Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(3):568-578. doi:10.7150/ijbs.29759 This issue Cite
Research Paper
Inhibition of Heme Oxygenase-1 enhances hyperthermia-induced autophagy and antiviral effect
Yang Yang1,3, He-Xiao Wang1, Lan Zhang1, Wei Huo1, Xiao-Dong Li2, Rui-Qun Qi1, Xiao-Yu Song3, Shi Wei4, Xing-Hua Gao1, Shuai Han5 ![]() , Liu Cao3
, Liu Cao3 ![]()
1. Department of Dermatology, No.1 Hospital of China Medical University and Key Laboratory of Immunodermatology, Ministry of Health and Ministry of Education, Shenyang 110001, China
2. Department of Dermatology, Central Hospital Affiliated to Shen Yang Medical College, Shenyang, 110001, China
3. Key Laboratory of Medical Cell Biology, China Medical University, Shenyang, 110122, China
4. Department of Pathology, the University of Alabama at Birmingham, Birmingham, Alabama 35249, United States
5. Department of Neurosurgery, No.1 Hospital of China Medical University, Shenyang 110001, China
Received 2018-9-6; Accepted 2018-11-15; Published 2019-1-1
Abstract

Hyperthermia has been clinically utilized as an adjuvant therapy in the treatment of cervical carcinoma. However, thermotolerance induced by heme oxygenase-1 (HO-1), a stress-inducible cytoprotective protein, limits the efficacy of hyperthermic therapy, for which the exact mechanism remains unknown. In the present study, we found that heat treatment induced HO-1 expression and decreased copy number of HPV16 in cervical cancer cells and tissues from cervical cancer and precursor lesions. Knockdown of HO-1 stimulated autophagy accompanied by downregulation of X-linked inhibitor of apoptosis protein. Furthermore, silencing of HO-1 led to cell intolerance to hyperthermia, as manifested by inhibition of cell viability and induction of autophagic apoptosis. Moreover, HO-1 modulated hyperthermia-induced, autophagy-dependent antiviral effect. Thus, the findings indicate that blockade of HO-1 enhances hyperthermia-induced autophagy, an event resulting in apoptosis of cervical cancer cells through an antiviral mechanism. These observations imply the potential clinical utility of hyperthermia in combination with HO-1 inhibition in the treatment of cervical cancer.
Keywords: Heme oxygenase-1, autophagy, hyperthermia, cervical cancer, human papillomavirus
Introduction
Cervical cancer is the fourth most common cancer in women globally, and the leading cause of cancer related deaths in developing countries, where it is the second most common cancer in women [1, 2]. Persistent infection with certain types of human papillomavirus (HPV) causes nearly all cervical cancers [3]. The papillomavirus E2 proteins exert many functions in the viral cycle, including transcription regulation and DNA replication, which, in concert with modulating the host cells through direct protein interaction, play a pivotal role in carcinogenesis [4]. We and others have previously shown that moderate heat treatment (hyperthermia) induces HPV E2 gene mutation and promotes host response in vivo, and induces apoptosis and cell cycle arrest of cervical cancer cells in vitro [5-8]. Hyperthermia has been applied clinically as a concomitant therapy for cervical carcinoma, and has been shown to improve the patient outcomes [7]. However, cervical carcinomas with the same histologic grade and HPV subtype may have significant differences in response to hyperthermia treatment [9]. Thus, further studies to identify potential molecular biomarkers for the effectiveness of thermal treatment are necessary.
On the other hand, heme oxygenase-1 (HO-1) is an inducible intracellular enzyme in response to oxidative stress, irradiation, ultraviolet light, hypoxia and hyperthermia. Its catalytic action on heme produces three active products, namely, carbon monoxide, biliverdin and ferrous iron [10]. HO-1 itself and the by-products of heme degradation play a crucial role in maintaining cellular homeostasis, protecting cells from oxidative damage, attenuating inflammatory response and inhibiting cytokine production [11]. The cytoprotective function of HO-1 has been regarded as a shied from radiotherapy, chemotherapy, photodynamic therapy and other adjuvant therapy in several malignant diseases [12-14]. The function of anti-proliferation, anti-migration, and anti-invasion of HO-1 has also been reported in a number of cancer cell lines [15, 16]. Moreover, modulation of HO-1 has demonstrated therapeutic benefits by sensitizing cancer cells to therapies [13, 17, 18].
HPV produces oncoproteins to inhibit autophagy, thus suggesting a possible mechanism that autophagic dysfunction induced by HPV infection is an adaptive response to hinder viral clearance [19]. HO-1 can promote autophagy in the development of chemo-resistance in cells derived from leukemia, breast cancer, and renal cell carcinoma [20-22]. On the contrary, HO-1 may also inhibit autophagy to protect against cisplatin cytotoxicity in acute kidney injury [23]. Given the paradoxicaleffects of HO-1 on autophagy, we sought to investigate the expression of HO-1 in the hyperthermic condition and to explore the function and regulation of autophagy by HO-1 in cervical cancer in this study.
Materials and Methods
Patient specimen
This study was approved by the Ethical Committee of China Medical University (reference: [2017]2016-207-2). A total of 56 cervical biopsy specimens from June to October, 2017, were collected from the Department of Obstetrics and Gynecology, No.1 Hospital and Shengjing Hospital of China Medical University. Written informed consent was secured for all study participants.
The average age of the patients was 46.16 year. The specimens included 12 cervical intraepithelial neoplasia (CIN) I, 6 CINII, 4 CINIII, 26 cervical carcinomas and 8 patients without CIN (NCIN). Forty-two specimens harbored HPV16, 16 from CIN lesions and 26 from cervical carcinomas.
The fresh cervical tissue remnants after pathologic diagnosis (approximately 0.3 × 0.3 cm each) were equally splitted into two parts: one treated with 37 ºC for 30 min, the other treated with 44ºC for 30 min. Both parts were then submerged in culture medium at 37ºC for 12 hours.
Cell culture
Human cervical cancer cell lines (HPV-negative C33A, HPV16 positive SiHa with 1-2 integrated HPV copies per cell, and HPV-positive CaSki containing 60-600 integrated HPV strands per cell [24]) were obtained from ATCC (Manassas, VA). Cells were maintained in RPIM-1640 medium (Biological Industries, CT, USA) with 10% fetal bovine serum (FBS, Biological Industries). All cell lines were incubated at 37ºC in a humidified incubator containing 5% CO2.
