on ADAM17 enzyme activity in HaCaT cells; (C) Western blots showing the expression of ADAM17 in CD9 silence or over-expression C57-MKs; (D) TACE activity assay kit measuring the effect of CD9 on ADAM17 enzyme activity in C57-MKs. Data were obtained from at least three independent experiments and shown as the mean ± SEM. **P< 0.01 vs. Vector group. #P< 0.01 vs. Mock group. Bar = 50μm.
3. ADAM17 involvement in keratinocyte migration regulated by CD9
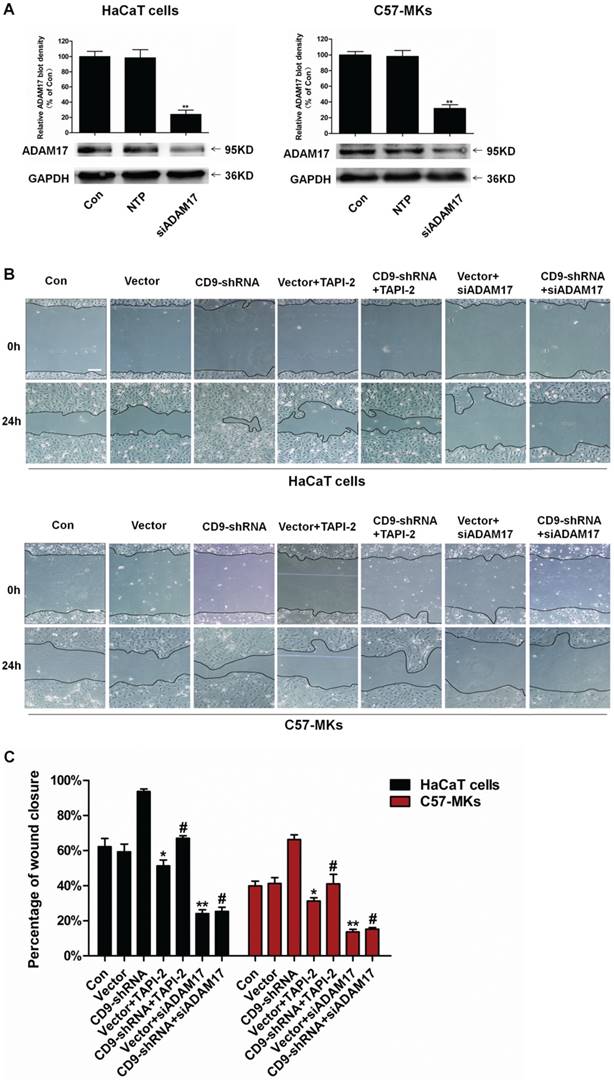
Our results suggested that CD9-regulated ADAM17 activity in keratinocytes. Then whether ADAM17 participated in the keratinocyte migration regulated by CD9 was determined. A cell scratch wound assay was used to evaluate keratinocyte migration taking the ADAM17 inhibitor TAPI-2 and siADAM17 as siRNA-mediated knockdown of ADAM17. As shown in Fig. 3A and 3B, untreated HaCaT cells or C57-MKs failed to heal wound after 24 h; however, CD9-silencing caused almost complete wound closure in HaCaT cells and 27% growth of wound closure in C57-MKs. Both TAPI-2 treatment and si-ADAM17 transfection significantly impaired keratinocyte migration (Fig. 3B). After TAPI-2 treatment, area of wound closure was reduced 27% in CD9-silenced HaCat cells, and 25% in CD9-silenced C57-MKs (Fig. 3B-C). Furthermore, down-regulation of CD9 promoted migration of HaCaT cells and C57-MKs could be blocked by si-ADAM17. After si-ADAM17 transfection, the area of wound closure was reduced 3.7-fold in CD9-silenced HaCat cells and 4.3-fold in CD9-silenced MKs (Fig. 3 B-C). These results suggest that ADAM17 plays a key role in CD9-regulated keratinocyte migration.
Involvement of ADAM17 in keratinocyte migration regulated by CD9. (A) Expression of ADAM17 is shown in HaCaT cells and C57-MKs after being transfected with NTP siRNA, or siRNA against either ADAM17; (B) The ADAM17 inhibitor- TAPI-2 or si-ADAM17 effect on the wound closure in CD9-silenced keratinocytes; (C) Quantification analysis the diminution of the wound closure area over time with Image J software. Data were obtained from at least three independent experiments and shown as the mean ± SEM. *, P<0.05; **P< 0.01 vs. Vector group. #P< 0.01 vs. CD9-shRNA group. Bar = 50μm.
