Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(3):668-679. doi:10.7150/ijbs.30168 This issue Cite
Research Paper
Attenuation of STAT3 Phosphorylation Promotes Apoptosis and Chemosensitivity in Human Osteosarcoma Induced by Raddeanin A
Zhuoying Wang1,2*, Chongren Wang1,2*, Dongqing Zuo1,2*, Tao Zhang2, Fei Yin1,2, Zifei Zhou2, Hongsheng Wang2, Jing Xu1,2, Min Mao2, Gangyang Wang2, Yingqi Hua1,2 ![]() , Wei Sun1,2
, Wei Sun1,2 ![]() , Zhengdong Cai1,2
, Zhengdong Cai1,2 ![]()
1. Department of Orthopaedics, Shanghai General Hospital, School of Medicine Shanghai Jiao Tong University, Shanghai, China;
2. Shanghai Bone Tumor Institution, Shanghai, China
* These authors contributed equally to this work.
Received 2018-9-24; Accepted 2018-12-7; Published 2019-1-24
Abstract

Osteosarcoma (OS) is the most common primary bone malignancy in adolescents. One major obstacle for current OS treatment is drug-resistance. Raddeanin A (RA), an oleanane-type triterpenoid saponin, exerts anti-tumor effects in several tumor models, but the effect of RA in human drug-resistant OS remained to be elucidated. In the present study, we investigated the anti-tumor effects of RA in both drug-sensitive and drug-resistant OS cells and its underlying mechanism. RA inhibited cell proliferation and colony formation and induced apoptotic cell death in a dose-dependent manner in both drug-sensitive and drug-resistant cells. Moreover, RA exposure resulted in the inhibition of interleukin-6 (IL-6)-induced JAK2/STAT3 signaling pathway activation and target gene expression in both drug-sensitive and drug-resistant cells. Meanwhile, we observed significantly increased MDR1 and STAT3 expression in drug-resistant OS cells compared with parental cells. STAT3 overexpression promoted chemo-resistance and MDR1 protein expression in both drug-sensitive OS cells and drug-resistant OS cells, while inhibiting STAT3 with siRNA sensitized OS cells to doxorubicin treatment. In addition, RA synergistically increased doxorubicin toxicity by increasing its cellular uptake, ablating efflux and downregulating MDR1 in drug-resistant cells with attenuation of STAT3 Phosphorylation. Finally, RA suppressed in vivo tumor growth and induced apoptosis in nude mouse using drug-resistant OS tibia orthotopic model. Taken together, RA is a promising potential therapeutic for the treatment of doxorubicin resistance in OS.
Keywords: MDR1, Raddeanin A, osteosarcoma, STAT3, drug-resistance
Introduction
Osteosarcoma (OS) is the most common primary bone sarcoma, affecting 1-3 people per million, and the estimated 5-year overall survival rate for OS patients is approximately 60%-70% [1]. One major obstacle for current OS treatment is drug resistance, either intrinsic or acquired, to OS chemotherapeutic agents such as doxorubicin, ifosfamide and cisplatin, leading to the recurrence of malignant tumors and, ultimately, patient relapse or death. Drug resistance is widely considered a poor prognostic indicator for patients with OS [2, 3]. To improve the efficacy of current chemotherapy, strategies to reverse OS drug resistance have been studied extensively over the past few decades. Members of the ATP-binding cassette transporter superfamily, including P-glycoprotein (MDR1) and multidrug resistance-associated protein 1 and 2 (MRP1/ABCC1 and MRP2/ABCC2), mediate the classical drug resistance mechanism and play crucial roles in OS resistance to doxorubicin. Among these proteins, MDR1 was shown to contribute to the tumor response to chemotherapy in an Asian population, especially in OS and breast cancer, and to patient prognosis in our previous study [4].
Signal transducer and activator of transcription-3 (STAT3) is primarily activated and regulated by interleukin-6 (IL-6) family cytokine receptor-associated Janus kinases (JAKs), and in turn, STAT3 regulates the transcriptional activation of several anti-apoptotic and pro-proliferative gene products, such as cyclins, B-cell lymphoma-2 (Bcl-2) and surviving [5]. We have previously shown that inhibition of the STAT3 pathway induces apoptotic cell death and blocks tumor growth in vitro and in vivo in OS [6-8]. Constitutive activation of STAT3 has been shown to confer resistance to chemotherapy-induced apoptosis in some malignancies [9-11]. Tang et al [12] confirmed that STAT3 activation by IL-6 regulates mesenchymal stem cells (MSC)-induced chemo-resistance and reported that blockade of STAT3 signaling re-sensitized drug-resistant OS Saos-2 cells to drug treatment. Duan et al [13] found that inhibiting the STAT3 pathway induces drug-resistant OS cell apoptosis. Thus, STAT3 may be a promising therapeutic target for overcoming drug resistance in OS. Some researchers [14, 15] have shown that STAT3 could participate in regulating the transcription of MDR1 and MDR1 could be a downstream target of STAT3. But the underlying mechanism is still need to be elucidated.
In our previous study, we have identified that ursolic acid (UA) derivative as potent anti-tumor agent for OS in preclinical studies [16, 17]. In this study, we show that Raddeanin A (RA), which shares similar active constituents with UA, also with anti-tumor activity in several tumor models [18-23], as a JAK/STAT3 pathway inhibitor in OS. Here we show RA could inhibit tumor proliferation and growth and induce apoptosis by modulating the STAT3 pathway and downstream target gene expression in both doxorubicin-sensitive and doxorubicin-resistant OS. Furthermore, RA synergistically increases doxorubicin toxicity in drug-resistant OS cells by inhibiting the STAT3/MDR1 signaling axis in vitro and in vivo.
Materials and Methods
Cell lines and culture
The human OS MG63, U2OS, HOS cell lines were obtained from American Type Culture Collection (ATCC). The human OS drug-resistant U2OSR and KHOSR cell lines were kind gift from Dr. Duan zhenfeng [24] (Sarcoma Biology Laboratory, Massachusetts General Hospital, Harvard University). All cell lines were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin in a humidified incubator at 37°C and 5% CO2.