HPV16 integration and copy number of E6 analysis
HPV16 integration and copy number of E6 were analyzed by using real-time fluorescence PCR with type-specific primers and probes as described (Table 1) [25, 26]. The amplication conditions were 10 min at 95ºC, followed by a two-step cycle of 95 ºC for 15 seconds and 60 ºC for 60 seconds, for 45 cycles. The total volume of 20 μl contained 20 pmol/μl of each primer, 10 pmol/μl of each probe, 10 μl volumn of ready-to-use hot start PCR mix (FastStart Essential DNA Probes Master, Roche Diagnostics, Mannheim, Germany), and 100 ng of target DNA from biopsy specimens or cervical cancer cells. Two standard curves were obtained by amplication of a dilution series of 500 thousand to 500 copies of a clone of HPV16 in PML2d (Addgene, Teddington, UK). Ratios of E2 to E6 of less than 0.93 indicate the presence of two states of HPV16. Values of greater than 1 indicate the predominance of the episomal state.
Primers and probes sequences of HPV16 E2 and E6
| Name | Sense (5'->3') |
|---|---|
| HPV16E2F | ACCGAAGTATCCTCTCCTGAAATTATTAG |
| HPV16E2R | CCAAGGCGACGGCTTTG |
| HPV16E2PRO | VIC-CACCCCGCCGCGACCCATA-MGBNFQ |
| HPV16E6F | GAGAACTGCAATGTTTCAGGACC |
| HPV16E6R | TGTATAGTTGTTTGCAGCTCTGTGC |
| HPV16E6PRO | 6FAM-CAGGAGCGACCCAGAAAGTTACCACAGTT-MGBNFQ |
F: forward primer; R: reverse primer; PRO: probe; MGBNFQ: quencher dye; 6-FAM: 6-carboxygluorescein
Western Blot
Proteins were separated by electrophoresis on 12% sodium dodecyl sulfate polyacrylamide gel and electrophoretically transferred onto polyvinylidene difluoride membranes, which were then incubated with antibodies raised in rabbit against microtubule-associated protein 1A/1B-light chain 3 (LC3), HO-1, X-linked inhibitor of apoptosis protein (XIAP), p62, beclin1, ATG5, ATG7, mTOR, p-mTOR (Ser 2448), cleaved PARP (Cell Signaling Technology, Bevery, MA, USA) and GAPDH (Proteintech, Wuhan, China). Subsequently, the membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit antibody (ZSGB-BIO, Beijing, China) and the protein bands were examined using ECL Western Blotting Substrate (Thermo Fisher Scientific, Waltham, MA).
Flow cytometry
Cellular apoptosis was detected by using APC (eBioscience, San Diego, CA, USA) or FITC Annexin-V Apoptosis Detection Kit (BD Biosciences, USA) in accordance with the manufacturer's instructions. The adherent cells were trypsinized and washed with cold Phosphate Buffered Saline (PBS). Cell suspension was incubated with 5 µl of APC or FITC Annexin-V, and 5 µl of propidium iodide (PI) for 15 min in the dark at room temperature. For cell cycle analysis, the cells were fixed with 75% cold ethanol overnight, and each sample was incubated with 5 µl of PI (40µg/ml) and 5µl of RNase A (10 µg/ml) at 37 ºC for 20 min. After staining, samples for apoptosis and cell cycle distribution were analyzed using BD LSRFortessa (BD Biosciences) and the percentage of cells at sub-G1, G1, S, and G2/M phase was analysed using ModFit sofeware (Becton Dickinson, USA).
RNA isolation and real-time PCR
Total RNA was isolated with miRNeasy mini kit (Qiagen, Stockach, Germany) and then converted to cDNA by using GoScript TM Reverse Transcription System (Promega, Madison, WI). Real-time PCR was performed to detect relative mRNA expression level using primers as described in Table 2.
Primer sequences for the mRNA quantification by real-time PCR
| Name | Sense (5'->3') | Antisense (5'->3') |
|---|---|---|
| HO-1 | GTCAGGCAGAGGGTGATAGAAG | GTGTAAGGACCCATCGGAGAAG |
| TP53 | GATGTTCCGAGAGCTGAATGAG | TATGGCGGGAGGTAGACTGA |
| OAS1 | ACTACCAGCAACTCTGCATCTA | CTCCACCACCCAAGTTTCCT |
| MX1 | CCTCGTGTTCCACCTGAAGA | CTCCAGCAGATCCCTGAAATATG |
| AP1S1 | TGTGCGAGCTGGACATCAT | CATCCTCCTCTTGCAGTAGGT |
| MYD88 | TAAGAAGGACCAGCAGAGCC | CATGTAGTCCAGCAACAGCC |
| AIP5 | CGACCTAGAACAGACCTTCAATC | GTTAGGGAGAACCTGCTCACA |
| CASP3 | ACAGACAGTGGTGTTGATGATG | GCACAAAGCGACTGGATGAA |
| CytC | ACTCTTACACAGCCGCCAAT | CTTCCTTCTTCTTAATGCCGACA |
| GAPDH | AAGAGCACAAGAGGAAGAGAGAGAC | GTCTACATGGCAACTGTGAGGAG |
RNA interference
HO-1 or XIAP validated small interfering RNA (siRNA) as well as scrambled siRNA were purchased from Life Technologies Corporation (Foster City, CA). The siRNA sequences are listed in Table S1. Cells were transfected with Silencer® Select Validated HO-1 siRNA or XIAP siRNA, and mixed with Lipofectamine™ RNAiMAX (Thermo Fisher scientific) as transfection reagent according to the manufacturer's protocol.
Cell viability assay
The cells were seeded in 96-well plates at a density of 4000 cells per well, following the treatment of the cells with 3-Methyladenine (3-MA)/Rapamycin (Sigma, St. Louis, MO, USA) for 2 hours or HO-1 siRNA for 24 hours. The cells were then treated with hyperthermia at 44ºC for 30 min. At day 0, 1, 2 and 3, 20 μl of MTS solution (Promega) was added to the medium and the cells were incubated at 37ºC for 3 hours. The absorbance was measured at 490nm using a spectrophotometer.
Tandem mass tag (TMT) labeling and liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis
CaSki cells were transfected with HO-1 siRNA or scrambled siRNA. At 48 hours, protein was extracted 48 hours after transfection by using lysis buffer with 1% protease inhibitor cocktail (Sigma), and reduced with 5 mM dithiothreitol for 30 min at 56ºC and 11 mM iodoacetamide for 15 min at room temperature in darkness. Peptides were labeled using TMT kit and then separated by high-performance liquid chromatography (HPLC), and combined into 18 fractions. The peptides were then subjected to NSI source followed by tandem mass spectrometry in Orbitrap FusionTM LumosTM TribridTM (Thermo) coupled online to the Ultra Performance Liquid Chromatography (UPLC). The resulting data were processed using Maxquant search engine (v.1.5.2.8). Each experiment was repeated three times.