4. CD9-regulated keratinocyte motility dependence on ADAM17
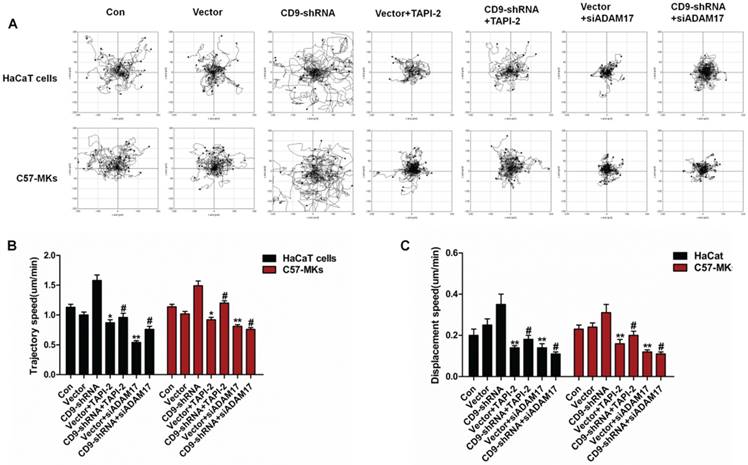
An in vitro cell motility assay was then performed to further confirm the regulatory role of ADAM17 in CD9-regulated keratinocyte motility. Notably, the ranges of cell migration and motility speeds were enhanced by CD9 down-regulation in HaCaT cells and C57-MKs (Fig. 4A-C). However, the improvement in cell motility by CD9-silenced was suppressed by TAPI-2 treatment, and was abolished by siADAM17 transfection (Fig. 4A). As shown in Fig. 4B, after TAPI-2 treatment, the trajectory speed of keratinocytes was reduced 1.7-fold in CD9-silenced HaCat cells, and 1.3-fold in CD9-silenced-MKs. The decreased amplitude of trajectory speed was 2.1-fold in CD9-silenced HaCat cells, and 2.0-fold in CD9-silenced-MKs after si-ADAM17 transfection. The displacement speed was analyzed to further confirm the effect of ADAM17 in CD9-regulated keratinocyte motility (Fig. 4C). Hence, these results suggest that ADAM17 plays a pivotal role in CD9-regulated keratinocyte motility.
CD9 regulates keratinocyte motility depends on ADAM17. (A) The effect of ADAM17 inhibitor-TAPI-2 or si-ADAM17 on cell motility trajectories in CD9-silenced keratinocytes; (B) Analysis of the trajectory speed of keratinocyte migration; (C) Analysis of the displacement speed of keratinocyte migration. Data were obtained from at least three independent experiments and shown as the mean ± SEM. *, P<0.05; **P< 0.01 vs. Vector group. #P< 0.01 vs. CD9-shRNA group. Bar = 50μm.
5. Down-regulation of CD9 drove shedding of AREG and HB-EGF via activation of ADAM17 shedding enzyme
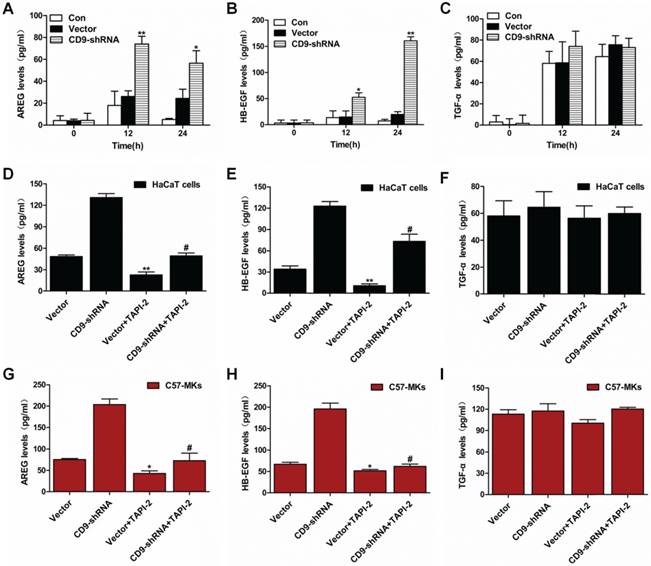
In the foregoing scratch assay, CD9-silenced keratinocytes presented a more obvious migration compared with the controls under no exogenous EGF (Fig. 4B). It is not clear which EGF molecules participated in keratinocyte migration regulated by CD9. Consequently, ADAM17's substrates were examined in term of shedding, including amphiregulin (AREG), HB-EGF and TGF-α, during the period that cells were cultured without exogenous EGF. At 12h of scratch assay, AREG and HB-EGF exhibited an obvious higher shedding in CD9-silenced keratinocytes than in controls (Fig. 5A-C). At 24h of the scratch assay, shedding of AREG in CD9-silenced keratinocytes fluctuated slight, but was significantly higher than of control. Shedding of HB-EGF in CD9-silenced keratinocytes showed a sharp increase that was significantly higher than that of control (Fig. 5A-B). Shedding of TGF-α increasing in CD9-silenced keratinocytes, but there was no significant difference compared with control at process of scratch assay (Fig. 5C). For AREG and HB-EGF, the elevated shedding significantly reduced by ADAM17 inhibitors-TAPI-2 in CD9-silenced keratinocytes (HaCaT cells and C57-MKs) (Fig. 5D-I). These results suggest that shedding of AREG and HB-EGF is promoted by down-regulation of CD9, dependent on activation of ADAM17 shedding enzyme.