Drugs and antibodies
RA (99.9% purity) was purchased from Yuanye Biotechnology, China, and dissolved in dimethyl sulfoxide (DMSO) to a 10mM stock solution that was stored in aliquots in the dark at -20°C. The following antibodies were used for immunoblotting: rabbit anti-actin (Santa Cruz Biotechnology, CA, USA) and anti-PCNA, anti-caspase-3, anti-Bcl-xl, anti-Bcl-2, anti- PARP, anti-STAT3, anti-phospho-STAT3 (Tyr705), anti-JAK2, anti-phospho-JAK2 (Tyr1007/1008), anti- Src, anti-phospho-Src (Tyr416), anti-MDR1, anti-MRP1 (Cell Signaling Technology, Inc., Danvers, MA, USA). Human IL-6 was purchased from Sigma (Sigma-Aldrich, Inc., MO, USA).
CCK8 cell viability assay
Cells were seeded into 96-well plates and treated with RA at the indicated concentrations for 48 h. Cells incubated with 0.1% DMSO in DMEM-h served as the vehicle control group. CCK8 (20 μl; 5 mg/ml, Dojindo Molecular Technologies, Inc.) was added to each well, and the plates were incubated for another 4 h at 37 °C according to the manufacturer's protocol. Then, the absorbance at 490 nm was measured using an ELX800 Micro Plate Reader (Bio-Tec Instruments, Inc.). Excel FORECAST (predict x based on known x and y values) was used to predict the drug IC50[25]. Three independent experiments were performed in triplicate.
Cell clonogenic assay
Colony formation assays were conducted as previously described [6]. Cells were plated at 500-1000 cells/well in six-well plates. After vehicle or RA treatment for 48 h, cells were maintained in fresh medium for 10-14 days until visible colonies were observed. Colonies were then washed, fixed, and stained with 0.1% crystal violet. The colony number was counted manually. Images were acquired with a digital camera.
Flow cytometry analysis
Cells were cultured in six-well plates (2.5×105/ well) and treated as indicated for 48 h. Apoptosis was detected and then analyzed using FlowJo software as previously described [26]. Dual parameter dot plots of Annexin V-FITC and PI staining revealed live cells in the lower-left quadrant (Annexin V-/PI-), early apoptotic cells in the lower-right quadrant (Annexin V+/PI-), late apoptotic cells in the upper-right quadrant (Annexin V-/PI+), and necrotic cells in the upper-left quadrant (Annexin V+/PI+).
Immunoblotting
Cells were washed twice with cold 1× PBS solution and lysed with RIPA lysis buffer (Beyotime, Shanghai, China) containing phosphatase and protease inhibitors (Beyotime, Shanghai, China). Equivalent amounts of total protein (30-60 μg) were separated in 8% or 12% polyacrylamide gels and transferred to nitrocellulose filter membranes. Membranes were blocked for 60 min with 5% milk in PBST (PBS and 0.1% Tween 20), incubated overnight at 4°C with 1:1000 dilutions of the primary antibodies, washed three times for 10 min each with PBST, and incubated for 1 h with the appropriate peroxidase-conjugated secondary antibody (1:5000 dilution). Membranes were then washed with PBST three times for 10 min each and then developed using the Odyssey two color infrared laser imaging system. The signal generated by β-actin was used as an internal control.
Reverse transcription polymerase chain reaction (RT-PCR)
RNA was collected and purified from treated cells using Trizol reagent (Invitrogen). RNA samples (1 μg) were subjected to RT-PCR using the TaKaRa RT-PCR kit (Takara, Shiga, Japan). Each 10 µL reaction contained 25 ng of sample cDNA. The following primer sequences were used: GAPDH F 5′-ATG TTC GTC ATG GGT GTG AA-3′, R 5′-TGT GGT CAT GAG TCC TTC CA-3′; and MDR1 F1, 5′-GGA AGA CAT GAC CAG GTA TGC C-3′, MDR1 R1, 5′-GGA GAC ATC ATC TGT AAG TCG GG-3′.
Drug uptake and efflux assay
Doxorubicin cellular uptake was evaluated based on the cellular distribution of red auto-fluorescence of the drug. Briefly, drug-resistant U2OSR cells (1×105 cells per well) were cultured in 12-well plates for 24 h. Triplicate wells were treated with indicated doxorubicin and the indicated concentration of RA for 6 h, and images were then acquired with a fluorescence microscope (Leica). Drug efflux assays were performed as described [24]. Drug-sensitive and drug-resistant cells (1×105 cells per well) were cultured in 12-well plates for 24 h. Triplicate wells were treated with the indicated concentration of RA for 6 h and then exposed to 50 nM calcein AM. After a 30-min incubation, the cells were washed and fixed with 4% paraformaldehyde. The nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Images were acquired with a fluorescence microscope (Leica). Cells were rinsed with DPBS, and total fluorescent emission in each well was measured with a SpectraMax® M5/M5e plate reader (Molecular Devices, US).
STAT3 transfection
OS cells were plated at a density of 1.5×105 cells per well in 6-well plates and allowed to attach overnight. Then, the cells were transfected with 4 μg of pcDNA3 or pGMSTAT3 using Lipofectamine 2000 (Invitrogen, MA, USA) for 48 h according to the manufacturer's protocol, transfected cells were then treated with 2 µg/ml puromycin for another 24-48 h and used in the following experiments.
Nude mouse tibia orthotopic tumor model
Four-week-old female BALB/c nude mice were housed under standard conditions with a 12-h light-dark cycle and were fed sufficient water and food. All the animal procedures were performed in accordance with a protocol approved by the Animal Care and Use Committee of Shanghai General Hospital and Shanghai Jiaotong University. KHOSR cells (106) were injected into the medullary cavity of the right tibia to establish an orthotopic OS model. Two weeks after tibial injection, each mouse in the RA groups received a weight-based dose of drug by intraperitoneal injection (ip) every 3 days. For the drug resistance orthotopic mouse models, 20 mice were randomized to four groups: vehicle (DMSO) (n=5), 5 mg/kg RA (n=5), 1 mg/kg doxorubicin (n=5), or 5 mg/kg RA plus 1 mg/kg doxorubicin (n=5). Mice in the vehicle group were injected with 100 µl of PBS containing 10% DMSO on the same schedule. After 7-10 treatments, the mice were euthanized. Tumors were dissected and stored in liquid nitrogen or fixed in formalin for further analysis.
TUNEL assay
Apoptosis in tumor samples was identified using a TUNEL Assay Kit (Beyotime, Beijing, China) according to the manufacturer's instructions. In brief, paraffin-embedded slides were deparaffinized with xylene, rehydrated with ethanol and incubated with proteinase K for antigen retrieval. After several washes with PBS, the sections were incubated with freshly prepared TUNEL reaction mixture for 1 h at 37 °C in a humidified chamber. Apoptotic cells in randomly chosen fields on the slides were observed using a light microscope (Leica).
