Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Methods and material
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(3):636-646. doi:10.7150/ijbs.29562 This issue Cite
Research Paper
SMARCAD1 Promotes Pancreatic Cancer Cell Growth and Metastasis through Wnt/β-catenin-Mediated EMT
Furao Liu1,2,#, Zebin Xia3,#, Meichao Zhang1,2,#, Jiping Ding1,2, Yang Feng1,2, Jianwei Wu1,2, Yun Dong1,2, Wei Gao1,2, Zengwei Han1,2, Yuanhua Liu4, Yuan Yao1,2, ![]() , Dong Li1,2,
, Dong Li1,2, ![]()
1. Department of Radiation Oncology, Hainan West Central Hospital (Shanghai Ninth People's Hospital, Hainan Branch), Shanghai Jiaotong University School of Medicine, Hainan, China.
2. Department of Radiation Oncology, Shanghai Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China.
3. Department of General Surgery, DaHua Hospital, Xuhui, Shanghai, China.
4. Department of Chemotherapy, Nanjing Medical University Affiliated Cancer Hospital, Cancer Institute of Jiangsu Province, Nanjing, Jiangsu, China
# These authors have contributed equally to this work
Received 2018-8-29; Accepted 2018-12-10; Published 2019-1-1
Abstract

Pancreatic cancer (PC) is one of the most lethal diseases, characterized by early metastasis and high mortality. Subunits of the SWI/SNF complex have been identified in many studies as the regulators of tumor progression, but the role of SMARCAD1, one member of the SWI/SNF family, in pancreatic cancer has not been elucidated. Based on analysis of GEO database and immunohistochemical detection of patient-derived pancreatic cancer tissues, we found that SMARCAD1 is more highly expressed in pancreatic cancer tissues and that its expression level negatively correlates with patients' survival time. With further investigation, it shows that SMARCAD1 promotes the proliferation, migration, invasion of pancreatic cancer cells. Mechanistically, we first demonstrate that SMARCAD1 induces EMT via activating Wnt/β-catenin signaling pathway in pancreatic cancer. Our results provide the role and potential mechanism of SMARCAD1 in pancreatic cancer, which may prove useful marker for diagnostic or therapeutic applications of PC disease.
Keywords: SMARCAD1, metastasis, EMT, Wnt/β-catenin
Introduction
Pancreatic cancer (PC) is the fourth leading cause of cancer-associated mortality in western countries and is predicted to be the second deadliest type of cancer in the year of 2030[1, 2]. PC is one of the cancer types with poorest prognosis, with low 5-year survival rate of 7.7% and a medium overall survival time of no more than one year [3]. Though many technologies and therapies have been applied to the diagnosis and treatment of pancreatic cancer, its death rate is still growing up by 0.3% a year [4]. There may be several reasons for this formidable situation. First, due to the lack of specific tumor markers and effective detecting tools, patients are diagnosed at advanced stages. Second, pancreatic cancer shows resistance to chemotherapy, radiotherapy and molecular targeted therapy. Furthermore, complex epigenetic changes pose a major challenge to the treatment of pancreatic cancer [5, 6].
Epithelial-Mesenchymal Transition (EMT) is a process that epithelial cells lose their junctions and gain mesenchymal traits, characterized by the downregulation of E-cadherin and upregulation of N-cadherin, vimentin, fibronectin and others [7, 8]. Numerous studies indicated that EMT plays a critical role in the progression and metastasis of pancreatic cancer [9-11]. Multiple signal pathways are involved in the regulation of EMT, among which the Wnt/β-catenin pathway is one of the typical impact factors [12, 13]. The canonical Wnt/β-catenin signal pathway has a crucial role in the development of pancreatic cancer [14]. Once Wnt ligands bind to their member receptors, the Wnt signal pathway would be triggered, along with the nuclear transposition of β-catenin. In the nuclei, β-catenin combines with the TCF/LEF transcription factors to co-activate a series of target genes including EMT markers [15, 16].
SWI/SNF was first described in yeast because of its critical role in responding to the mating type SWItch (SWI) and sucrose fermentation (SNF) [17, 18]. It is a large complex composed of more than 15 subunits, including SMARCA2, SMARCA4, SMARCAB1, SMARCAD1 etc., which are essential for regulating gene expression in mammalians [19]. SWI/SNF subunits are closely related to human diseases, especially cancer [20]. They have been found to mutate in nearly 20% of human tumors [21]. Among these, SMARCA2 and SMARCA4, two ATPase of SWI/SNF complex, were frequently linked to malignant cancer, e.g. pancreatic cancer. Recent study showed that SMARCA2 (BRM) promote growth but inhibit chemosensitivity of pancreatic cancer cells in vitro and vivo [22]. A series of investigations have confirmed that SMARCA4 (Brg1) promote differentiation, growth and chemosensitivity of PC, as well as inducing EMT-like phenotype [23-25].
SMARCAD1 is a chromatin remodeling ATPase of SNF2 helicase superfamily [26], which is similar to SMARCA2 and SMARCA4. SMARCAD1 contains an ATP-binding motif, two DEAD/H boxes and a CUE1 domain, which is required for the localization to nucleus [27, 28]. It plays a significant role in the maintenance of chromatin stability, most notably in DNA double-strand breaks (DSB) repair [29]. Loss of SMARCAD1 impairs the recombination of DSB and results in accumulation of acetylated histones [30-34]. In addition, SMARCAD1 can also bind to chromatin and DNA, affecting gene expression. Depletion of SMARCAD1 leads to an open chromatin conformation in normally transcriptionally repressed regions [28]. The gene coding for SMARCAD1 is mapped to human chromosome 4q22-q23, a region that is involved in several human diseases due to break points and deletion mutants of genes [27], which may indicate its biological importance. SMARCAD1 is expressed throughout the development, but its expression level is significantly higher in embryonal, mammary, and lymphoid tumors [35]. It was predicted to be associated with some human diseases, such as skin cancer susceptibility [36], Basan syndrome [37], bladder cancer [38] and breast cancer [39]. But the precise role of SMARCAD1 in PC and the underlying mechanism have not yet been explored.