Shedding of ADAM17 substrates in keratinocytes. (A) The shedding of ADAM17 substrates include AREG, HB-EGF as well as TGF-α in a scratch assay of 24h in CD9-silenced keratinocytes; (B-C) This increased shedding was significantly reduced by ADAM17 inhibitor-TAPI-2 in HaCaT cells(B) and C57-MKs(C). Data were obtained from at least three independent experiments and shown as the mean ± SEM. *, P<0.05; **P< 0.01 vs. Vector group. #P< 0.01 vs. CD9-shRNA group. Bar = 50μm.
6. Neutralizing anti-HB-EGF mAbs inhibited the migration of CD9-silenced keratinocytes
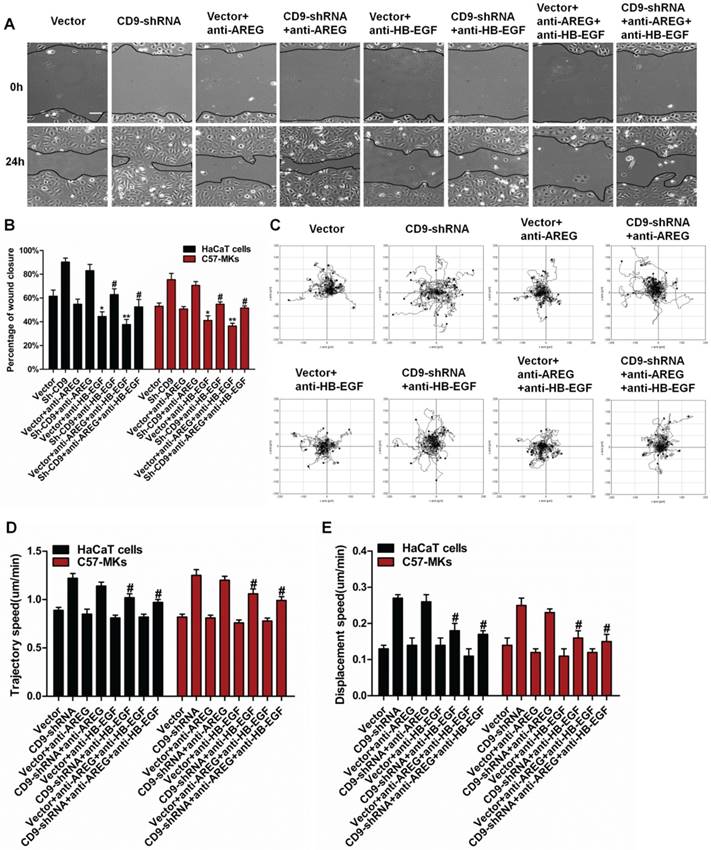
Scratch wound assays were carried out on keratinocyte monolayers with or without EGFR neutralizing antibody (Ab), aiming at making sure how AREG and HB-EGF greatly affect the keratinocyte migration. As shown in Figure 6A, the presence of anti-HB-EGF and anti-AREG helped to reduce the scratch wound-induced migration. However, incubation of keratinocytes with anti-AREG, had only modest effects on area of wound closure, it was reduced 7.3% in CD9-silenced HaCaT cells, and 4.9% in CD9-silenced C57-MKs (Fig. 6B) (p>0.05). Anti-HB-EGF displayed stronger inhibition of keratinocyte migration, area of wound closure was reduced 27.4% in CD9-silenced HaCat cells (p<0.01), and 21% in CD9-silenced C57-MKs (Fig. 6B) (p<0.01). The strongest inhibition of keratinocyte migration in cell scratch wound assay occurred with a cocktail of two neutralizing Abs (AREG and HB-EGF), area of wound closure was reduced 37.7% in CD9-silenced HaCat cells (p<0.01), and 23.9% in CD9-silenced C57-MKs (Fig. 6B) (p<0.01). Both AREG and HB-EGF were massively released in CD9-silenced keratinocytes, yet neutralizing antibodies against HB-EGF were more effective in blocking keratinocyte migration than were AREG mAbs. The analysis of motility was used to further confirm the effect of HB-EGF in CD9-regulated keratinocyte motility. The cell motility assay showed that neutralizing anti-HB-EGF mAbs significantly suppressed the motility speed of CD9-silenced keratinocytes (Fig. 6C-E). No differences in the range of cell migration and motility speeds were detected in the presence of neutralizing anti-AREG mAbs (Fig. 6C-E). These results suggest that the release of HB-EGF depending on ADAM17 plays a pivotal role in CD9-regulated keratinocyte migration.