Statistical analysis
Data are presented as the mean ± S.D. Student's t-test was used to compare two groups (P<0.05 was considered significant) unless otherwise indicated. All experiments were performed at least three times.
Results
RA suppresses cell proliferation and colony formation in chemosensitive and chemoresistant OS cell lines
To evaluate the anti-proliferative effect of RA, we performed CCK8 viability assays using a panel of OS cells. CCK8 cell viability assays conducted 48 h after vehicle (0.1% DMSO) or RA treatment showed a dose-dependent inhibition of cell survival in OS cell lines (Fig. 1A). As shown in Table 1, the 48 h IC50 values for HOS, U2OS, MG63, cells were 1.512 ± 0.034, 3.527 ± 0.018, 3.977 ± 0.055, 3.584 ± 0.045 µM, respectively. The corresponding IC50 values for KHOSR and U2OSR cells were 2.053 ± 0.086 and 6.510 ± 0.062 µM, respectively. The IC50 values of RA were generally higher in the second group chemoresistant cells than in the first group corresponding sensitive cells. Colony formation assays are commonly used to study the survival and proliferation of adherent cells. In the current study, significantly fewer colonies were observed after HOS and KHOSR OS cells were treated with the indicated of RA, confirming the inhibitory role of RA on OS proliferation (Fig. 1B). In addition, PCNA activity, a cellular marker of proliferation, was significantly suppressed after RA treatment for 48 h (Fig. 1C). Cell viability was inhibited in time-dependent manner after RA treatment using the indicate concentration (Supplementary Figure 1A), and the expression of PCNA was significantly gradually decreased after RA treatment in a certain concentration for a serial time (Supplementary Figure 1B). These results showed that RA inhibits the proliferation of chemo sensitive and chemo resistant OS cells in a dose- and time-dependent manner.
RA blocks cell proliferation and colony formation in both drug-sensitive and drug-resistant human OS cells. (A) The human OS HOS, KHOSR, U2OS, U2OSR and MG63 cell lines were treated with vehicle (0.1% DMSO) or the indicated concentration of RA for 48 h, and cell viability was measured by CCK8 assay. (B) OS cells (HOS and KHOSR) were plated in six-well plates and treated with vehicle or the indicated concentration of RA for 48 h, cells were then maintained in fresh medium for 10-14 days. Colonies were fixed and stained with 0.1% crystal violet. (C) HOS, KHOSR, U2OS, and U2OSR cells were treated with the indicated concentration of RA for 48 h, and the expression of the proliferation marker PCNA was analyzed by immunoblotting. β-actin was used as a loading control. *P<0.05, ** P<0.01 compared with vehicle control.
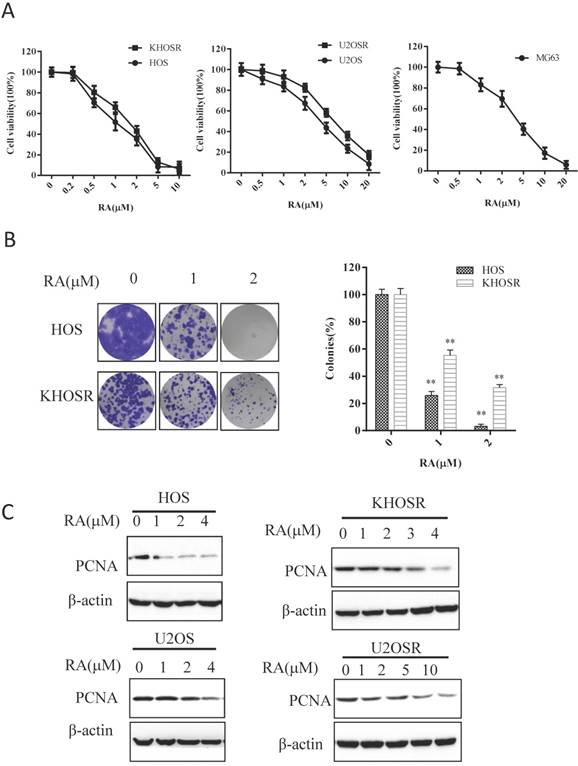
IC50s of OS cell lines
| Cell line | IC50(µM)* |
|---|---|
| HOS | 1.512 ± 0.034 |
| KHOSR | 2.053 ± 0.086 |
| U2OS | 3.527 ± 0.018 |
| U2OSR | 6.510 ± 0.062 |
| MG63 | 3.977 ± 0.055 |
*IC50 is the concentration of drug (μM) that produced 50% inhibition of cell growth.
RA induces apoptosis in chemosensitive and chemoresistant OS cell lines
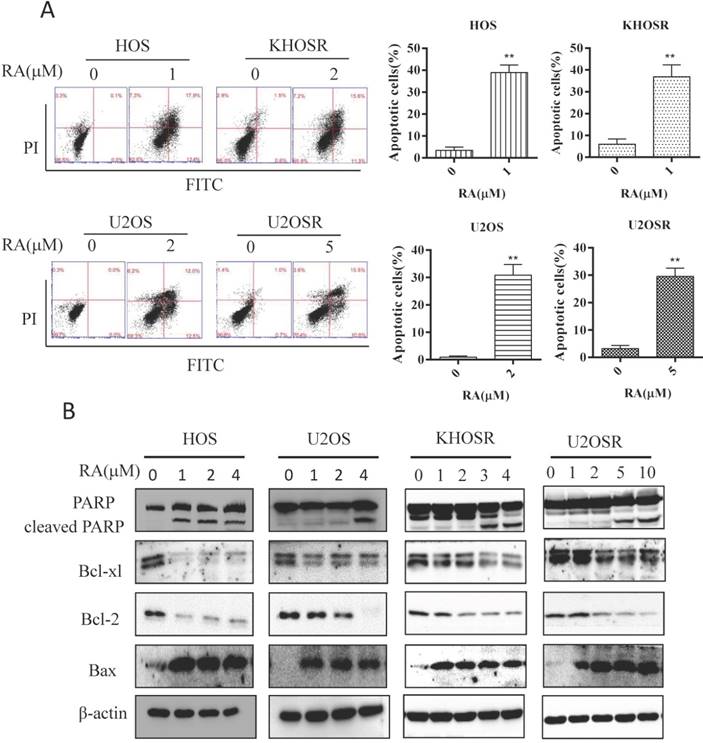
Apoptosis is a programmed cell death mechanism characterized by depolymerization of the cytoskeleton, cell shrinkage, chromatin condensation, nuclear fragmentation and translocation of phosphatidylserine to the cell surface. It is a common mechanism by which anti-cancer drugs kill cancer cells. As shown in Fig. 2A, RA induced significant apoptosis in both HOS, U2OS, and HOSR, U2OSR cells using Flow cytometry analysis. Moreover, RA markedly activated PARP cleavage and decreased the expression of the anti-apoptotic genes Bcl-2 and Bcl-xl in a dose dependent manner (Fig. 2B). And the proapoptotic gene Bax, a member of the Bcl-2 gene family, was decreased after RA treatment in a dose dependent manner (Fig. 2B). These data imply that RA induces dose -dependent apoptosis in chemosensitive and chemoresistant OS cell lines.