In this study, we found by analyzing the GEO database and pancreatic cancer tissue chips that SMARCAD1 is upregulated in pancreatic cancer tissues and correlates with tumor progression. We investigated the role of SMARCAD1 in the proliferation, migration, invasion in pancreatic cancer cells. Mechanistically, we first demonstrate that SMARCAD1 induces EMT via activating Wnt/β-catenin signaling pathway in pancreatic cancer cell.
Results
SMARCAD1 is highly expressed in pancreatic cancer tissues
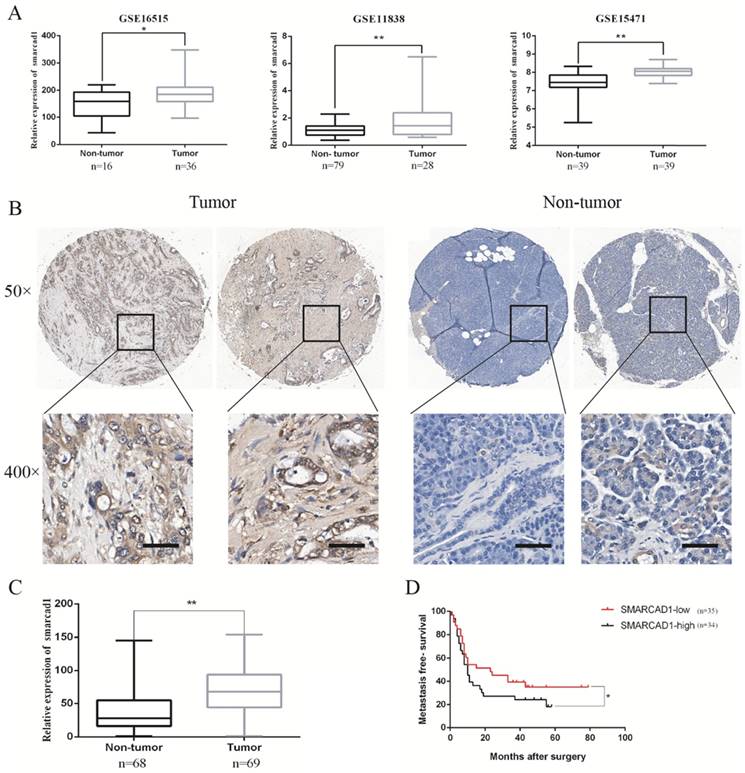
To determine the main members of SWI/SNF family`s expression level in pancreatic cancer tissues, we examined the open Gene Expression Omnibus (GEO) database and excavated the data from three mRNA expression profile datasets (GSE 16515, GSE 11838, GSE 15471). Through analyzing differences in gene expression between pancreatic cancer and adjacent tissues in these date sets, we found that the expression of SMARCA2 or SMARCA4 in pancreatic cancer tissues has changed only in one or two data sets (Fig. S1). However, the expression of SMARCAD1 in pancreatic cancer tissues was increased consistently in all the three data sets (Fig. 1). As gene mutations or amplification can lead to changes in gene expression, we queried cBioPortal data platform to check the mutation status and amplification of SMARCAD1 in pancreatic cancer. The mutation rate of SMARCAD1 in pancreatic cancer was found to be less than 0.5% (Fig. S2). But the amplification frequency was higher than 2.5%, which may contribute to the upregulation of SMARCAD1 in pancreatic cancer (Fig. S2). To further characterize the role of SMARCAD1 in pancreatic cancer, we examined SMARCAD1 protein expression by immunohistochemistry in 69 PC tissues and 68 adjacent normal pancreatic tissues. Comparing the SMARCAD1 H-scores in the cancer tissue and the normal tissue from 69 pancreatic cancer patients, we found that SMARCAD1 protein levels were much higher in pancreatic cancer tissues (P<0.01) (Fig. 1B, C). Kaplan-Meier survival analysis also revealed that patients with high level SMARCAD1 proteins in tumor displayed poorer metastasis-free survival than those with low levels SMARCAD1 proteins (P<0.05) (Fig. 1D). These data suggest that SMARCAD1 may be a potential regulator for the development and progression of pancreatic cancer.
The expression of SMARCAD1 is significantly higher in pancreatic cancer and positively correlated with poor prognosis. A. The mRNA expression level of SMARCAD1 in pancreatic cancer is much higher than that of normal tissues from GSE16515, GSE11838 and GSE15471. B-C. Expression levels of SMARCAD1 by immunohistochemistry performed with tissue microarray of PC (n=69) and adjacent normal tissues (n=68). Representative images showed positive expression of SMARCAD1 in PC and negative expression in paired normal tissues, respectively. Scale bars=50μm. D. Kaplan-Meier analysis shows the correlation between SMARCAD1 expression and overall survival in patients. Patients with high SMARCAD1 expression had poorer overall survival than those with low expression. *p<.05, **p<.01.
SMARCAD1 promotes proliferation of PANC-1 cells
In order to investigate the correlation between SMARCAD1 expression and pancreatic cancer development, CCK8 and colony-forming assay were conducted in pancreatic cancer cell line PANC-1. First, we depleted the SMARCAD1 gene with shRNAs in PANC-1 cells, and the efficiency of SMARCAD1 knockdown was confirmed by western blot (Fig. 2A). Then viability of cells was measured by CCK8 assay, which showed that SMARCAD1 knockdown decreased the growth rate of PANC-1 (P<0.05) (Fig. 2C). In addition, we analyzed cell-cycle distribution by using flow cytometry and discovered the slight accumulation of PC cells in G1 phase and reduction in S-phase following SMARCAD1 knockdown (Fig. S3). Above results were also proved by clone formation assay, in which SMARCAD1 knockdown reduced the number of clones by 70%, compared with control (P<0.01) (Fig. 2E). These data indicated that SMARCAD1 may play a positive role in controlling the proliferation of pancreatic cancer cell.