Statistical analysis
All experiments were repeated at least three times and quantitative values were expressed as mean ± SD. Statistical significance was determined by one-way analysis of variance (ANOVA) or paired or unpaired t-test for comparisons of group means. A p<0.05 was considered statistically significant.
Results
Hyperthermia induces HO-1expression in cervical cancer cells and tissues from cervical cancer and its precursors
We first examined the effect of hyperthermia on a number of key genes influencing cell fate in 3 human cervical carcinoma lines. Cells were treated with 37 ºC and 44 ºC by water baths for 30 minutes, respectively. As shown in Figure 1A, there was a sustained increase in HO-1 mRNA expression 6 hours after heat-treatment at 44ºC, with C33A being the most thermo-sensitive line that showed a near 30-fold increase. There was only an approximately 2-5 fold increase in HO-1 mRNA expression in CaSki and SiHa cells (Figure S1). In contrast, there were no significant changes in the expression of the other genes examined. In keeping with this finding, western blot analysis showed hyperthermia-induced HO-1 protein expression in a time-dependent manner, peaked at 12-24 hours (Figure 1B).
Hyperthermia induces HO-1 expression in cervical cancer cells and tissue. (A) Hyperthermia induces HO-1 mRNA expression in a time-dependent manner in C33A cells, but does not alter the expression of other genes examined. (B) Hyperthermia induces HO-1 protein expression in a time-dependent manner in all three cervical cancer cell lines. (C) Hyperthermia induces HO-1 mRNA expression in cervical cancer and precursor tissues ex vivo. (D) Hyperthermia significantly induces HO-1 mRNA expression in CINI lesions ex vivo. All data were normalized against the endogenous control of GAPDH (*p<0.05, ***p<0.001, by paired t-test). Abbreviations: HO-1, Heme oxygenase-1; TP53, cellular tumor antigen p53; OAS1, 2'-5'-oligoadenylate synthase 1; MX1, myxovirus resistant 1; AP1S1, AP-1 complex subunit sigma-1A; MYD88: myeloid differentiation factor 88; AIP5, atrophin-interacting protein 5; CASP, caspase; CytC, cytochrome C.
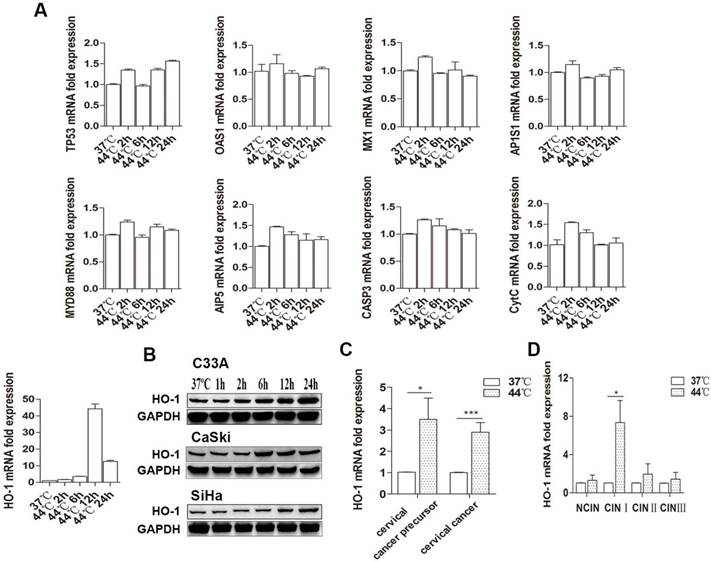
To further validate these observations ex vivo, we expanded our investigation into cervical biopsies. Forty-seven fresh samples from 21 patients with and without cervical cancer precursor lesions [7 CINI, 5 CINII, 4 CIN III, and 5 NCIN] and 26 patients with cervical cancer were equally splitted and treated with 37ºC and 44ºC, respectively, by water bath for 30 minutes. In concert with the findings in cervical cancer cell lines, hyperthermia induced an average of 3.5- and 2.9-fold increase in HO-1 mRNA expression in cervical cancer precursor and cervical cancer tissues, respectively (Figure 1C). In regard to different grades of CIN lesions, hyperthermia drastically induced HO-1 mRNA expression of in CINI, with an average of 7 fold, while no significant changes were found in HO-1 expression in CINs II and III (Figure 1D).
Identification of dysregulated proteins in HO-1 silenced cervical cancer cells
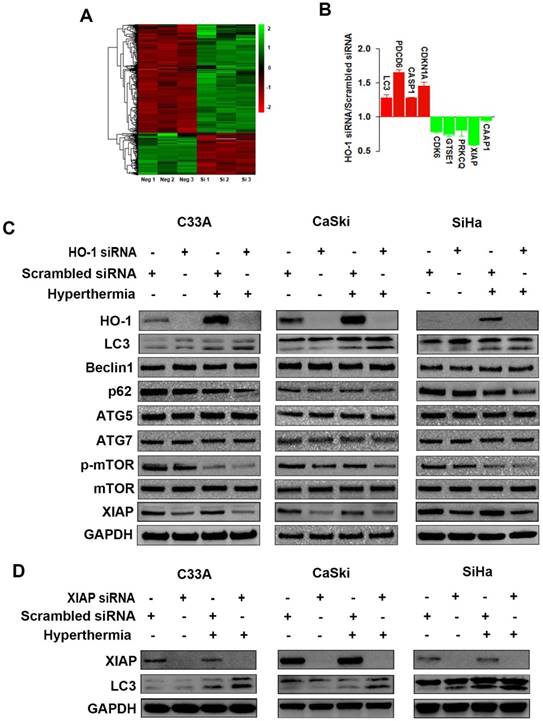
To explore the dysregulated proteins regulated by HO-1 in cervical cancer cells, tandem mass tag (TMT) labelling and liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis were performed to identify the differentially expressed proteins between the HO-1 siRNA and scrambled siRNA expressing CaSki cells. To that end, a total of 6918 molecules were identified. Based on the criteria of p<0.05 and fold change ≥1.2, 191 upregulated and 496 downregulated proteins were displayed in HO-1 silenced cells (Figure 2A). It was of substantial interest that a number of cell cycle-, apoptosis- and phagocytosis-related molecules were identified among these 687 differentially expressed proteins, including XIAP, caspase activity and apoptosis inhibitor 1 (CAAP1), programmed cell death protein 6 (PDCD6), cyclin-dependent kinase inhibitor 1A (CDKN1A), cyclin-dependent kinase 6 (CDK6), and LC3 (Figure 2B).