The effect of AREG and HB-EGF on keratinocyte migration and motility regulated by CD9. (A) The effects of AREG or HB-EGF on wound closure in CD9-silenced keratinocytes; (B) Quantification analysis of the diminution of wound closure area over time using Image J software; (C) The effect of AREG or HB-EGF on cell motility trajectories in CD9-silenced keratinocytes. (D) Analysis of the trajectory speed of keratinocyte migration; (E) Analysis of the displacement speed of keratinocyte migration. Data were obtained from at least three independent experiments and shown as the mean ± SEM. *, P<0.05; **P< 0.01 vs. Vector group. #P< 0.01 vs. CD9-shRNA group. Bar = 50μm.
7. HB-EGF promoted keratinocyte migration via activation of EGFR/ERK
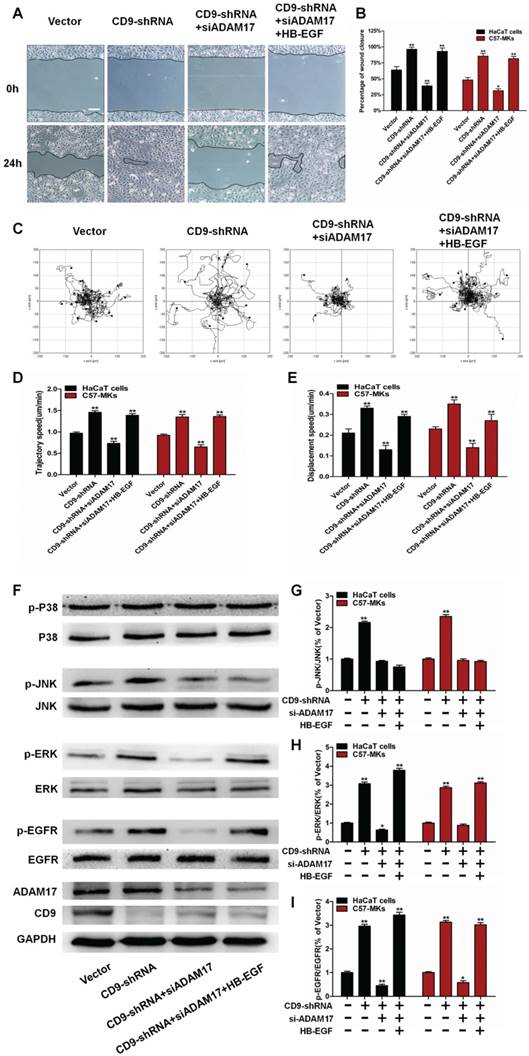
To make an evaluation on the functional correlation of HB-EGF-dependent EGFR/MAPK signaling in CD9-regulated keratinocyte migration, we performed in vitro scratch wounding assay of HaCaT cells or C57-MKs in the presence or absence of siADAM7 or recombinant HB-EGF. CD9-stimulated migration of keratinocytes was blocked by si-ADAM17 transfection, also resulting in decreased motility of CD9-silenced keratinocytes (Fig. 7A-C). The inhibition of CD9-regulated keratinocyte migration by siADAM17 was rescued by addition of recombinant HB-EGF, including wound healing and keratinocyte motility (Fig. 7A-E). These results suggest that HB-EGF is indispensable to CD9-regulated keratinocyte migration. A Western blot analysis of similarly treated cultures demonstrated that down-regulation of CD9 increased phosphorylation of EGFR, ERK and JNK in keratinocytes, this was prevented by si-ADAM17 transfection (Fig. 7F-I). Importantly, adding the recombinant HB-EGF to cultures treated with si-ADAM17 of CD9-silenced keratinocytes greatly stimulated the EGFR/ERK phosphorylation (Fig. 7F-I). As HB-EGF shows requirement for CD9-regulated migration of keratinocytes, HB-EGF is more likely to be a key intermediate in the EGFR/ERK signaling pathway.
The role of HB-EGF-dependent EGFR/MAPK signaling in CD9-regulated keratinocyte migration. (A) The effect of recombinant HB-EGF on the wound closure in CD9-silenced keratinocytes treated with siADAM17; (B) Quantification analysis on the diminution of the wound closure area over time with Image J software; (C) The effect of recombinant HB-EGF on cell motility trajectories in CD9-silenced keratinocytes treated with siADAM17. (D) Analysis of the trajectory speed of keratinocyte migration; (E) Analysis of the displacement speed of keratinocyte migration. (F-I) Representative Western blot results showing the effect of recombinant on phosphorylation of EGFR/ERK in CD9-silenced keratinocytes treated with siADAM17 (F), with no alteration to JNK pathway(G), increased phosphorylation of the EGFR(H) and ERK pathway(I). Data were obtained from at least three independent experiments and shown as the mean ± SEM. *, P<0.05; **P< 0.01 vs. Vector group. Bar = 50μm.