SMARCAD1 enhances proliferation of PANC-1 cells. A-B. The efficiency of SMARCAD1 knockdown (A) or overexpression (B) in PANC-1 cells was detected by western blotting. β-actin was used as an internal control. C-D. CCK8 assay was performed to determine the proliferation of PANC-1 cells with SMARCAD1 knockdown (C) or overexpression (D) at the indicated time points after plated. Cell viability was measured at 450nm. E-F. The effect of SMARCAD1 knockdown (E) or overexpression (F) on Colony-forming of PANC-1 cells was shown in the top panels. Number of foci was counted as shown in the bottom panels. All data were presented as mean ±SEM. *p<.05, **p<.01.
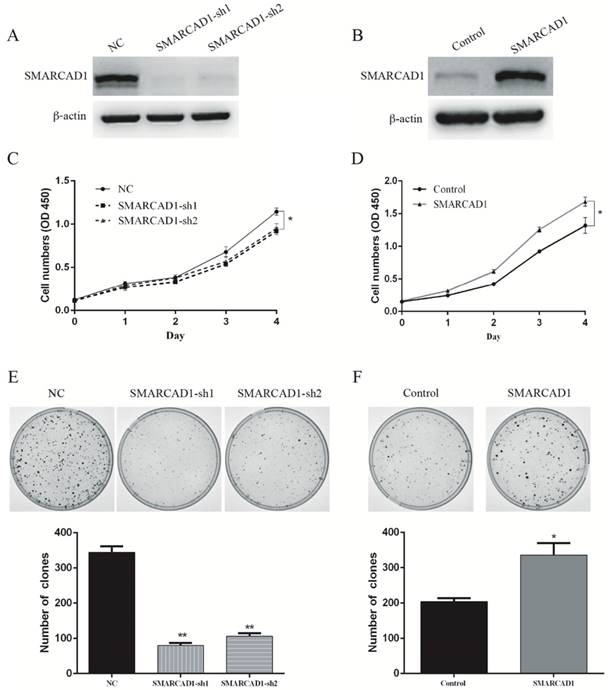
To further confirm the effect of SMARCAD1 on the proliferation of PANC-1 cell line, SMARCAD1-lentivirus was constructed and the expression was validated by western blot (Fig. 2B). In keeping with the results from SMARCAD1 depletion, ectopic expression of SMARCAD1 increased viability of PANC-1 cells (P<0.01) (Fig. 2D). Moreover, colony-forming ability of PANC-1 cells with SMARCAD1 overexpression was almost 1.5 fold higher than that of the control (P<0.01) (Fig. 2F). Taken together, these results identified that SMARCAD1 serves as a critical regulator in controlling the proliferation of pancreatic cancer cell.
SMARCAD1 enhances migration and invasion in PANC-1 cells
To identify the function of SMARCAD1 on migration and invasion of pancreatic cancer, we performed wound-healing and transwell assay with PANC-1 cells, which showed that the motility of PANC-1 cells was affected by the expression level of SMARCAD1. Compared with the control cells, the cells with SMARCAD1 depletion presented a prominent reduction in wound formation capacity (P<0.01) (Fig. 3A). This inhibiting effect was also verified by transwell migration assay. The migration rate of the cells with SMARCAD1 depletion was much lower than that of the control cells (P<0.01) (Fig. 3C). In contrast with the results from SMARCAD1 knockdown, overexpression of SMARCAD1 significantly promoted ability of wound formation and migration in PANC-1 cells (P<0.01) (Fig. 3B, 3D).
SMARCAD1 promotes migration and invasion of PANC-1 cells. A-B. Effect of SMARCAD1 knockdown (A) or SMARCAD1 overexpression (B) on cell migration was detected by wound healing at indicated time points after scratching. The wound healing was measured by ImageJ software. C-D. Motility ability of PANC-1 cells with SMARCAD1 depletion (C) or overexpression (D) was assessed by transwell assay at 24h. Representative images of migration were photographed at 24h (Top panel). The number of migrated cells was counted from 5 randomly selected fields under microscope (Bottom panel). E-F. Invasion ability of PANC-1 cells with SMARCAD1 depletion (E) or overexpression (F) was assessed by transwell assay at 48h. Representative images of invasion were photographed at 48h (Top panel). The number of invaded cells was counted from 5 randomly selected fields under microscope (Bottom panel). Scale bars=150um. Data were presented as mean ±SEM. *p<.05, **p<.01.
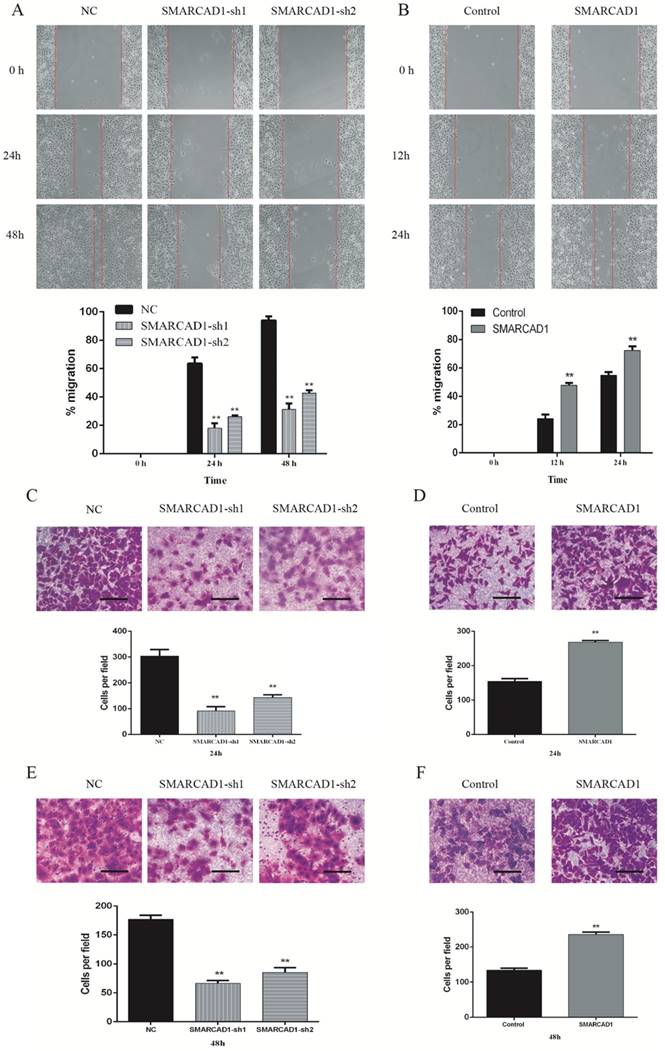
We further analyses the effects of SMARCAD1 on metastatic ability of PANC-1 cells by invasion assay. Compared with the negative group, SMARCAD1 knockdown decreased cells' invasion rate significantly (P<0.01) (Fig. 3E), while SMARCAD1 overexpression increase the cells' invasion ability (P<0.01) (Fig. 3F). Based on these findings, we could conclude that SMARCAD1 enhances the migration and invasion of pancreatic cancer cell.