Hyperthermia and HO-1 regulate key molecules in autophagy. (A) Differentially expressed proteins between HO-1 siRNA and scrambled siRNA CaSki cells by LC-MS/MS analysis. (B) Illustration of differentially expressed cycle-, apoptosis- and phagocytosis-related molecules. (C) Hyperthermia enhances downregulation of XIAP, p62, and p-mTOR and upregulation of LC3 induced by HO-1 silencing in three cervical cancer cell lines. The cells were transfected with HO-1 siRNA or scrambled siRNA for 24 hours prior to exposure to hyperthermia, and the proteins were analyzed by western blot. (D) Hyperthermia enhances conversion of LC3-I to LC3-II induced by XIAP silencing. The cells were transfected with XIAP siRNA or scrambled siRNA for 24 hours prior to exposure to hyperthermia, and the proteins were analyzed by western blot. Abbreviations: LC3, microtubule-associated protein 1 light chain 3; XIAP, X-linked inhibitor of apoptosis; PDCD6, programmed cell death protein 6; CDKN1A, cyclin-dependent kinase inhibitor 1; CDK6, cyclin-dependent kinase 6; GTSE1, G2 and S phase-expressed protein 1; PRKCQ, protein kinase C theta type; CAAP1, caspase activity and apoptosis inhibitor 1; CASP, caspase.
Inhibition of HO-1 promotes hyperthermia-induced autophagy
HO-1 may act as either a positive regulator of the cytoprotective autophagy or a negative mediator to inhibit cytotoxic autophagy in different therapeutic chemical- or physical-induced responses [22, 23]. Western blot analysis showed that both knockdown of HO-1 and hyperthermia at 44 ºC promoted the conversion of LC3-I (cytosolic form) to LC3-II (membrane-bound), a process to initiate formation and lengthening of the autophagosome, currently considering as a simple, quick procedure to verify the presence of autophagy [27]. Meanwhile, either silencing HO-1 or hyperthermia reduced the expression of p62 and p-mTOR Furthermore, there was a further increase in the LC3-II/LC3-I ratio and a further downregulation of p62 and p-mTOR with both hyperthermia and HO-1 silencing, suggesting a superior therapeutic effect with the combined strategy over the single agent-induced autophagy (Figure 2C).
XIAP, an anti-apoptotic molecule, has been recently recognized as a negative regulator of autophagy [28]. Our research to that end revealed that silencing of HO-1 inhibited the expression of XIAP. Thus, we next sought to examine whether XIAP plays a role in regulating autophagy. As shown in Figure 2D, knockdown of XIAP by siRNA led to increased conversion of LC3-I to LC3-II in three cervical cancer cell lines, an event further enhanced by hyperthermia. These findings indicate that HO-1 inhibits autophagy via downregulation of XIAP.
Knockdown of HO-1 inhibits the viability of cervical cancer cells
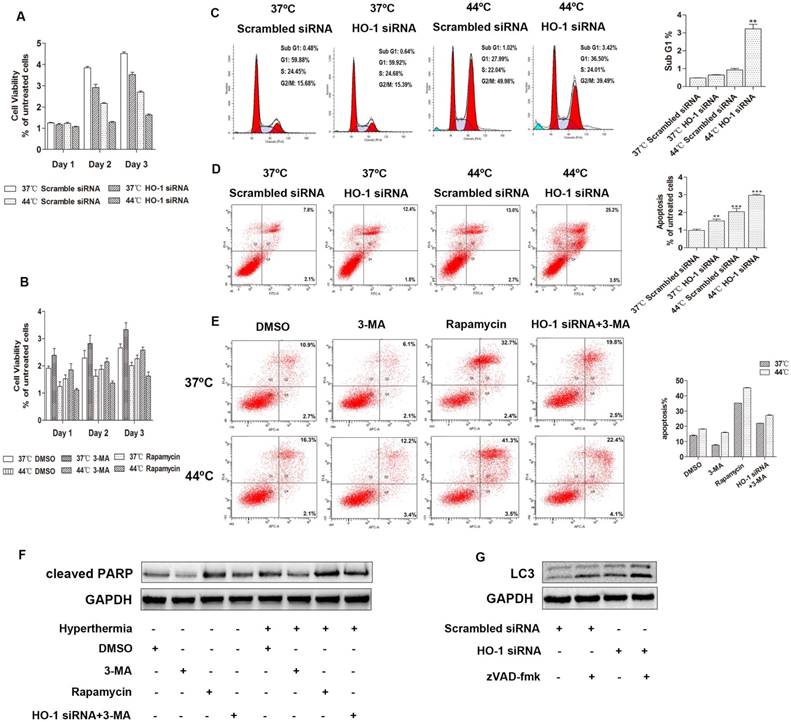
To examine whether hyperthermia and HO-1 affect the viability of cervical cancer cells, MTS cell proliferation assay was carried out. Compared to the control group, the cell viability was reduced two days after heat treatment or HO-1 knockdown. The combination of hyperthermia and HO-1 silencing further diminished the cell viability, suggesting that inhibition of HO-1 facilitates heat-mediated down-regulation of cervical cancer cell proliferation (Figure 3A, Figure S2A). In a parallel study, blockage of autophagy induction by 3-MA increased the cell viability, while induction of autophagy by rapamycin significantly sensitized cervical cancer cells to heat-induced cytotoxicity (Figure 3B, Figure S2B). These data indicate that upregulation of autophagy by HO-1 knockdown and hyperthermia provides a synergistic effect in inhibiting cervical cancer cell viability.