RA induces apoptosis in drug-sensitive and drug-resistant human OS cells. HOS, KHOSR, U2OS, and U2OSR OS cells were treated with vehicle or the indicated concentration of RA for 48 h. (A) The apoptosis index was determined by flow cytometry with Annexin V-FITC and PI staining. (B) Expression levels of cleaved PARP, Bax, Bcl-2 and Bcl-xl were determined by immunoblotting. *P<0.05, ** P<0.01 compared with vehicle control.
RA inhibits interleukin-6 (IL-6)-induced STAT3 Tyr705 phosphorylation in chemosensitive and chemoresistant OS cell lines
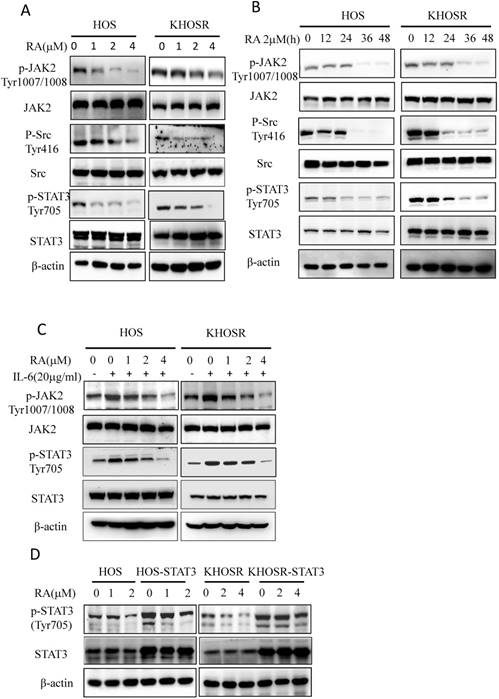
Cytokines such as IL-6, IL-21, IL-23 and IL-27 activate the STAT3 pathway, and the stimulation of IL-6R leads to the activation of several transcription factors, most notably STAT3[27]. Our results showed that RA treatment blocked not only p-STAT3 Tyr705 levels but also p-JAK2 Tyr1007/1008 expression in HOS and KHOSR OS cells (Fig. 3A, B) in dose- and time- dependent manner. To further determine the upstream pathways involved, we treated OS cells with the indicated concentration of RA with or without 20 µg/ml IL-6. As shown in Fig. 3C, IL-6 increased the both p-JAK2 Tyr1007/1008 and p-STAT3 Tyr705 levels, which could be quenched by subsequent RA treatment (Fig. 3C). Moreover, we established a STAT3-overexpressing OS cell line, HOS-STAT3 and KHOSR-STAT3. As shown in Fig. 3D, overexpression of STAT3 increased p-STAT3 Tyr705 levels by augmenting total STAT3 levels, which could also be inhibited by RA treatment. These data indicate that RA inhibits STAT3 Tyr705 phosphorylation and target gene expression in both chemosensitive and chemoresistant OS cells.
RA inhibits interleukin-6 (IL-6)-induced STAT3 Tyr705 phosphorylation and target gene expression. (A, B) drug-sensitive HOS and drug-resistant KHOSR OS cells were treated with the indicated RA concentration for 48 h or with 2.0·M RA for different periods of time (0, 12, 24, 36, 48 h). The expression of STAT3, p-STAT3Tyr705, JAK2, p-JAK2Tyr1007/1008, Src, and p-SrcTyr416 were determined by immunoblotting. (C) HOS and KHOSR OS cells were treated with the indicated RA concentration for 48 h with or without subsequent 20 µg/ml IL-6 treatment for 30 min. Then, the levels of STAT3, p-STAT3Tyr705, JAK2, and p-JAK2Tyr1007/1008 were measured by immunoblotting. (D) HOS and KHOSR cells were transfected with pCDNA or pGMSTAT3 for 48 h and selected with 2 g/ml puromycin for 48 h, STAT3 were over expressed in the HOS-STAT3 and KHOSR-STAT3 cells. (E) the resulting HOS-STAT3 compared with HOS and KHOSR-STAT3 compared with KHOSR cells were then treated with the indicated concentration of RA, and STAT3 and p-STAT3Tyr705 expression levels were detected by immunoblotting. β-actin was used as a loading control. *P<0.05, ** P<0.01 compared with vehicle control.
RA suppresses MDR1 expression through inhibiting STAT3 in chemoresistant OS cells
Recent studies have shown that inhibiting STAT3 effectively enhances multidrug sensitivity by blocking STAT3-mediated MDR1 gene expression in both leukemia and breast cancer cells [14,15], indicating that MDR1 could be a downstream target of STAT3. we examined doxorubicin resistance related ABC family genes and STAT3 expression in U2OSR, KHOSR compared with its parental cells. Our results showed markedly elevated expression of MDR1, MRP1, STAT3Tyr705 phosphorylation and STAT3 expression in chemoresistant cells compared with the corresponding parental OS cells (Fig.4A). Then to investigate the possible mechanism, we explored how MDR1 expression is affected by RA treatment. After being incubated with the indicated RA for 48 h, KHOSR and U2OSR cells exhibited a dose-dependent decrease in MDR1 and MRP1 expression (Fig. 4B). To further investigated the MDR1 expression in level of mRNA, we used real-time PCR assay, and then found that MDR1 mRNA expression was decreased with RA treated in two human chemo-resistant OS cells (Fig. 4C).