SMARCAD1 has no significant effect on sensitivity of PANC-1 cells to gemcitabine
It has been proved that SMARCAD1 played an important role in the maintenance of chromatin stability [33, 34]. O'Donnell, P. H. et.al suggested that SMARCAD1 was associated with capecitabine/5-FU susceptibility [40]. Therefore, we attempted to investigate whether SMARCAD1 has an effect on the chemosensitivity of PANC-1 cells. We treated PANC-1 cells with different concentrations of gemcitabine for 48 hours. The results of CCK8 assay showed that knockdown of SMARCAD1 had little effect on the sensitivity of PANC-1 to gemcitabine (Fig. S4). Above result indicated that SMARCAD1 has no obvious effect on gemcitabine susceptibility of PANC-1 cells.
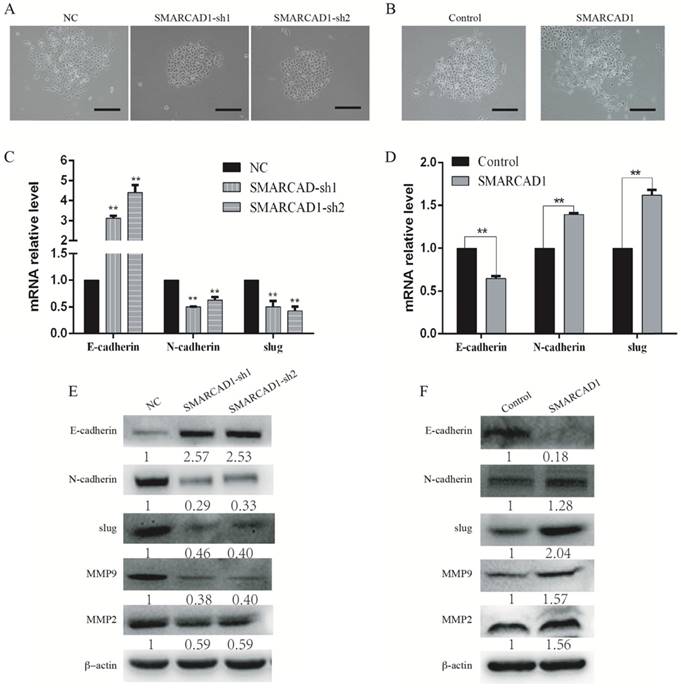
SMARCAD1 regulates EMT in PANC-1 cells
With SMARCAD1 depletion or ectopic expression, we observed significantly changes in cell morphology compared to that of the control cells (Fig. 4A-B). The PANC-1 cells with SMARCAD1 knockdown presented more characteristics of epithelium and gained more contacts with each other (Fig. 4A). However, with SMARCAD1 overexpression, PANC-1 cells acquired more fibroblast-like mesenchymal traits: cells lost polarity, and the distribution is more dispersed (Fig. 4B). Those cell morphological changes were consistent with characteristics of Epithelial-Mesenchymal Transition (EMT). Given that EMT is associated with cell motility [7], we then examined whether EMT was responsible for SMARCAD1-mediated changes in migration and invasion. E-cadherin, N-cadherin and Slug are recognized hallmarks which indicated the existence of EMT [8]. We next measured the expression of E-cadherin, N-cadherin and Slug in PANC-1 cells by using western blotting and Quantitative Real-Time PCR. The epithelial marker E-cadherin was upregulated significantly in SMARCAD1 knockdown cells, while the mesenchymal markers N-cadherin and Slug were downregulated (Fig. 4C, E). In addition, members of the matrix metalloproteinase family, MMP2 and MMP9, are closely related to cell invasion and migration [41]. Then we tested the expression changes of MMP2 and MMP9 in this study. Compared with the control group, SMARCAD1 knockdown lead to significantly reduction of MMP2 and MMP9 (Fig. 4E). In contrast, overexpression the SMARCAD1 resulted in the downregulation of E-cadherin, accompanied with upregulation of N-cadherin, Slug, MMP2 and MMP9 (Fig. 4D, 4F). Those findings validate the role of SMARCAD1 in EMT induction.
SMARCAD1 induces EMT in PANC-1 cells. A-B. The morphology changes of PANC-1 cells: cells lose contact with each other with SMARCAD1 depletion (A) or gain more contact with SMARCAD1 overexpression (B), Scale bars=250μm. C-D. Changes in mRNA level of EMT relative markers were tested by Quantitative real-time PCR in SMARCAD1 knockdown (C) or overexpression (D) cells. The results were presented as mean ±SEM. All values were normalized to the level (=1) in NC or control cells. *p<.05, **p<.01. (D). E-F. The protein levels of EMT relative markers in SMARCAD1 knockdown (E) or overexpression (F) cells were assessed by western blotting. β-actin was used as an internal control.