Downregulation of HO-1 promotes autophagic apoptosis in cervical cancer cells. (A) HO-1 knockdown facilitates hyperthermia-mediated down-regulation of cervical cancer cell proliferation. C33A cells transiently transfected with scramble and HO-1 siRNA were incubated for 24 hours at 37ºC or 44ºC, cell viability was measured by MTS assay on day 1, 2 and 3. (B) Autophagy inhibitor 3-Methyladenine (3-MA) attenuates cervical cancer cells to hyperthermia-induced cytotoxicity, and autophagy inducer rapamycin sensitizes cervical cancer cells to hyperthermia-induced cytotoxicity. C33A cells were pretreated with DMSO, 3-MA (10 mM), or rapamycin (100nM) for 2 hours and then incubated at 37ºC or 44ºC prior to MTS assay. (C) Comparison of the sub G1 phases in C33A cells treated with hyperthermia and/or HO-1 siRNA by flow cytometry analysis. (D) Flow cytometric analysis of apoptosis of C33A cells treated with hyperthermia and/or HO-1 siRNA. (E) Flow cytometric analysis of apoptosis of C33A cells treated with 3-MA, rapamycin, or HO siRNA, with or without hyperthermia. Cells were incubated with DMSO, 3-MA (10 mM), or rapamycin (100 nM), or transfected with HO-1 siRNA 24 hour prior to exposure to 3-MA, for 2 hours before treated with hyperthermia. Cleaved PARP was detected by western blot (F). (G) C33A cells transfected with HO-1 siRNA alone or in combination with zVAD-fmk (20 μM) for 24 h, and the proteins were analyzed by western blot. The experiment was replicated three times. **p<0.01, ***p<0.001 compared to the control group (one way ANOVA). Each point represents the mean ± SD.
Absence of HO-1 augments hyperthermia-induced autophagic apoptosis
We have previously shown that local hyperthermia induces apoptosis of keratinocytes infected with HPV and melanoma cells [8,29]. Apoptotic cells are characterized by DNA fragmentation, among other typical features, that can be analysed by PI flow cytometric assay [30]. Apoptosis can be determined by quantifying the sub-G1 peak [31]. To explore the potential effect of HO-1 knockdown and hyperthermia on apoptosis of cervical cancer cells, the sub-G1 assay was first carried out. After transfection with either HO-1 siRNA or scrambled siRNA, cells were subjected with hyperthermia at 44ºC for 30 minutes. While the sub-G1 peak was negligible in the control cells (0.48% in C33A), there was a 1.33-fold increase in the cells treated with HO-1 siRNA, and a 1.92-fold increase in the cells treated with hyperthermia. The proportion of sub-G1 peak further increased to an average of 6.7-fold in the cells treated with both HO-1 siRNA and hyperthermia (p<0.01; Figure 3C). The data obtained using CaSki and SiHa cells were shown in Figure S2C.
Apart from the sub-G1 assay, the total number of apoptotic cells in each condition was further quantitated by flow cytometry analysis. To that end, there were a 2.04-fold increase with hyperthermia, a 1.52-fold increase with HO-1 siRNA, and a 2.97-fold increase with their combination in C33A cells (p<0.001; Figure 3D). Similar observations were obtained when using CaSki and SiHa cells (Figure S2D).
Autophagy is involved in the process of various forms of regulated or unregulated cell death [32]. We next asked if autophagy plays a role in the apoptosis of cervical cancer cells induced by hyperthermia and HO-1 silencing. To that end, 3-MA attenuated heat-induced apoptosis and the expression of cleaved PARP, whereas rapamycin, an autophagy agonist, increased the proportion of apoptotic cells and upregualted cleaved PARP. Furthermore, 3-MA decreased HO-1 knockdown-induced apoptosis and cleaved PARP expression, with or without hyperthermia (Figure 3E and F). Likewise, similar observations were obtained in CaSki and SiHa cells (Figure S2E and S2F). Thus, the collective findings indicate that autophagy is involved in heat-induced apoptosis.
In order to further clarify the crosstalk between apoptosis and autophagy, our data revealed that zVAD-fmk, an inhibitor of pan-caspase, led to increased conversion of LC3-I to LC3-II in three cervical cancer cell lines, with or without HO-1 silencing (Figure 3G and S3G).
Hyperthermia reduces the copy number of HPV16 E6 in cervical biopsy specimens
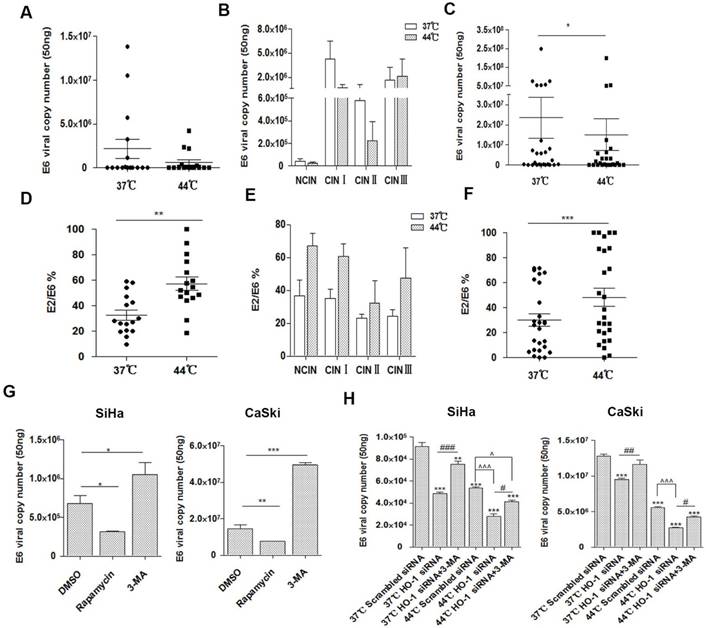
In addition to viral load, a number of other HPV-related factors are also thought to predispose cervical cancer, of which viral integration status is a key parameter and has been widely investigated [25, 33]. We thus examined HPV16 copy numbers and integration status over hyperthermia treatment in the CIN and cervical cancer tissue samples. The average viral loads from different grades of CIN lesions varied greatly (ranged 1.252×103 to 1.3825×107 copies per 50 ng DNA), and the copy number declined after hyperthermia treatment (1.102×103 to 4.2586×106 copies), especially in CINI and CINII, although it did not reach a statistical significance (Figure 4A and 4B). The average viral loads in cervical cancer samples decreased significantly, from 2.35×107 to 1.51×107 copies, with an average reduction of 35.7% (p<0.05; Fig. 4C). Five samples (1 CINI and 4 cervical cancer) with mixed forms (integrated and episomal) of HPV changed to pure episomal form, where equivalent amounts of E2 and E6 were detected. Four cervical cancer samples harbored integrated form of HPV, where the E2/E6 ratios were low (0.1) [25], either before or after hyperthermia treatment. Four cervical cancer samples altered from purely integrated form to mixed form after treatment. In the 14 cancer samples harbouring mixed forms of HPV16, the integration degree increased in 2, and significantly reduced in 12 (average E2/E6 ranging 39.38% to 60.1%) after hyperthermia treatment. While no alteration in the HPV16 copy number was observed in CIN lesions with hyperthermia, the integration degree was markedly decreased (average E2/E6 ranging 31.58% to 57.23%) (Figure 4D-4F).