RA inhibits the STAT3 phosphorylation and reduces MDR1 expression in drug-resistant OS. (A) MDR1, MRP1, STAT3, and p-STAT3Tyr705 levels were determined by immunoblotting in drug-resistant cells compared with drug-sensitive cells (KHOSR and HOS, U2OSR and U2OS). (B) KHOSR and U2OSR cells were treated with vehicle or the indicated concentration of RA for 48 h. MDR1 and MRP1 expression were detected by immunoblotting. (C) MDR1 expression was determined by real-time qPCR in KHOSR and U2OSR after RA treated. (D) U2OSR and MG63 cells were transfected with pCDNA or pGMSTAT3 for 48 h and then treated with puromycin to establish a STAT3-overexpressing cell line (MG63-STAT3 and U2OSR-STAT3). MDR1 expression was detected by immunoblotting. (E) STAT3, p-STAT3Tyr705, and MDR1 levels were determined by immunoblotting in STAT3-overexpressing cell lines treated with series concentrations of RA. (F) U2OSR cells were treated with the indicated concentration of RA with or without STAT3 siRNA transfection for 48 h, and MDR1, STAT3, and p-STAT3Tyr705 expression were determined by immunoblotting. (G) MG63 cells were transfection with NC/STAT3 siRNA or pcDNA3/pGMSTAT3 for 48 h, treated with or without doxorubicin for 48h. The cell viability was detected by CCK8 assay.
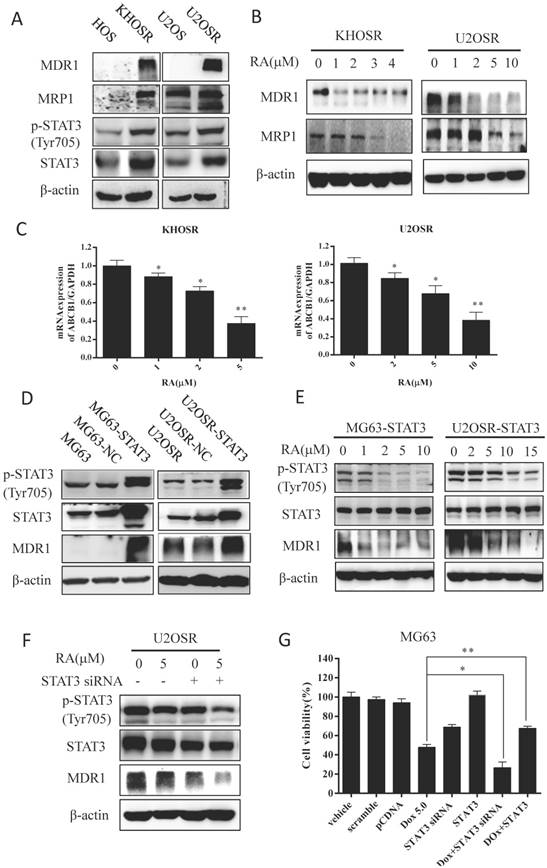
Surprisingly, overexpression of STAT3 increased MDR1 expression in chemosensitive MG63 and chemoresistant U2OSR OS cells (Fig. 4D), and the increased STAT3Tyr705 phosphorylation and MDR1 expression mediated by STAT3 overexpression could be abated by RA treatment again (Fig. 4E). Moreover, STAT3 siRNA and RA treatment synergistically downregulated MDR1 expression in U2OSR cells (Fig. 4F).
Further CCK8 proliferation assays showed overexpression of STAT3 increased doxorubicin resistance in MG63 cells, whereas STAT3 siRNA sensitized MG63 cells to doxorubicin treatment (Fig. 4G). Taken together, those results imply that RA could suppressed MDR1expression in level of protein and mRNA through inhibiting the STAT3 activation in chemicoresistance OS cells to restore the drug sensitivity.
RA restores sensitivity to doxorubicin in chemo-resistant OS cells
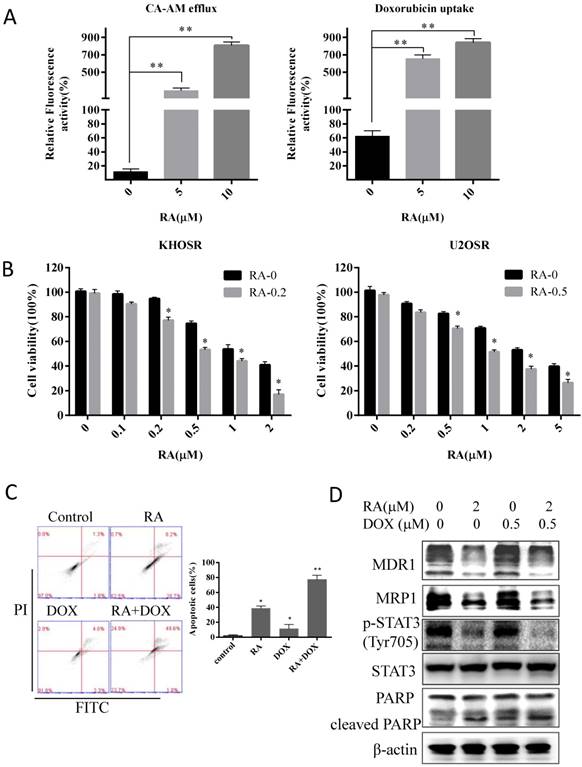
Drug resistance is a major obstacle to the successful treatment of OS with first-line chemotherapy such as doxorubicin, cisplatin and methotrexate. Herein, two chemoresistant OS cell lines were used to explore the effects and mechanism of RA on OS drug resistance. As shown in Fig. 3A and B, RA exposure induced dose- and time- dependent apoptosis in chemoresistant cell lines, as well as we observed in chemosensitive OS cells (Fig. 2). Moreover, U2OSR cells exhibited less doxorubicin uptake after been treated with 5 µM doxorubicin for 2 h, evaluated by fluorescence microscopy imaging, which showed cellular red fluorescence in drug-resistant cells compared with its parental U2OS cell line (supplementary Figure 2). We nextly measured the efflux of the non-toxic live cell dye calcein AM and the uptake of doxorubicin in drug-resistant cells upon RA treatment for 2 hours. Calcein AM was a known substrate of the multidrug resistance protein P-glycoprotein (MDR1/ABCB1) or other ABC membrane pump proteins, and it has been employed for fluorescent substrate efflux assays. RA significantly increased the accumulation of fluorescent calcein, which hydrolyzed from the retainment of intracellular calcein AM by intracellular esterase, indicating RA could decrease drug efflux in drug-resistant cells U2OSR (Fig. 5A). Meanwhile RA promoted doxorubicin uptake by facilitating the red fluorescence doxorubicin transportation in U2OSR cells (Fig. 5A). As shown in supplementary Figure 3, the accumulation of fluorescent calcein (green) demonstrated the decrease of calcein AM efflux, and the accumulation of fluorescent doxorubicin (red) demonstrated the increase of doxorubicin uptake in U2OSR treated with RA in a dose-dependent manner.