SMARCAD1 regulates EMT by modulating Wnt/β-catenin pathway
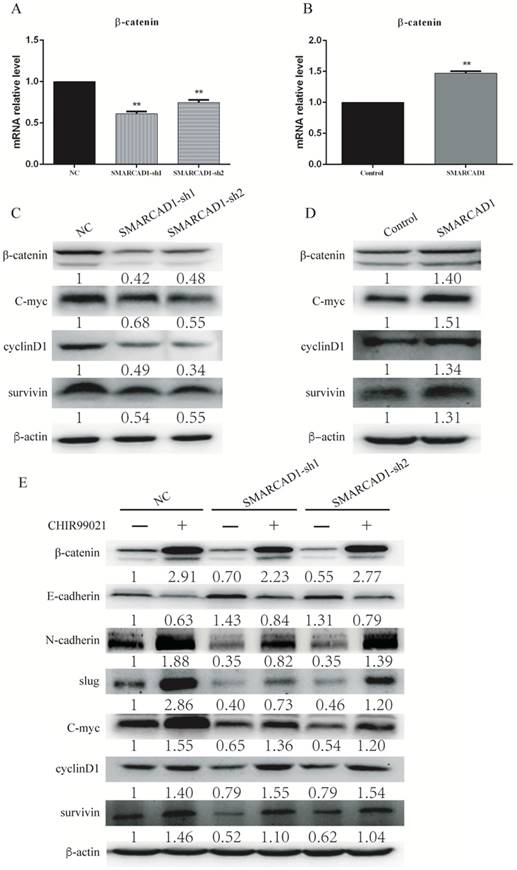
β-catenin is a core molecule of the Wnt/β-catenin signaling pathway, playing a significant role in tumorigenesis [42]. In addition, elevated expression of β-catenin could induce epithelial-mesenchymal transition in most cases [43]. SMARCAD1 is known as a chromatin remodeler involving the regulation of gene expression [44]. To gain an insight into the mechanism by which SMARCAD1 mediated EMT, we examined the expression of β-catenin and its direct downstream molecules, including c-Myc, Cyclin D1 and survivin [43, 45]. With SMARCAD1 knockdown, both mRNA and protein levels of β-catenin in PANC-1 cells were downregulated (Fig. 5A, 5C). Conversely, overexpression of SMARCAD1 led to the upregulation of β-catenin in mRNA and protein levels (Fig. 5B, 5D). These results indicate that SMARCAD1 regulates β-catenin expression at the transcriptional level, however, the more detailed mechanisms need to be further explored. Next, we assessed the expression of the downstream targets of β-catenin by using western blot. Upon SMARCAD1 knockdown, the expression of c-Myc, Cyclin D1 and survivin in PANC-1 cells was significantly reduced (Fig. 5C), which may be the reason for the change of cell cycle. In contrast, overexpression of SMARCAD1 enhanced the expression of these genes (Fig. 5D). To further confirm that SMARCAD1 regulated EMT through the Wnt/β-catenin pathway, we performed rescue experiment in PANC-1 cell line by using CHIR99021, an activator specific for Wnt/β-catenin pathway [45]. As shown in Fig. 5E, CHIR-99021 could rescue the suppression of genes caused by SMARCAD1 knockdown. Together, these results suggest that SMARCAD1 regulates EMT by targeting the Wnt/β-catenin pathway.
SMARCAD1-induced EMT was regulated by Wnt/beta-catenin signaling pathway. A-B. The mRNA level of β-catenin was detected by Quantitative real-time PCR in PANC-1 cells with SMARCAD1 knockdown (A) or overexpression (B) respectively. The data were presented as mean ±SEM. All values were normalized to the level (=1) in NC or control cells. *p<.05, **p<.01. C-D. β-catenin, cyclin-D1, c-Myc and survivin protein levels were assayed by western blotting in PANC-1 cells with SMARCAD1 knockdown (C) or overexpression (D) respectively. E. PANC-1 cells with SMARCAD1 depletion were treated with CHIR99021 (6μM/ml) for 24h. The protein levels of EMT markers and Wnt/β-catenin target genes (β-catenin, cyclin-D1, c-Myc and survivin) were detected by western blotting. β-actin was used as an internal control.
The sequences of primers for Quantitative Real-Time PCR
| Gene names | Primer Sequence |
|---|---|
| β-actin-F | 5'-GGCTGTATTCCCCTCCATCG-3 |
| β-actin-R | 5'-CCAGTTGGTAACAATGCCATGT-3' |
| Slug-F | 5'-GGGGAGAAGCCTTTTTCTTG-3' |
| Slug-R | 5'-TCCTCATGTTTGTGCAGGAG-3' |
| β-catenin-F | 5'-CATCTACACAGTTTGATGCTGCT-3' |
| β-catenin-R | 5'-GCAGTTTTGTCAGTTCAGGGA-3' |
| E-cadherin-F | 5'-GAGTGCCAACTGGACCATTCAGTA-3' |
| E-cadherin-R | 5'-AGTCACCCACCTCTAAGGCCATC-3' |
| N-cadherin-F | 5'-GTCAGCAGAAGTTGAAGAAATAGTG-3' |
| N-cadherin-R | 5'-GCAAGTTGATTGGAGGGATG-3' |
Discussion
This study presented evidence for the role and mechanism of SMARCAD1 in the progression of pancreatic cancer cell's migration and invasion for the first time. Through analysis of GEO database and pancreatic cancer tissue chip, we confirmed that SMARCAD1 expression was upregulated in pancreatic cancer and correlated with tumor progression. Furthermore, a series of functional assays demonstrated that SMARCAD1 induced EMT by regulating the transcription of β-catenin to facilitate distant metastasis of pancreatic cancer cell.