Inhibition of HO-1 enhances hyperthermia-induced, autophagy-dependent antiviral effect. (A) Hyperthermia decreases the total number of HPV16 E6 (episomal and integrated) copies in NCIN-CIN III lesions. Horizontal bars represent means. (B) Hyperthermia modestly decreases HPV16 E6 copy numbers in CINI and CINII lesions. (C) Hyperthermia significantly decreases HPV16 E6 copy numbers in cervical cancer tissue. (D) Hyperthermia significantly alters HPV16 integration in CIN lesions, although no statistical significance is reach when compared to the individual grade (E). (F) Hyperthermia significantly affects HPV16 integration in cervical cancer samples. (G) Pharmacologic induction of autophagy by rapamycin significantly decreases, and inhibition of autophagy by 3-MA significantly increases the HPV16 copy numbers in cervical cancer cells. SiHa cells and CaSki cells were pretreated with DMSO, 3-MA (10 mM), or rapamycin (100 nM) for 12 hours and DNA was extracted to analyze the copy numbers of HPV16 E6. (H) Hyperthermia and HO-1 knockdown significantly decreased HPV16 E6 copy numbers in cervical cancer cells. HPV16 E6 copy numbers were analyzed in SiHa cells and CaSki cells treated with HO-1 siRNA transfection, hyperthermia, or their combination, in the presence or absence of 3-MA. *p<0.05, **p<0.01, ***p<0.001 (paired t-test in A-F, one way ANOVA in G and H). # and ^ p<0.05, ##p<0.01, ### and ^^^ p<0.001 (unpaired t-test).
HO-1 modulates autophagy-induced HPV16 E6 copy number alteration
Recent studies have demonstrated that inhibition of autophagy results in enhancement of HPV infectivity and prevention of viral clearance [34, 35]. These findings prompted us to investigate if altered autophagy affects the HPV infectivity in cervical cancer cells. As illustrated in Figure 4G, pharmacologic induction of autophagy by rapamycin significantly decreased, and inhibition of autophagy by 3-MA significantly increased the HPV16 copy numbers, respectively.
The role of HO-1 in antiviral activity remains a matter of debate. Thus, we next examined if HO-1 was involved in the modulation of HPV16 E6 copy number in cervical cancer cells. As shown in Figure 4H, both hyperthermia and HO-1 knockdown resulted in markedly decreased HPV16 E6 copy numbers in SiHa cells (41.3% and 46.8%, respectively) and CaSki cells (56.6% and 25.7%, respectively). The combination of hyperthermia and HO-1 silencing reduced the viral load by 69.3% in SiHa cells and 78.9% in CaSki cells, respectively, when compared to the control group.
To further verify whether HO-1 modulates autophagy-induced antiviral effect, we examined the viral copy numbers in the HO-1 knockdown group treated with 3-MA simultaneously. To that end, inhibition of autophagy by 3-MA significantly dampened the HO-1 silencing-induced viral load reduction (Figure 4H). Thus, upregulation of autophagy by HO-1 knockdown effectively reduces HPV16 E6 viral load in cervical cancer cells.
Discussion
In this study, we demonstrated that HO-1 was overexpressed in HPV infected cells and tissue subjected to hyperthermia, especially in low grade CIN lesions. The reason for this result is probably that the small sample size limits to some extent the generalization of the finding or that low grade CIN lesions are more sensitive to hyperthermia, so further study is needed in the future. While HO-1 is considered as the first line of cellular defense of cancer cells against the insults, the exact mechanism underlying this protective effect remains unclear. The present study explored the function and regulatory mechanism of autophagy by HO-1, in hyperthermia-stimulated conditions.
To discern the regulatory molecules of HO-1 in cervical cancer cells, we first carried out proteomic analysis to identify differentially expressed proteins. The research to that end has identified a number of cell cycle and apoptosis-related molecules among the 687 abnormally expressed proteins, including XIAP, CAAP1, CDK6, PDCD6, CASP1 and CDKN1A. Furthermore, upregulation of LC3 indicated a potential role of LC3-associated phagocytosis in this event. Inhibition of phagocytosis reportedly leads to severe virus infection and early death, which may partly explain the observation of viral load reduction induced by HO-1 knockdown in our study [35-38]. There is considerable evidence that HO-1 induction may provide a potential therapeutic effect by stimulating an innate cellular response, decreasing viral replication and inflammation against a variety of virus, such as Ebola virus, human immunodeficiency virus, hepatitis B and C virus, enterovirus 71, influenza virus, dengue virus and respiratory syncytial virus [39-41]. Similarly, inhibition of HO-1 provides a promising therapeutic strategy against human herpesvirus-8-related malignancies [42]. The controversial topic of the effect of HO-1 on antiviral ability remains in dispute. Our findings indicate that silencing HO-1 reduces viral copy number, a process likely autophagy-dependent.
Previous studies have suggested the disruption status of viral E2 gene concomitant with viral load as a potential mechanism for HPV16 positive cervical cancer pathogenesis [43]. Concurrent hyperthermia and chemoradiation has been utilized clinically given its effects in disrupting cell cycle, inhibiting cell proliferation, inducing apoptosis and mutating HPV [5-7, 44]. In the present study, we found that hyperthermia not only reduced the copy number, but also changed the physical state of HPV16, decreased the degree of viral integration in most cervical cancer and precursor lesions. Meanwhile, knockdown of HO-1 increased heat-induced apoptosis and inhibited cell viability. Inactivation of HO-1 also exacerbated the autophagic response and decreased viral copy number. These findings, along with previous observations, suggest that inhibition of HO-1 is an effective modality in sensitizing cancer cells to therapies [13, 17].
Autophagic and apoptotic signalling pathways almost always intertwine to affect cell fate [45]. The research to that end has resulted in conflicting observations. Some authorities consider that autophagy augments pro-apoptotic function of chemotherapeutic agents, while others believe that autophagy may enhance survival of tumor cells [46-48]. We found that autophagy promoted heat-stimulated autophagic apoptosis in cervical cancer cells. An early study has demonstrated that in addition to accelerating cell death induced by hyperthermia, autophagy can prevent virus duplication and infection in accordance with our finding that autophagy induced by inhibition of HO-1 decreases virus load in cervical cancer cells [49].