Discussion
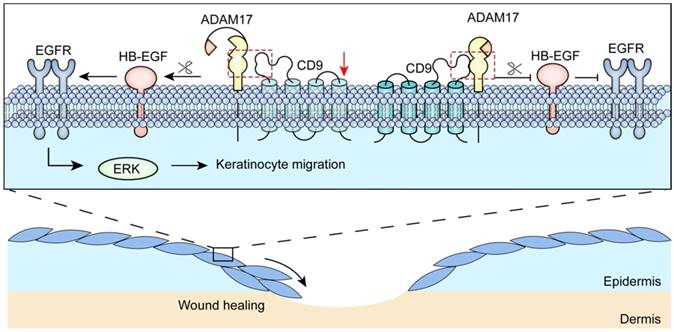
Several studies have shown that tetraspanin CD9 form complexes by interacting with other transmembrane molecules, that are required for cell migration and adhesion [27, 28]. Nevertheless, the mechanisms of regulation of interaction between CD9 and transmembrane molecules in keratinocyte migration remain largely unknown. In this study, we demonstrated that CD9 interacted primarily with the mature form (i.e. catalytically active) of ADAM17 in migratory keratinocytes, localizing the major site of ADAM17 activity at the plasma membrane. Furthermore, we found that shedding of HB-EGF depended on ADAM17 sheddase activity; that was regulated by CD9 down-regulation, promoting keratinocyte migration and wound healing via the EGFR/ERK signaling pathway (Fig. 8).
Schematic illustrating that CD9/ADAM17 axis plays a key role in keratinocyte migration via activation of EGFR/ERK signaling pathway.
Tetraspanins can be combined with integrins, signaling receptors as well as ADAMs, thereby generating microdomains on the cell surface enriched with tetraspanin, which serves as one of its unique feature [29]. The association of CD9 with specific integrins, including α2β1, α3β1 and α6β4, are implicated in development, cell adhesion, migration [30]. However, CD9 forms a complex with α2β1 or α3β1 and does not participate in keratinocyte migration [11, 31]. Indeed, CD9 association with α6β4 is involved in keratinocyte motility, whereas CD9 does not co-localize with α6β4 in vivo [32]. Therefore, CD9 may regulate keratinocyte migration by interacting with other non-integrins transmembrane molecules. Importantly, ADAM17 is a key factor for defensing against injury, including by regulating keratinocyte migration and promoting cutaneous wound re-epithelialization [33]. Although ADAM17 has significant biological function, the interaction between CD9 and ADAM17 has not be elucidated in migratory keratinocytes. Different approaches have been adopted, such as co-localization and co-immunoprecipitation, to demonstrate the emergence of these complexes. In our studies, important clues to ADAM17 regulation by CD9 were the findings that ADAM17 co-immunoprecipitated with CD9 in HaCaT cell lines and C57-MKs. The association of CD9 with ADAM17 was also observed in cell types, include monocytic and endothelial cells [34]. To our knowledge, this study was the first to report an interaction between CD9 and ADAM17 in keratinocytes. Furthermore, the expression of CD9 and ADAM17 could be observed on the cell surface as well as in the cytoplasm. Interestingly, cell-cell contact areas saw a particularly obvious co-localization of ADAM17 and CD9. The subcellular localization of CD9 and ADAM17 complexes is intimately involved in ADAM17-regulatory function [34, 35]. According to these studies, there is physical association as well as direct functional linkage between CD9 and ADAM17 on the cell membrane of keratinocytes.
It is well established that most tetraspanins participate in ADAMs maturation, transport, localization or activation [7]. For example, the maturation process and surface expression of ADAM10 were significantly changed due to over-expression or siRNA-mediated knockdown of tetraspanin15, regulating the activity of ADAM10 in HeLa cells [36]. In human umbilical vein endothelial cells, knockdown of tetraspanin14 significantly reduces ADAM10 surface expression and activity [37]. However, another special case about the regulatory mechanisms is that ADAM17 association with CD9 to modulate sheddase activity of ADAM17. Our experiments, using over-expression and down-regulation of CD9 in HaCaT cell lines and C57-MKs, found a negative influence of CD9 on ADAM17 sheddase activity, whereas CD9 did not affect the expression and maturation of ADAM17. Several lines of evidence suggest that the conformational changes between tetraspanins and ADAMs are responsible for ADAMs sheddase activity [7, 38]. Tetraspanins-ADAMs interactions are greatly mediated by major extracellular region of the tetraspanins [35]. In fact, the large extracellular loop (LEL) of CD9 is imposed to ADAM17, which inhibits the proteolytic activity of ADAM17 in endothelial cells [34, 39, 40]. ADAM17 sheddase activity release from the inhibitory effect conferred through CD9-LEL would prove that CD9 can negatively affect the ADAM17 activity in keratinocytes. However, some questions regarding the exact regions between ADAM17 and CD9-LEL required to mediate ADAM17 sheddase activity have yet to be answered. It is therefore possible that the conformational change between CD9 and ADAM17 is emerging as a key regulatory mechanism of ADAM17 sheddase activity in keratinocytes.