The progression of tumor is a complex and multi-step process, in which genetic and epigenetic changes are the two most important factors leading to tumorigenesis [46, 47]. It has been identified that the SNF2 helicase superfamily are subjected to epigenetic alterations in human malignancies [19, 48, 49]. SMARCAD1 is a member of the SNF2 helicase superfamily, potentially involved in the soft tissue malignancies like leiomyosarcoma, hepatocellular carcinoma, and hematologic malignancies [27]. However, to date, there are few studies on the biological function and related mechanism of SMARCAD1 in cancers. A previous study showed that SMARCAD1 knockdown markedly reduces metastasis of breast cancer [39]. In present study, we observed that the expression level of SMARCAD1 was significantly increased in pancreatic cancer. Our study also shows that the prognosis of patient with high expression of SMARCAD1 is poor. A variety of in vitro assays, including CCK8, clone formation, wound healing and transwell, have demonstrated that SMARCAD1 knockdown decreased malignancy of PANC-1 cells by inhibiting cell proliferation and metastasis. Consistently, ectopic expression of SMARCAD1 led to an increase in PANC-1 malignancy by promoting cells' proliferation, migration and invasion. Therefore, we hypothesized that SMARCAD1 is an important factor during the progression of pancreatic cancer cell.
EMT is a process in which epithelial cells escape from the structural constrains by altering cytoskeleton and cell types. The activation of EMT is considered to be the causative factor leading to metastasis cascade [50-53]. Andrew D. Rhim et al showed that EMT is associated with the metastasis of pancreatic cancer [11]. Thus, discovery of the essential mechanisms that induce EMT is crucial for the prevention and treatment for tumor invasion and migration. We found that decreased expression of SMARCAD1 led to upregulation of E-cadherin and downregulation of N-cadherin, slug, MMP2 and MMP9. In contrast, ectopic expression of SMARCAD1 upregulates N-cadherin, slug, MMP2 and MMP9, while downregulating E-cadherin. Those findings were also supported by morphological changes of PANC-1. The results show that SMARCAD1 induces EMT in PANC-1 cells.
Among all the signaling pathways, Wnt/β-catenin pathway has a major function in EMT [45]. In this study, we confirmed that expression of β-catenin, c-Myc, cyclin D1 and survivin were enhanced or inhibited with SMARCAD1 knockdown and overexpression. Moreover, the expression changes of β-catenin and its downstream molecules caused by SMARCAD1 depletion can be rescued by CHIR99021, a Wnt/β-catenin pathway activator. These results demonstrated that SMARCAD1 regulates Wnt/β-catenin signaling pathway in PANC-1 cells. To gain insights into the regulatory relationship between SMARCAD1 and Wnt pathway, we pay attention to the transcription changes of β-catenin, which is a sign of Wnt signaling pathway and critical for triggering EMT. By Real-Time PCR, we found that SMARCAD1 knockdown decreased mRNA level of β-catenin, while SMARCAD1 overexpression increased it. SMARCAD1 is known as a subunit of SWI/SNF chromatin remodeling complex, which is famous for its function on maintaining chromatin structure and gene expression [54]. SWI/SNF complex make use of the energy provided by ATP to change the position of the nucleosome, thereby adjusting chromatin architecture and ultimately regulating the gene transcription [55, 56]. And fun30, a homologue of SMARCAD1 in Saccharomyces cerevisiae, has also been proved to bind chromatin and DNA directly [28, 57]. Thus we consider that SMARCAD1 may regulate the transcription level of β-catenin through its function of chromatin remodeling. Along with transcriptional alteration of β-catenin, the expression of c-Myc, cyclin D1, survivin and EMT markers were changed, which then lead to a shift in biological behavior of PANC-1 cells. However, the more detailed mechanisms still need to be further explored in the future.
In conclusion, this study demonstrated that SMARCAD1 plays a significant effect in the survival and distant metastasis of pancreatic cancer cell. SMARCAD1 activates Wnt/β-catenin signaling pathway by altering the expression of β-catenin. Activated Wnt signaling pathway induces EMT and promotes cancer metastasis.
Methods and material
Bioinformatics analysis
We first searched for data sets containing pancreatic cancer and normal tissues in the GEO database, excluding the data that had been treated with chemotherapy or radiotherapy. In order to increase the credibility of the statistical results, we have abandoned data with less than 50 overall samples or less than 20 cancer samples. Finally, we got three data sets (GSE16515, GSE11838, and GSE15471), which contain 36 tumor samples and 16 normal samples, 28 tumor samples and 79 normal samples, 39 pairs of tumor and normal samples respectively. All the data were analyzed by GraphPad prism 6 software.
Cell line and cultures
Human pancreatic cancer cell line PANC-1 and Human embryonic kidney epithelial cell line 293T were cultured in DMEM (Gibco company), supplemented with 10% fetal bovine serum (FBS), 100U/mL Penicilin, 100µg/mL Streptomycin and 2mM L-Glutamine. Cells were cultured in a 37˚C incubator containing 95% O2 and 5% CO2.
Human tissue microarray
Tissue microarray that contains 69 primary pancreatic cancers and 68 adjacent normal pancreatic tissues were purchased from Shanghai Xinchao Company (Shanghai, China). No patients enrolled in the study had received preoperative treatment. The specimens were incubated with anti-SMARCAD1 antibody (1:50), and staining results were observed with a Nikon ECLIPSETs2R microscope.
Lentivirus infection for the construction of SMARCAD1-knockdown and over-expression
Lentiviral constructs expressing SMARCAD1 shRNAs and negative control were obtained from Sigma. The sequences of target shRNAs were as follows:
Sh1 5'-CCAGCACCTTATGACAATTAA-3'.
Sh2 5'-GCCTTATTTGACAATGCTTAT-3'.
Lentivirus was produced in 293T cells using the transfection reagent (QIAGEN) following the manufacturer's instructions. Supernatant containing virus was collected after 48h transfection. PANC-1 cells were infected with collected virus containing polybrene (10µg/ml), followed by selection with puromycin.
For over-expression of SMARCAD1, pLX304-V5-Blast was constructed and used. PANC-1 cells are transfected at the confluence between 30% and 50% and were selected with Blasticidin S (10µg/mL) after 48 hours of transfection.