To further verify this finding, we examined the combination effect of doxorubicin in a serial concentration of 0, 0.1, 0.2, 0.5, 1, and 2 µM (or 0, 0.2, 0.5, 1, 2 and 5 µM) with RA in drug resistance OS cell KHOSR (or in U2OSR) using CCK8 viability assays (Fig. 5B). The serial concentration of doxorubicin was 0, 0.05, 0.1, 0.2, 0.5, 1, and 2 µM in KHOSR (and the serial concentration was 0, 0.1, 0.2, 0.5, 1, 2, and 5 µM in U2OSR), plusing RA with indicated concentration, to analyze the respective IC50 of doxorubicin plusing with RA of the indicated concentration using the CalcuSyn system. As shown in Table 2, the IC50s of doxorubicin alone in both KHOSR and U2OSR cell lines were 1.820 ± 0.071 and 3.202 ± 0.083 µM, whereas the IC50 plus RA (0.2 µM) was 0.630 ± 0.141 µM in KHOSR and in U2OSR the IC50 plus RA (0.5 µM) was 1.256 ± 0.092 µM. RA was capable of reversing drug resistant at concentrations 19.36- and 57.17-fold lower than that required for doxorubicin in KHOSR and U2OSR respectively.
Ability of RA to promote chemosensitivity in osteosarcoma MDR cell lines
| KHOSR IC50(µM) | U2OSR IC50(µM) | |
|---|---|---|
| Doxorubicin | 1.820 ± 0.071 | 3.202 ± 0.083 |
| plus RA 0.2µM | 0.630 ± 0.141 | 1.433 ± 0.012 |
| plus RA 0.5µM | 0.601 ± 0.085 | 1.256 ± 0.092 |
| plus RA 1µM | 0.396 ± 0.016 | 0.981 ± 0.016 |
| plus RA 2µM | 0.094 ± 0.014 | 0.374 ± 0.073 |
| plus RA 5µM | NA | 0.065 ± 0.059 |
*IC50 is the concentration of drug (μM) that produced 50% inhibition of cell growth.
To demonstrate the combination effect, we investigated the apoptotic cells using Flow cytometry analysis and immunoblotting assay. The percent of apoptotic cells was significantly higher in 0.5 µM doxorubicin combined with 2 µM RA than control or in either 0.5 µM doxorubicin or 2 µM RA alone (Fig. 5C). As showed in Fig. 5D, the expression of MDR1 and MRP1 was significantly reduced by RA, and when combined with RA, the expression of cleaved PARP was increased compared with doxorubicin or RA respectively. In short, those results indicate RA could modulate the drug uptake and efflux, induces apoptosis and sensitizes chemo-resistant cells to doxorubicin treatment in human OS cells.
RA inhibits tumor growth in orthotopic chemoresistant OS animal model
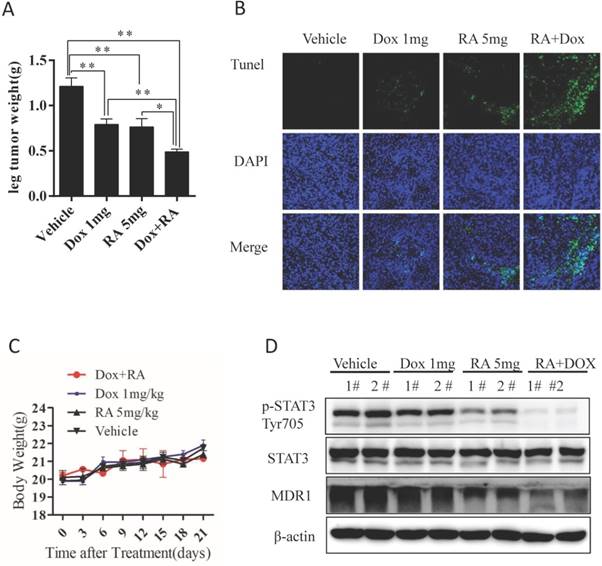
To assess whether the biologic effect of RA on chemo-resistant OS is potentially effective in vivo, we established a nude mouse orthotopic model using chemo-resistant KHOSR cells as described previously [29]. Mice were randomized into four groups and then treated by ip injection with vehicle, 5 mg/kg RA, 1 mg/kg doxorubicin and RA plus doxorubicin. As shown in Fig. 6A, 5 mg/kg RA, 1 mg/kg doxorubicin or RA plus doxorubicin significantly decreased tumor weight compared with vehicle. Interestingly, RA showed a significant synergistic effect with doxorubicin, which correlated with the in vitro findings as we indicated in Fig 5B, and 5C. However, there were no differences in mouse body weight, indicating that RA treatment have tolerable toxicity in vivo (Fig. 6C). The apoptosis index in tumor samples was analyzed by TUNEL assay. In agreement with the in vitro study finding, treatment with RA plus doxorubicin caused significantly more apoptosis than the other treatments (Fig. 6B). Furthermore, RA downregulated STAT3Tyr705 phosphorylation and MDR1 expression in tumor samples (Fig. 6D). These results indicate that RA inhibits in vivo tumor growth in an orthotopic chemoresistance model of human OS.
RA reverses doxorubicin resistance in human OS cells by inhibiting STAT3 phosphorylation. (A) Cells were then treated with the indicated concentration of RA for 2 hours and then incubated with calcein AM for 30 min, calcein AM efflux was evaluated by green fluorescence observed using a fluorescence microscope and quantified by SpectraMax® M5/M5e plate reader. Cells were treated with the indicated concentrations of RA for 2 hours and doxorubicin, and doxorubicin uptake was evaluated by red fluorescence observed in fluorescence images and quantified by SpectraMax® M5/M5e plate reader. The cell nucleuses were stained by DAPI, which produced blue fluorescence. Relative fluorescence activity meaned the ratio of green (or red) quantity related to blue quantity. (B) KHOSR and U2OSR cells were treated with RA in combination with the indicated concentration of doxorubicin for 48 h, and cell viability was determined by CCK8 assay. (C) U2OSR cells were treated with or without doxorubicin pretreated with or without of RA for 2 h and then subjected to Annexin V-FITC/PI staining and flow cytometry analysis. (D) MDR1, MRP1, STAT3 phosphorylation, total STAT3, and cleaved-PARP expression were detected by immunoblotting in U2OSR cells treated with doxorubicin in presence or absence of RA. β-actin was used as a loading control. *P<0.05, **P<0.01 compared with vehicle control.