In various cell tapes, the sheddase activity of ADAM17 modulates several cellular processes, including cell invasion, motility, and migration [41, 42]. It was previously demonstrated that ADAM17 contributed to tumor cell migration and invasion in vitro [43, 44]. Moreover, Huang et al. reported that knockdown of ADAM17 significant inhibited the migration of squamous cell carcinoma [41]. The work has proven that ADAM17 inhibitor-TAPI-2 and ADAM17 siRNA can block the keratinocyte migration as well as motility induced by CD9 silencing, suggesting that ADAM17 sheddase activity can greatly affect the CD9-regulated keratinocyte migration. ADAM17 acting as an ectodomain sheddase appears to have many substrates, activated by various stimuli to activate the shedding and release of the EGFR ligands, including AREG, HB-EGF, and TGF-α [7, 33]. In the course of wound healing, shedding of EGFR ligands mediated by ADAM can crucially affect the cell proliferation and migration [18, 45]. In particular, the release level of AREG and HB-EGF were upregulated in CD9-silenced keratinocytes. To determine which EGF molecules participated in the CD9-regulated migration in keratinocytes, neutralizing antibodies were used to block AREG or HB-EGF released from supernatant in a wound closure assay, and found that the inhibiting effect of anti-HB-EGF and anti-AREG antibody on the CD9 silencing-induced migration reached 45% and 9%, respectively. Which is in line with another study, where AREG is an effective growth promoting factor for human keratinocytes [46]. Importantly, HB-EGF provides a significant contribution in human keratinocytes migration and wound re-epithelialization [47]. Further studies found that the inhibition of CD9-regulated keratinocyte migration by siADAM17 could be rescued by addition of recombinant HB-EGF. These results suggested that in comparison with other factors, the HB-EGF could remarkably promote CD9-silenced keratinocytes regarding the migratory activity. Moreover, adding the recombinant HB-EGF to cultures treated with siADAM17 of CD9-silenced keratinocytes greatly stimulated the EGFR/ERK phosphorylation. Collectively, according to these results, shedding of HB-EGF is decided by the ADAM17 sheddase activity; that is itself regulated by CD9 down-regulation, emerging as a central link in regulating keratinocyte migration via activation of EGFR/ERK.
In conclusion, our findings demonstrate that down-regulation of CD9 activates the ADAM17 sheddase activity in keratinocytes, which is critical for cell migration and EGFR ligands shedding (Fig. 8). Moreover, CD9 associates with of ADAM17 (i.e. catalytically active) on the surface of keratinocytes, which negatively regulated sheddase activity of ADAM17. Importantly, CD9/ADAM17 axis plays a key role in keratinocyte migration via activation of EGFR/ERK signaling pathway. On that account, activating the pathway is likely to be an attractive target to improve the acute and chronic cutaneous wound healing.
Acknowledgements
This work was supported by National Nature Science Foundation of China (NSFC No. 81571900 and No. 81601683), Science Foundation for The Excellent Youth Scholars of Third Military Medical University (2017MPRC-05) and Innovation Program of Southwest Hospital (SWH2016JSYB-02).