Western blotting
Proteins were lysed by lysis buffer (20mM Tris, pH 7, 0.5% NP-40, 250mM NaCl, 3mM EDTA, 3mM EGTA, 2mM DTT, 0.5mM PMSF, 20mM β-glycerol phosphate, 1mM sodium vanadate, and 1 mg/mL of leupeptin). Anti-bodies used are as follows:
Anti-SMARCAD1 (CST, 12458), anti-β-actin (Transgen Biotech, Hc201), anti-E-cadherin (CST, 14472), anti-N-cadherin (BD, 610920), anti-slug (CST, 9585), anti-β-catenin (CST, 9562),anti-MMP9 (CST, 2270), anti-MMP2 (ABclonal, A6247), anti-c-Myc (CST, 9402), anti-Cyclin D1 (CST, 2978), anti-survivin (Sigma, ab8228).
RNA extraction and Quantitative real-time PCR
RNA samples were extracted with TRIzol reagent (9018; Takara) and reversed by PrimeScript RTreagent Kit with gDNA Eraser (RR037A, TAKARA). Genes of interest were detected using SYBR Green Master Mix Reagent (11201ES03, Yeasen) using Roche Light Cycler480 Real-Time PCR detector. All reagents were used accordingly to the manufacturers' instructions.
Cell viability assay
Cells (3×103 per well) were planted in 96-well plates. The viability of cell was measured at 0h, 24h, 48h, 72h, 96h by CCK8 (DOJINDO,CK04) assay at 450nm.
Clone formation assay
About two or three thousand cells were seeded in 60mm dish. After culturing 10-14 days, clones (≥50 cells/clone) were counted using Nikon ECLIPSETs2R microscope.
Wound healing assay
Scratch was made by 200ul pipette tip when the cell confluence grown to 90%. Then cells were incubated without FBS for three days. Gap size was measured at indicated time points respectively.
Transwell migration and invasion assays
The transwell filter chambers were pre-coated by with or without matrigel (BD Bioscience). 200ul cell suspension (5×104 cells) were seeded into the upper chamber, while the lower chamber was filled with 600ul medium containing 10%FBS. After 24-48h incubation, cells invaded the lower chamber were fixed and stained with 0.1% crystal violet.
Statistics
The t-test was used to determine statistical significance, and the measurement data was shown as the mean ± standard deviation. The results were considered statistically significant while P<0.05.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by National Natural Science Foundation of China (grant81370600, D.L.); The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (TP2015022, D.L.); Shanghai Pujiang Program (15PJ1404800, D.L.) and Innovation Program of Shanghai Municipal Education Commission (15ZZ056, D.L.).
Authorship
LFR, XZB, ZMC designed and performed research, analyzed data, and prepared the manuscript. DJP, FY, DY, WJW, GW, HZW and LYH performed research. LFR, LD and YY designed research, analyzed data, and prepared the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. NAT REV GASTRO HEPAT. 2018;15:333-48
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. CANCER RES. 2014;74:2913-21
3. Ko J, Bhagwat N, Black T, Yee SS, Na YJ, Fisher SA. et al. miRNA profiling of magnetic nanopore-isolated extracellular vesicles for the diagnosis of pancreatic cancer. CANCER RES. 2018;78:3688-97
4. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
5. He J, Ahuja N, Makary MA, Cameron JL, Eckhauser FE, Choti MA. et al. 2564 resected periampullary adenocarcinomas at a single institution: trends over three decades. HPB. 2014;16:83-90
6. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646-74
7. Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438-49
8. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-8
9. Wang S, Huang S, Sun YL. Epithelial-Mesenchymal Transition in Pancreatic Cancer: A Review. Biomed Res Int. 2017;2017:1-10
10. Gaianigo N, Melisi D, Carbone C. EMT and Treatment Resistance in Pancreatic Cancer. Cancers (Basel). 2017:9
11. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F. et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349-61
12. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. SCI SIGNAL. 2014;7:re8
13. Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973-81
14. Nakamoto M, Hisaoka M. Clinicopathological Implications of Wingless/int1 (WNT) Signaling Pathway in Pancreatic Ductal Adenocarcinoma. J UOEH. 2016;38:1-8
15. Lien WH, Fuchs E. Wnt some lose some: transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 2014;28:1517-32
16. Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP. et al. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245-56
17. GARVIK JEHAB. <A new gene affecting the efficiency of mating-type interconversions in homothallic strains of Saccharomyces cerevisiae.pdf>. Genetics. 1977;87:33-50
18. LENORE NEIGEBORN, CARLSON M. <GENES AFFECTING THE REGULATION OF SUCZ GENE EXPRESSION BY GLUCOSE REPRESSION IN SACCHAROMYCES CEREVISIAE.pdf>. Genetics. 1984;108:845-58
19. Savas S, Skardasi G. The SWI/SNF complex subunit genes: Their functions, variations, and links to risk and survival outcomes in human cancers. Crit Rev Oncol Hematol. 2018;123:114-31
20. Lu C, Allis CD. SWI/SNF complex in cancer. Nat Genet. 2017;49:178-9
21. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J. et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592-601
22. Zhang Z, Wang F, Du C, Guo H, Ma L, Liu X. et al. BRM/SMARCA2 promotes the proliferation and chemoresistance of pancreatic cancer cells by targeting JAK2/STAT3 signaling. Cancer Lett. 2017;402:213-24
23. Nilotpal Roy SM, Karina E. Villanueva, Atsushi Urano, Xinyuan Lu, Guido, Von Figura ESS, David W. Dawson, Eric A. Collisson, and Matthias Hebrok. <Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation.pdf>. GENES & DEVELOPMENT. 2015;29:658-71
24. Liu X, Tian X, Wang F, Ma Y, Kornmann M, Yang Y. BRG1 promotes chemoresistance of pancreatic cancer cells through crosstalking with Akt signalling. Eur J Cancer. 2014;50:2251-62
25. Rosson GB, Bartlett C, Reed W, Weissman BE. BRG1 loss in MiaPaCa2 cells induces an altered cellular morphology and disruption in the organization of the actin cytoskeleton. J Cell Physiol. 2005;205:286-94