In summary, the data in the present study suggest a possible biologic mechanism for thermosensitization of cervical cancer cells by silencing of stress-inducible enzyme, HO-1. These findings provide evidence that patients with cervical cancer may benefit from concurrent HO-1 silencing and hyperthermia treatment given their additive effect in activating autophagic apoptosis and antiviral properties. Whether targeting downstream molecules of HO-1 may overcome hyperthermia resistance is worth further exploring. Moreover, HO-1 has been recognized to have major immunomodulatory and anti-inflammatory properties, while hyperthermia may induce antiviral activity by endogenous interferon-dependent pathway [50]. Thus, other potential mechanisms by which inhibition of HO-1 promotes hyperthermia-induced viral clearance in cervical cancer cells remain to be investigated.
Abbreviations
HPV: human papillomavirus; HO-1: Heme oxygenase-1; LC3: microtubule-associated protein 1 light chain 3; XIAP: X-linked inhibitor of apoptosis; TMT: Tandem mass tag; PI: propidium iodide; HIV: Human Immunodeficiency Virus; KSHV: Kaposi's Sarcoma-associated Herpesvirus; IFN: interferon; siRNA: small interfering RNA; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; MTS: 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium; TP53: cellular tumor antigen p53; OAS1: 2'-5'-oligoadenylate synthase 1; MX1: myxovirus resistant 1; AP1S1: AP-1 complex subunit sigma-1A; MYD88: myeloid differentiation factor 88; AIP5: atrophin-interacting protein 5; CASP: caspase; CytC: cytochrome C; PDCD6: programmed cell death protein 6; CDKN1A: cyclin-dependent kinase inhibitor 1; CDK6: cyclin-dependent kinase 6; GTSE1: G2 and S phase-expressed protein 1; PRKCQ: protein kinase C theta type; CAAP1: caspase activity and apoptosis inhibitor 1; PBS: Phosphate Buffered Saline.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We thank Mingzhe Wu, MD, for her contribution in the clinical sample collection. This work was supported by the National Natural Science Foundation of China [grant number 81803148]; Clinical Research Center of Liaoning Province under Grant [number 2015225012]; Supports by National Key R&D Program of China under Grant [2016YFC1302400]; and The Ministry of education innovation team development plan The Ministry of education innovation team development plan to Liu Cao under Grant [IRT_17R107&IRT13101].
Author contributions
Y.Y. H.-X. W. and L.Z. performed biochemical experiments, and participated in data analysis. W.H., X.-D. L. and R.-Q. Q. conceived the idea, participated in the design of the study and interpretation of data. X.-Y. S., W.S. and X.-H. G. participated in the intellectual input and supervision. S.H. and L.C. drafted the manuscript with contributions from other authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen G, Huang P, Xie J. et al. microRNA211 suppresses the growth and metastasis of cervical cancer by directly targeting ZEB1. Mol Med Rep. 2018;17:1275-82
2. Akinfolarin AC, Olusegun AK, Omoladun O. et al. Age and Pattern of Pap Smear Abnormalities: Implications for Cervical Cancer Control in a Developing Country. J Cytol. 2017;34:208-11
3. Bosch FX, Lorincz A, Munoz N. et al. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol. 2002;55:244-65
4. Bellanger S, Tan CL, Xue YZ. et al. Tumor suppressor or oncogene? A critical role of the human papillomavirus (HPV) E2 protein in cervical cancer progression. Am J Cancer Res. 2011;1:373-89
5. Piguet V. Heat-Induced Editing of HPV Genes to Clear Mucocutaneous Warts? J Invest Dermatol. 2017;137:796-7
6. Yang Y, Wang H, Zhang X. et al. Heat Increases the Editing Efficiency of Human Papillomavirus E2 Gene by Inducing Upregulation of APOBEC3A and 3G. J Invest Dermatol. 2017;137:810-8
7. Oei AL, van Leeuwen CM, ten Cate R. et al. Hyperthermia Selectively Targets Human Papillomavirus in Cervical Tumors via p53-Dependent Apoptosis. Cancer Res. 2015;75:5120-9
8. Wang X, Gao XH, Li X. et al. Local hyperthermia induces apoptosis of keratinocytes in both normal skin and condyloma acuminata via different pathways. Apoptosis. 2009;14:721-8
9. Liefers GJ, Tollenaar RA. Cancer genetics and their application to individualised medicine. Eur J Cancer. 2002;38:872-9
10. Prawan A, Kundu JK, Surh YJ. Molecular basis of heme oxygenase-1 induction: implications for chemoprevention and chemoprotection. Antioxid Redox Signal. 2005;7:1688-703
11. Lv H, Yu Z, Zheng Y. et al. Isovitexin Exerts Anti-Inflammatory and Anti-Oxidant Activities on Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting MAPK and NF-κB and Activating HO-1/Nrf2 Pathways. Int J Biol Sci. 2016;12:72-86
12. Kongpetch S, Kukongviriyapan V, Prawan A. et al. Crucial role of heme oxygenase-1 on the sensitivity of cholangiocarcinoma cells to chemotherapeutic agents. PLoS One. 2012;7:e34994
13. Berberat PO, Dambrauskas Z, Gulbinas A. et al. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin Cancer Res. 2005;11:3790-8
14. Miyake M, Ishii M, Kawashima K. et al. siRNA-mediated knockdown of the heme synthesis and degradation pathways: modulation of treatment effect of 5-aminolevulinic acid-based photodynamic therapy in urothelial cancer cell lines. Photochem Photobiol. 2009;85:1020-7
15. Zou C, Zhang H, Li Q. et al. Heme oxygenase-1: a molecular brake on hepatocellular carcinoma cell migration. Carcinogenesis. 2011;32:1840-8
16. Hill M, Pereira V, Chauveau C. et al. Heme oxygenase-1 inhibits rat and human breast cancer cell proliferation: mutual cross inhibition with indoleamine 2,3-dioxygenase. FASEB J. 2005;19:957-68
17. Alaoui-Jamali MA, Bismar TA, Gupta A. et al. A novel experimental heme oxygenase-1-targeted therapy for hormone-refractory prostate cancer. Cancer Res. 2009;69:8017-24
18. Xu J, Li J, Wang J. et al. Heme oxygenase1 protects H2O2-insulted glomerular mesangial cells from excessive autophagy. Mol Med Rep. 2016;13:5269-75
19. Rademacher BL, Matkowskyj KA, Meske LM. et al. The role of pharmacologic modulation of autophagy on anal cancer development in an HPV mouse model of carcinogenesis. Virology. 2017;507:82-8