Author Contributions
J.P.Z. developed the initial concept and supervised the study; J.P.Z. and J.L. designed the experiments; J.L., G.Q.Z., N.X.J and Y.W. performed the experiments and analyzed the data; M.F.Y., X.P.J. and Y.S.H. contributed to the reagents, materials, and analysis tools. J.L. and J.P.Z. co-wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Park S, Gonzalez DG, Guirao B, Boucher JD, Cockburn K, Marsh ED. et al. Tissue-scale coordination of cellular behaviour promotes epidermal wound repair in live mice (vol 19, pg 155, 2017). Nature cell biology. 2017;19:407 -
2. Shaw TJ, Martin P. Wound repair at a glance. Journal of cell science. 2009;122:3209-13
3. Zhang JP, Dong JD, Gu H, Yu S, Zhang XH, Gou YL. et al. CD9 Is Critical for Cutaneous Wound Healing through JNK Signaling. J Invest Dermatol. 2012;132:226-36
4. Jiang XP, Zhang DX, Teng M, Zhang Q, Zhang JP, Huang YS. Downregulation of CD9 in keratinocyte contributes to cell migration via upregulation of matrix metalloproteinase-9. PLoS One. 2013;8:e77806
5. Powner D, Kopp PM, Monkley SJ, Critchley DR, Berditchevski F. Tetraspanin CD9 in cell migration. Biochemical Society transactions. 2011;39:563-7
6. Penas PF, Garcia-Diez A, Sanchez-Madrid F, Yanez-Mo M. Tetraspanins are localized at motility-related structures and involved in normal human keratinocyte wound healing migration. J Invest Dermatol. 2000;114:1126-35
7. Matthews AL, Noy PJ, Reyat JS, Tomlinson MG. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets. 2017;28:333-41
8. Charrin S, Jouannet S, Boucheix C, Rubinstein E. Tetraspanins at a glance. Journal of cell science. 2014;127:3641-8
9. Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nature reviews Cancer. 2014;14:49-60
10. Jiang X, Teng M, Guo X, Zhang D, Zhang Q, Zhang J. et al. Switch from alphavbeta5 to alphavbeta6 integrin is required for CD9-regulated keratinocyte migration and MMP-9 activation. FEBS letters. 2014;588:4044-52
11. Grenache DG, Zhang Z, Wells LE, Santoro SA, Davidson JM, Zutter MM. Wound healing in the alpha2beta1 integrin-deficient mouse: altered keratinocyte biology and dysregulated matrix metalloproteinase expression. J Invest Dermatol. 2007;127:455-66
12. Furuya M, Kato H, Nishimura N, Ishiwata I, Ikeda H, Ito R. et al. Down-regulation of CD9 in human ovarian carcinoma cell might contribute to peritoneal dissemination: morphologic alteration and reduced expression of beta1 integrin subsets. Cancer research. 2005;65:2617-25
13. Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13:649-65
14. Klein T, Bischoff R. Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: biological function and structure. Journal of proteome research. 2011;10:17-33
15. Franzke CW, Cobzaru C, Triantafyllopoulou A, Loffek S, Horiuchi K, Threadgill DW. et al. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation (vol 209, pg 1105, 2012). J Exp Med. 2012;209:2517 -
16. Shimoda M, Horiuchi K, Sasaki A, Tsukamoto T, Okabayashi K, Hasegawa H. et al. Epithelial Cell-Derived a Disintegrin and Metalloproteinase-17 Confers Resistance to Colonic Inflammation Through EGFR Activation. EBioMedicine. 2016;5:114-24
17. Brooke MA, Etheridge SL, Kaplan N, Simpson C, O'Toole EA, Ishida-Yamamoto A. et al. iRHOM2-dependent regulation of ADAM17 in cutaneous disease and epidermal barrier function. Human molecular genetics. 2014;23:4064-76
18. Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2008;16:585-601
19. Cha D, O'Brien P, O'Toole EA, Woodley DT, Hudson LG. Enhanced modulation of keratinocyte motility by transforming growth factor-alpha (TGF-alpha) relative to epidermal growth factor (EGF). J Invest Dermatol. 1996;106:590-7
20. Tolino MA, Block ER, Klarlund JK. Brief treatment with heparin-binding EGF-like growth factor, but not with EGF, is sufficient to accelerate epithelial wound healing. Biochimica et biophysica acta. 2011;1810:875-8
21. Shirakata Y, Kimura R, Nanba D, Iwamoto R, Tokumaru S, Morimoto C. et al. Heparin-binding EGF-like growth factor accelerates keratinocyte migration and skin wound healing. Journal of cell science. 2005;118:2363-70
22. Zuidscherwoude M, Gottfert F, Dunlock VM, Figdor CG, van den Bogaart G, van Spriel AB. The tetraspanin web revisited by super-resolution microscopy. Scientific reports. 2015;5:12201
23. Jiang X, Guo X, Xu X, Teng M, Huang C, Zhang D. et al. Hypoxia regulates CD9-mediated keratinocyte migration via the P38/MAPK pathway. Scientific reports. 2014;4:6304
24. Ma X, Xu D, Ai Y, Zhao S, Zhang L, Ming G. et al. Angiotensin-(1-7)/Mas Signaling Inhibits Lipopolysaccharide-Induced ADAM17 Shedding Activity and Apoptosis in Alveolar Epithelial Cells. Pharmacology. 2016;97:63-71
25. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nature protocols. 2007;2:329-33
26. Dang I, Gautreau A. Random Migration Assays of Mammalian Cells and Quantitative Analyses of Single Cell Trajectories. Methods in molecular biology. 2018;1749:1-9
27. Hemler ME. Tetraspanin functions and associated microdomains. Nature reviews Molecular cell biology. 2005;6:801-11
28. Kotha J, Longhurst C, Appling W, Jennings LK. Tetraspanin CD9 regulates beta 1 integrin activation and enhances cell motility to fibronectin via a PI-3 kinase-dependent pathway. Experimental cell research. 2008;314:1811-22
29. Hemler ME. Targeting of tetraspanin proteins-potential benefits and strategies. Nature reviews Drug discovery. 2008;7:747-58
30. Berditchevski F. Complexes of tetraspanins with integrins: more than meets the eye. Journal of cell science. 2001;114:4143-51
31. Berditchevski F. Complexes of tetraspanins with integrins: more than meets the eye. Journal of cell science. 2001;114:4143-51
32. Baudoux B, Castanares-Zapatero D, Leclercq-Smekens M, Berna N, Poumay Y. The tetraspanin CD9 associates with the integrin alpha6beta4 in cultured human epidermal keratinocytes and is involved in cell motility. European journal of cell biology. 2000;79:41-51
33. Le Gall SM, Maretzky T, Issuree PD, Niu XD, Reiss K, Saftig P. et al. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. Journal of cell science. 2010;123:3913-22
34. Gutierrez-Lopez MD, Gilsanz A, Yanez-Mo M, Ovalle S, Lafuente EM, Dominguez C. et al. The sheddase activity of ADAM17/TACE is regulated by the tetraspanin CD9. Cellular and molecular life sciences: CMLS. 2011;68:3275-92
35. Noy PJ, Yang J, Reyat JS, Matthews AL, Charlton AE, Furmston J. et al. TspanC8 Tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) Interact via Their Extracellular Regions: EVIDENCE FOR DISTINCT BINDING MECHANISMS FOR DIFFERENT TspanC8 PROTEINS. The Journal of biological chemistry. 2016;291:3145-57
36. Prox J, Willenbrock M, Weber S, Lehmann T, Schmidt-Arras D, Schwanbeck R. et al. Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cellular and molecular life sciences: CMLS. 2012;69:2919-32
37. Haining EJ, Yang J, Bailey RL, Khan K, Collier R, Tsai S. et al. The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. The Journal of biological chemistry. 2012;287:39753-65
38. Noy PJ, Yang J, Reyat JS, Matthews AL, Charlton AE, Furmston J. et al. TspanC8 Tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) Interact via Their Extracellular Regions: EVIDENCE FOR DISTINCT BINDING MECHANISMS FOR DIFFERENT TspanC8 PROTEINS. The Journal of biological chemistry. 2016;291:3145-57
39. Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nature reviews Cancer. 2014;14:49-60
40. Hemler ME. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu Rev Cell Dev Biol. 2003;19:397-422
41. Huang Y, Benaich N, Tape C, Kwok HF, Murphy G. Targeting the sheddase activity of ADAM17 by an anti-ADAM17 antibody D1(A12) inhibits head and neck squamous cell carcinoma cell proliferation and motility via blockage of bradykinin induced HERs transactivation. International journal of biological sciences. 2014;10:702-14
42. Rossello A, Nuti E, Ferrini S, Fabbi M. Targeting ADAM17 Sheddase Activity in Cancer. Current drug targets. 2016;17:1908-27
43. Kondo N, Ishiguro Y, Kimura M, Sano D, Fujita K, Sakakibara A. et al. Antitumor effect of gefitinib on head and neck squamous cell carcinoma enhanced by trastuzumab. Oncology reports. 2008;20:373-8
44. Zheng X, Jiang F, Katakowski M, Zhang ZG, Lu Q-e, Chopp M. ADAM17 promotes breast cancer cell malignant phenotype through EGFR-PI3K-AKT activation. Cancer Biol Ther. 2009;8:1045-54
45. Tokumaru S, Higashiyama S, Endo T, Nakagawa T, Miyagawa JI, Yamamori K. et al. Ectodomain shedding of epidermal growth factor receptor ligands is required for keratinocyte migration in cutaneous wound healing. The Journal of cell biology. 2000;151:209-20
46. Piepkorn M, Lo C, Plowman G. Amphiregulin-dependent proliferation of cultured human keratinocytes: autocrine growth, the effects of exogenous recombinant cytokine, and apparent requirement for heparin-like glycosaminoglycans. Journal of cellular physiology. 1994;159:114-20
47. Hashimoto K, Higashiyama S, Asada H, Hashimura E, Kobayashi T, Sudo K. et al. Heparin-binding epidermal growth factor-like growth factor is an autocrine growth factor for human keratinocytes. The Journal of biological chemistry. 1994;269:20060-6
Author contact
![]() Corresponding author: Jiaping Zhang, Tel: 023-68754353. Email address: 1980475546com.
Corresponding author: Jiaping Zhang, Tel: 023-68754353. Email address: 1980475546com.