26. Terui R, Nagao K, Kawasoe Y, Taki K, Higashi TL, Tanaka S. et al. Nucleosomes around a mismatched base pair are excluded via an Msh2-dependent reaction with the aid of SNF2 family ATPase Smarcad1. Genes Dev. 2018;32:806-21
27. Adra CN, Donato JL, Badovinac R, Syed F, Kheraj R, Cai H. et al. SMARCAD1, a novel human helicase family-defining member associated with genetic instability: cloning, expression, and mapping to 4q22-q23, a band rich in breakpoints and deletion mutants involved in several human diseases. Genomics. 2000;69:162-73
28. Ding D, Bergmaier P, Sachs P, Klangwart M, Ruckert T, Bartels N. et al. The CUE1 domain of the SNF2-like chromatin remodeler SMARCAD1 mediates its association with KRAB-associated protein 1 (KAP1) and KAP1 target genes. J Biol Chem. 2018;293:2711-24
29. Chakraborty S, Pandita RK, Hambarde S, Mattoo AR, Charaka V, Ahmed KM. et al. SMARCAD1 Phosphorylation and Ubiquitination Are Required for Resection during DNA Double-Strand Break Repair. iScience. 2018;2:123-35
30. Jasencakova Z, Groth A. Broken Silence Restored—Remodeling Primes for Deacetylation at Replication Forks. Mol Cell. 2011;42:267-9
31. Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B. et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature. 2012;489:581-4
32. Rother MB, van Attikum H. DNA repair goes hip-hop: SMARCA and CHD chromatin remodellers join the break dance. Philos Trans R Soc Lond B Biol Sci. 2017;372:1-13
33. Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, Daza-Martin M. et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol. 2016;23:647-55
34. Rowbotham SP, Barki L, Neves-Costa A, Santos F, Dean W, Hawkes N. et al. Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol Cell. 2011;42:285-96
35. Okazaki N, Ikeda S, Ohara R, Shimada K, Yanagawa T, Nagase T. et al. The Novel Protein Complex with SMARCAD1/KIAA1122 Binds to the Vicinity of TSS. J Mol Biol. 2008;382:257-65
36. Günther C, Lee-Kirsch MA, Eckhard J, Matanovic A, Kerscher T, Rüschendorf F. et al. SMARCAD1 Haploinsufficiency Underlies Huriez Syndrome and Associated Skin Cancer Susceptibility. J INVEST DERMATOL. 2018;138:1428-31
37. Li M, Wang J, Li Z, Zhang J, Ni C, Cheng R. et al. Genome-wide linkage analysis and whole-genome sequencing identify a recurrent SMARCAD1 variant in a unique Chinese family with Basan syndrome. EUR J HUM GENET. 2016;24:1367-70
38. Tapak L, Saidijam M, Sadeghifar M, Poorolajal J, Mahjub H. Competing risks data analysis with high-dimensional covariates: an application in bladder cancer. GENOM PROTEOM BIOINF. 2015;13:169-76
39. Al Kubaisy E, Arafat K, De Wever O, Hassan AH, Attoub S. SMARCAD1 knockdown uncovers its role in breast cancer cell migration, invasion, and metastasis. Expert Opin Ther Targets. 2016;20:1035-43
40. O'Donnell PH, Stark AL, Gamazon ER, Wheeler HE, McIlwee BE, Gorsic L. et al. Identification of novel germline polymorphisms governing capecitabine sensitivity. Cancer. 2012;118:4063-73
41. M.W. ROOMI JCM, T. KALINOVSKY, M. RATH, A. NIEDZWIECKI. In vitro modulation of MMP-2 and MMP-9 in human cervical and ovarian cancer cell lines by cytokines, inducers and inhibitors. ONCOL REP. 2010;23:605-14
42. Karl Willert RN. β-catenin: a key mediator of Wnt signaling. CURR OPIN GENET DEV. 1998;8:95-102
43. Yang S, Liu Y, Li MY, Ng CSH, Yang SL, Wang S. et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/beta-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. 2017;16:124
44. Doiguchi M, Nakagawa T, Imamura Y, Yoneda M, Higashi M, Kubota K. et al. SMARCAD1 is an ATP-dependent stimulator of nucleosomal H2A acetylation via CBP, resulting in transcriptional regulation. Sci Rep. 2016;6:20179
45. Liu CC, Cai DL, Sun F, Wu ZH, Yue B, Zhao SL. et al. FERMT1 mediates epithelial-mesenchymal transition to promote colon cancer metastasis via modulation of β-catenin transcriptional activity. Oncogene. 2017;36:1779-92
46. D.F. Alonso GVR, J. Garona, N.B. Iannucci and D.E. Gomez. Metastasis: Recent Discoveries and Novel Perioperative Treatment Strategies with Particular Interest in the Hemostatic Compound Desmopressin. CURR PHARM BIOTECHNO. 2011;12:1974-80
47. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679-95
48. Glaros S, Cirrincione GM, Muchardt C, Kleer CG, Michael CW, Reisman D. The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene. 2007;26:7058-66
49. Karnezis AN, Wang YM, Ramos P, Hendricks WP, Oliva E, D'Angelo E. et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J Pathol. 2016;238:389-400
50. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21-45
51. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED. et al. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374-83
52. Valastyan S, Weinberg RA. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell. 2011;147:275-92
53. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488-94
54. Pulice JL, Kadoch C. Composition and Function of Mammalian SWI/SNF Chromatin Remodeling Complexes in Human Disease. Cold Spring Harb Symp Quant Biol. 2016;81:53-60
55. Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273-304
56. Dykhuizen EC, Hargreaves DC, Miller EL, Cui K, Korshunov A, Kool M. et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIalpha. Nature. 2013;497:624-7
57. Lee J, Choi ES, Seo HD, Kang K, Gilmore JM, Florens L. et al. Chromatin remodeller Fun30Fft3 induces nucleosome disassembly to facilitate RNA polymerase II elongation. Nat Commun. 2017;8:14527
Author contact
![]() Corresponding authors: Department of Radiation Oncology, Shanghai Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China; E-mail: lidongedu.cn (Dong Li); yaoyuanedu.cn (Yuan Yao)
Corresponding authors: Department of Radiation Oncology, Shanghai Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China; E-mail: lidongedu.cn (Dong Li); yaoyuanedu.cn (Yuan Yao)