RA inhibits the in vivo growth of drug-resistant OS. (A) Macroscopic appearance of OS tumors in the tibia of BALB/c nude mice after treatment. Tumor weight quantification in BALB/c nude mice after treatment. (B) TUNEL assay of tumor samples from the leg in different treatment groups. (C) Body weight was measured every 3 days. (D) Tumor samples were lysed and subjected to immunoblotting with the indicated antibodies. β-actin was used as a loading control. *P<0.05, **P<0.01 compared with vehicle control.
Discussion
The introduction of biologic agents and the use of additional cytotoxic chemotherapy has not definitively improved the survival of patients with OS in the past two decades. Therefore, there is an unmet need to discover more effective agents to overcome chemotherapy drug resistance. Our previous studies found that oleanolic acid derivates or analogues exert anti-tumor effect in OS preclinical models [16, 17], and might be potential new drug for OS clinical trials. While our other studies support the hypothesis that activated STAT3 might be target for anti-tumor therapy and natural compounds targeting the STAT3 pathway could be used for inhibiting OS [6-8]. Herein, we identified RA, a triterpenoid saponin extracted from the root of Anemone raddeana Regel, which shares similar active form with Oleanolic acid, as effective anti-tumor agent in OS, and the anti-proliferative and pro-apoptotic effects of RA could be modulated by the STAT3 pathway through its target genes regulation.
Our study showed that STAT3 expression is higher in chemoresistant cell lines (KHOSR and U2OSR) than its parental cells. Moreover, RA, attenuated MDR1 mRNA and protein expression in a dose-dependent manner in all the tested OS cell lines. Thus, it is possible that STAT3 contributes to doxorubicin resistance in OS. Suppressing STAT3 increased the sensitivity of chemo-resistant cells to doxorubicin by increasing its uptake and ablating drug efflux in human OS. These finding prompted the discovery that inhibiting STAT3 could effectively inhibit tumor progression and reverse doxorubicin resistance in OS. Yun et al [30] found that cinnamaldehyde derivative (CB-PIC) sensitizes drug-resistant cancer cells to drug-induced apoptosis by suppressing MDR1, although CB-PIC did not directly attenuate MDR1 activity but rather inhibited MDR1 mRNA and protein expression by suppressing the STAT3 and AKT signaling pathways. While the study by Zhang et al [14] showed that STAT3 could bind the +64~+72 region of the MDR1 promoter and thus initiate its transcription in leukemia cells, the blockade of STAT3 activation by a STAT3 decoy oligodeoxynucleotide (ODN) promoted adriamycin accumulation and increased the cellular sensitivity to adriamycin by downregulating the transcription and protein expression of MDR1. In our study, RA inhibited MDR1 mRNA and protein expression in a dose-dependent manner, and the overexpression of STAT3 promoted doxorubicin resistance and MDR1 expression in chemo-sensitive MG63 cells and chemo-resistant U2OSR cells. Moreover, STAT3 siRNA and RA treatment synergistically downregulated MDR1 expression in U2OSR cells. Hence, we concluded that MDR1 is a downstream target of STAT3 in OS. A recent study by Peng et al [31] showed RA could reverse STAT3/NFIL3 mediated chemotherapy resistance by methotrexate (MTX), 5-FU and etoposide (VP16) through inducing apoptosis in choriocarcinoma cells. They have identified STAT3/NFIL3 axis as a new mechanism for choriocarcinoma drug resistance, but the weakness of their study is they only presented the in vitro proof without showing the reversal effect in animal models. Whereas our study providing strong evidence for the interaction of STAT3 and MDR1, indicating inhibition of STAT3/MDR1 axis by RA might be key to reverse doxorubicin induced chemo-resistance in OS.
Regarding the interplay between STAT3, its target genes and MDR1, Ji et al [32] showed that a novel triazolonaphthalimide derivative LSS-11 could hinder the binding of STAT3 to the MDR1 and MRP1 promoters by conducting chromatin coimmunoprecipitation (ChIP) assay. Indicating MDR1 is regulated by the STAT3 at the transcription level. Our study provide evidence that STAT3 regulate MDR1 expression in vitro and in vivo. However, how STAT3 target genes are associated with MDR1 function remains to be determined. Thus, more efforts are still needed to elucidate the possible mechanisms in the future.