20. Tan Q, Wang H, Hu Y. et al. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015;106:1023-32
21. Cao L, Wang J, Ma D. et al. Heme oxygenase-1 contributes to imatinib resistance by promoting autophagy in chronic myeloid leukemia through disrupting the mTOR signaling pathway. Biomed Pharmacother. 2016;78:30-8
22. Banerjee P, Basu A, Wegiel B. et al. Heme oxygenase-1 promotes survival of renal cancer cells through modulation of apoptosis- and autophagy-regulating molecules. J Biol Chem. 2012;287:32113-23
23. Bolisetty S, Traylor AM, Kim J. et al. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J Am Soc Nephrol. 2010;21:1702-12
24. Ostrowska KM, Malkin A, Meade A. et al. Investigation of the influence of high-risk human papillomavirus on the biochemical composition of cervical cancer cells using vibrational spectroscopy. Analyst. 2010;135:3087-93
25. Peitsaro P, Johansson B, Syrjanen S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J Clin Microbiol. 2002;40:886-91
26. Lorenzi A, Rautava J, Kero K. et al. Physical state and copy numbers of HPV16 in oral asymptomatic infections that persisted or cleared during the 6-year follow-up. J Gen Virol. 2017;98:681-9
27. Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861-73
28. Huang X, Xu Z, Mei Y. et al. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013;32:2204-16
29. Wang HX, Yang Y, Guo H. et al. HSPB1 deficiency sensitizes melanoma cells to hyperthermia induced cell death. Oncotarget. 2016;7:67449-62
30. Ryu DS, Kim SH, Lee DS. Anti-proliferative effect of polysaccharides from Salicornia herbacea on induction of G2/M arrest and apoptosis in human colon cancer cells. J Microbiol Biotechnol. 2009;19:1482-9
31. Ryu DS, Kim SH, Kwon JH. et al. Orostachys japonicus induces apoptosis and cell cycle arrest through the mitochondria-dependent apoptotic pathway in AGS human gastric cancer cells. Int J Oncol. 2014;45:459-69
32. Ryter SW, Mizumura K, Choi AM. The impact of autophagy on cell death modalities. Int J Cell Biol. 2014;2014:502676
33. Li W, Wang W, Si M. et al. The physical state of HPV16 infection and its clinical significance in cancer precursor lesion and cervical carcinoma. J Cancer Res Clin Oncol. 2008;134:1355-61
34. Surviladze Z, Sterk RT, DeHaro SA. et al. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J Virol. 2013;87:2508-17
35. Griffin LM, Cicchini L, Pyeon D. Human papillomavirus infection is inhibited by host autophagy in primary human keratinocytes. Virology. 2013;437:12-9
36. Mattoscio D, Casadio C, Miccolo C. et al. Autophagy regulates UBC9 levels during viral-mediated tumorigenesis. PLoS Pathog. 2017;13:e1006262
37. Tay SK, Jenkins D, Maddox P. et al. Tissue macrophage response in human papillomavirus infection and cervical intraepithelial neoplasia. Br J Obstet Gynaecol. 1987;94:1094-7
38. Nainu F, Shiratsuchi A, Nakanishi Y. Induction of Apoptosis and Subsequent Phagocytosis of Virus-Infected Cells As an Antiviral Mechanism. Front Immunol. 2017;8:1220
39. Huang H, Konduru K, Solovena V. et al. Therapeutic potential of the heme oxygenase-1 inducer hemin against Ebola virus infection. Curr Trends Immunol. 2016;17:117-23
40. Espinoza JA, Gonzalez PA, Kalergis AM. Modulation of Antiviral Immunity by Heme Oxygenase-1. Am J Pathol. 2017;187:487-93
41. Espinoza JA, León MA, Céspedes PF. et al. Heme Oxygenase-1 Modulates Human Respiratory Syncytial Virus Replication and Lung Pathogenesis during Infection. J Immunol. 2017;199:212-23
42. Dai L, Qiao J, Nguyen D. et al. Role of heme oxygenase-1 in the pathogenesis and tumorigenicity of Kaposi's sarcoma-associated herpesvirus. Oncotarget. 2016;7:10459-71
43. Das D, Bhattacharjee B, Sen S. et al. Association of viral load with HPV16 positive cervical cancer pathogenesis: causal relevance in isolates harboring intact viral E2 gene. Virology. 2010;402:197-202
44. Harima Y, Ohguri T, Imada H. et al. A multicentre randomised clinical trial of chemoradiotherapy plus hyperthermia versus chemoradiotherapy alone in patients with locally advanced cervical cancer. Int J Hyperthermia. 2016;32:801-8
45. Mariño G, Niso-Santano M, Baehrecke EH. et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81-94
46. Liu H, Zhang L, Zhang X. et al. PI3K/AKT/mTOR pathway promotes progestin resistance in endometrial cancer cells by inhibition of autophagy. Onco Targets Ther. 2017;10:2865-71
47. Liu L, Liao JZ, He XX. et al. The role of autophagy in hepatocellular carcinoma: friend or foe. Oncotarget. 2017;8:57707-22
48. Hippert MM, O'Toole PS, Thorburn A. Autophagy in cancer: good, bad, or both? Cancer Res. 2006;66:9349-51
49. Wang HY, Yang GF, Huang YH. et al. Reduced expression of autophagy markers correlates with high-risk human papillomavirus infection in human cervical squamous cell carcinoma. Oncol Lett. 2014;8:1492-8
50. Zhu LL, Gao XH, Qi R. et al. Local hyperthermia could induce antiviral activity by endogenous interferon-dependent pathway in condyloma acuminata. Antiviral Res. 2010;88:187-92
Author contact
![]() Corresponding authors: Liu Cao, Key Laboratory of Medical Cell Biology, Ministry of Education; Institute of Translational Medicine, China Medical University; Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning Province, 110122, China. Tel: +86 24 31939636, Fax: +86 24 23264417, e-mail: lcaoedu.cn. Shuai Han, Department of Neurosurgery, No.1 Hospital of China Medical University, Shenyang, Liaoning Province, 110001, China. Tel: +86 138 4056 9777, e-mail: hanshuai537197com.
Corresponding authors: Liu Cao, Key Laboratory of Medical Cell Biology, Ministry of Education; Institute of Translational Medicine, China Medical University; Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning Province, 110122, China. Tel: +86 24 31939636, Fax: +86 24 23264417, e-mail: lcaoedu.cn. Shuai Han, Department of Neurosurgery, No.1 Hospital of China Medical University, Shenyang, Liaoning Province, 110001, China. Tel: +86 138 4056 9777, e-mail: hanshuai537197com.