Abbreviations
OS: osteosarcoma; RA: Raddeanin A; STAT3: Signal transducers and activators of transcription; MDR1: multi-drug resistance 1; MRP1: Multidrug Resistance-Associated Protein.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank animal Center of Shanghai General Hospital, for all the animal holding and care. This project was supported by the NSFC (81502604, 81501584, and 81702973), the Shanghai Science and Technology Commission (14140904000), the Doctoral Innovation Fund of Shanghai Jiaotong University School of Medicine (No. BXJ201732), the Shanghai Municipal Commission of Health and Family Planning (No. 20164Y0270), and a Research Grant from the Shanghai Hospital Development Center (SHDC12013107).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Isakoff MS, Bielack SS, Meltzer P. et al. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J Clin Oncol. 2015;33(27):3029-3035
2. Li S, Sun W, Wang H. et al. Research progress on the multidrug resistance mechanisms of osteosarcoma chemotherapy and reversal. Tumour Biol. 2015;36(3):1329-1338
3. Serra M, Pasello M, Manara MC. et al. May P-glycoprotein status be used to stratify high-grade osteosarcoma patients? Results from the Italian/Scandinavian Sarcoma Group 1 treatment protocol. Int J Oncol. 2006;29(6):1459-1468
4. Zhou Z, Chen Q, Zuo D. et al. ABCB1 (rs1128503) polymorphism and response to chemotherapy in patients with malignant tumors-evidences from a meta-analysis. Int J Clin Exp Med. 2015;8(1):265-272
5. Zhao C, Li H, Lin HJ. et al. Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism. Trends Pharmacol Sci. 2016;37(1):47-61
6. Zuo D, Zhou Z, Wang H. et al. Alternol, a natural compound, exerts an anti-tumour effect on osteosarcoma by modulating of STAT3 and ROS/MAPK signalling pathways. J Cell Mol Med. 2017;21(2):208-221
7. Zhang T, Li J, Yin F. et al. Toosendanin demonstrates promising antitumor efficacy in osteosarcoma by targeting STAT3. Oncogene. 2017;36(47):6627-6639
8. Zhang T, Li S, Li J. et al. Natural product pectolinarigenin inhibits osteosarcoma growth and metastasis via SHP-1-mediated STAT3 signaling inhibition. Cell death & disease. 2016;7(10):e2421
9. Peng J, Wang Q, Liu H. et al. EPHA3 regulates the multidrug resistance of small cell lung cancer via the PI3K/BMX/STAT3 signaling pathway. Tumour Biol. 2016;37(9):11959-11971
10. Zhang F, Wang Z, Fan Y. et al. Elevated STAT3 Signaling-Mediated Upregulation of MMP-2/9 Confers Enhanced Invasion Ability in Multidrug-Resistant Breast Cancer Cells. Int J Mol Sci. 2015;16(10):24772-24790
11. Zhu H, Chen X, Chen B. et al. Activating transcription factor 4 mediates a multidrug resistance phenotype of esophageal squamous cell carcinoma cells through transactivation of STAT3 expression. Cancer letters. 2014;354(1):142-152
12. Tu B, Zhu J, Liu S. et al. Mesenchymal stem cells promote osteosarcoma cell survival and drug resistance through activation of STAT3. Oncotarget. 2016;7(30):48296-48308
13. Ryu K, Susa M, Choy E. et al. Oleanane triterpenoid CDDO-Me induces apoptosis in multidrug resistant osteosarcoma cells through inhibition of Stat3 pathway. BMC cancer. 2010;10:187
14. Zhang X, Xiao W, Wang L. et al. Deactivation of signal transducer and activator of transcription 3 reverses chemotherapeutics resistance of leukemia cells via down-regulating P-gp. PloS one. 2011;6(6):e20965
15. Bourguignon LY, Peyrollier K, Xia W. et al. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem. 2008;283(25):17635-17651
16. Hua Y, Zhang Z, Li J. et al. Oleanolic acid derivative Dex-OA has potent anti-tumor and anti-metastatic activity on osteosarcoma cells in vitro and in vivo. Invest New Drugs. 2011;29(2):258-265
17. Chen J, Fu H, Wang Z. et al. A new synthetic ursolic acid derivative IUA with anti-tumor efficacy against osteosarcoma cells via inhibition of JNK signaling pathway. Cell Physiol Biochem. 2014;34(3):724-733
18. Gu G, Qi H, Jiang T. et al. Investigation of the cytotoxicity, apoptosis and pharmacokinetics of Raddeanin A. Oncology letters. 2017;13(3):1365-1369
19. Li JN, Yu Y, Zhang YF. et al. Synergy of Raddeanin A and cisplatin induced therapeutic effect enhancement in human hepatocellular carcinoma. Biochem Biophys Res Commun. 2017;485(2):335-341
20. Teng YH, Li JP, Liu SL. et al. Autophagy Protects from Raddeanin A-Induced Apoptosis in SGC-7901 Human Gastric Cancer Cells. Evid Based Complement Alternat Med. 2016;2016:9406758
21. Guan YY, Liu HJ, Luan X. et al. Raddeanin A, a triterpenoid saponin isolated from Anemone raddeana, suppresses the angiogenesis and growth of human colorectal tumor by inhibiting VEGFR2 signaling. Phytomedicine. 2015;22(1):103-10
22. Xue G, Zou X, Zhou JY. et al. Raddeanin A induces human gastric cancer cells apoptosis and inhibits their invasion in vitro. Biochem Biophys Res Commun. 2013;439(2):196-202
23. Wang MK, Ding LS, Wu FE. Antitumor effects of raddeanin A on S180, H22 and U14 cell xenografts in mice. Ai zheng. 2008;27(9):910-913
24. Ye S, Zhang J, Shen J. et al. NVP-TAE684 reverses multidrug resistance (MDR) in human osteosarcoma by inhibiting P-glycoprotein (PGP1) function. Br J Pharmacol. 2016;173(3):613-626
25. Tong ZG, Liu N, Song HS. et al. Cytochalasin B inhibits the proliferation of human glioma U251 cells through cell cycle arrest and apoptosis. Genet Mol Res. 2014;13(4):10811-10822
26. Fu Z, Deng B, Liao Y. et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC cancer. 2013;13:580
27. Heinrich PC, Behrmann I, Muller-Newen G. et al. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334( Pt 2):297-314
28. Wang H, Li W, Xu J. et al. NDRG1 inhibition sensitizes osteosarcoma cells to combretastatin A-4 through targeting autophagy. Cell death & disease. 2017;8(9):e3048
29. Wang Z, Wang C, Zhou Z. et al. CD151-mediated adhesion is crucial to osteosarcoma pulmonary metastasis. Oncotarget. 2016;7(37):60623-60638
30. Yun M, Lee D, Park MN. et al. Cinnamaldehyde derivative (CB-PIC) sensitizes chemo-resistant cancer cells to drug-induced apoptosis via suppression of MDR1 and its upstream STAT3 and AKT signalling. Cell Physiol Biochem. 2015;35(5):1821-1830
31. Peng Z, Zhang C, Zhou W. et al. The STAT3/NFIL3 signaling axis-mediated chemotherapy resistance is reversed by Raddeanin A via inducing apoptosis in choriocarcinoma cells. J Cell Physiol. 2018;233(7):5370-5382
32. Ji L, Liu X, Zhang S. et al. The Novel Triazolonaphthalimide Derivative LSS-11 Synergizes the Anti-Proliferative Effect of Paclitaxel via STAT3-Dependent MDR1 and MRP1 Downregulation in Chemoresistant Lung Cancer Cells. Molecules. 2017;22(11):1822
Author contact
![]() Corresponding authors: Yingqi Hua, E-mail: yhuaedu.cn; Or Wei Sun, Email: viv-suncom; Or Zhengdong Cai, Email: caizhengdongedu.cn
Corresponding authors: Yingqi Hua, E-mail: yhuaedu.cn; Or Wei Sun, Email: viv-suncom; Or Zhengdong Cai, Email: caizhengdongedu.cn