Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Contribution of Mitochondrial...
The Framework of...
Future Considerations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(3):680-687. doi:10.7150/ijbs.30464 This issue Cite
Review
Reactive Oxygen Species Related Noncoding RNAs as Regulators of Cardiovascular Diseases
Yanhan Dong1* ![]() , Wenhua Xu1*, Cuiyun Liu1, Peijun Liu2, Peifeng Li1
, Wenhua Xu1*, Cuiyun Liu1, Peijun Liu2, Peifeng Li1 ![]() , Kun Wang1
, Kun Wang1 ![]()
1. Qingdao University, Deng Zhou Road 38, Qingdao 266021, China
2. Biochemistry Department No.2 Middle School Qingdao Shandong P.R. China 266000
*The authors contributed equally to this work.
Received 2018-10-7; Accepted 2018-12-19; Published 2019-1-24
Abstract

Reactive oxygen species (ROS) are a class of reactive molecules that have been implicated in a variety of cardiovascular diseases, accompanied by disorder of multiple signaling events. As cardiomyocytes maintain abundant of mitochondria, which supply the major source of endogenous ROS, oxidative damage to mitochondria often drives apoptotic cell death and initiates cardiac pathology. In recent years, non-coding RNAs (ncRNAs) have received much attention to uncover their roles in regulating gene expression during those pathological events in the heart, such as myocardial infarction, cardiac hypertrophy, and heart failure. Emerging evidences have highlighted that different ROS levels in response to diverse cardiac stresses result in differential expression of ncRNAs, subsequently altering the expression of pathogenetic genes. However, the knowledge about the ncRNA-linked ROS regulatory mechanisms in cardiac pathologies is still largely unexplored. In this review, we summarize the connections that exist among ROS, ncRNAs, and cardiac diseases to understand the interactions among the molecular entities underlying cardiac pathological events in the hopes of guiding novel therapies for heart diseases in the future.
Keywords: ROS, ncRNAs, heart diseases, mitochondria, cardiomyocyte death
Introduction
Reactive oxygen species (ROS) are oxygen-containing molecules that usually harbor unpaired electrons rendering them highly reactive and potentially dangerous oxidants [1]. Major ROS sources in the heart are constituted with superoxide anion (O2.-), hydrogen peroxide (H2O2), and hydroxyl radical (HO ). As highly metabolic cells, cardiomyocytes maintain abundant of mitochondria to support the cellular store of adenosine triphosphate (ATP), which is required for cardiac function [2]. And the mitochondria could supply the major source of endogenous ROS, in which oxygen is converted into superoxide anions, hydroxyl radicals and hydrogen peroxide through the electron transport chain (ETC) [3]. Besides, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and nitric oxide (NO) synthases are another two critical sources of ROS production [4]. Under physiological conditions, there is a balance between ROS generation and elimination. In view of the critical role in cell signaling, dysregulated generation of ROS usually contributes to the pathogenesis of cardiovascular diseases, including atherosclerosis, heart failure, cardiomyopathy, and cardiac hypertrophy.
In recent years, emerging evidence indicated that non-coding RNAs (ncRNAs) play important roles in various biological processes, as well as in disease progression [5-7]. Noncoding RNAs are originally from genome and generally not translated into proteins, and they are currently classified into two subgroups according to their transcript size: short ncRNAs (<30 nucleotides), including miRNAs, piwi-interacting RNAs (piRNAs), and short interfering RNAs (siRNAs); and long ncRNAs longer than 200 nucleotides (lncRNAs) [8]. Most of the non-coding RNAs show tissue specific distribution pattern and thus participate in gene expression regulation. Researches on miRNAs indicated that some miRNAs are perturbed and localized to mitochondria, regulating mitochondrial-mediated cardiac cell survival/death when exposed to ROS stress [9-11]. For instance, mitochondria overproduce ROS during ischemic-reperfusion (I/R), while miRNAs's expression are altered after I/R injury [12]. Increasing evidence suggests that not only miRNAs, but also lncRNAs are related to increased ROS in cardiomyocytes. There are complex interactions between ROS metabolism and ncRNAs levels, and further studies need be provided to reveal the molecular mechanisms of ncRNAs regulating myocardial diseases under ROS-related stress conditions. In this review, we will focus on the ROS-related ncRNAs that alter the mitochondrial homeostasis and involve in the context of the cardiovascular system.
Contribution of Mitochondrial ROS to Cardiovascular Diseases
Physiological ROS are highly reactive molecules that play a critical role in intracellular signaling as second messengers; however, when ROS production is exacerbated or scavenging insufficient, disorder of many biological processes occurs [13]. Furthermore, the redox imbalance induced by altered ROS generally influences signaling pathways that contribute to cardiomyocytes' apoptosis, ER stress, and autophagy, which ultimately contribute to a wide range of cardiovascular diseases, such as myocardial infarction, stroke, atherosclerosis and heart failure [14, 15]. In addition, mitochondrial formation of ROS is increased particularly during cell injury, causing damage to tissue and cellular compartments [16, 17]. One of the most rapid and severe inducers of oxidative stress in the cardiovascular system is ischemia-reperfusion (IR) injury (Fig. 1), which is associated with a burst of ROS production, leading to the opening of the mitochondrial permeability transition pore (mPTP) [18, 19]. The mPTP is a high conductance non-specific channel at the inner mitochondrial membrane, and it remains closed during cardiac ischemic, but opens approximate 2 min after reperfusion [20]. When reperfusion after a period of ischemic produces oxidative stress, mPTP opening takes place accompanied by high matrix Ca2+ concentration, elevated phosphate concentration and ATP depletion [21]. Cyclophilin D, a prolyl isomerase located in the mitochondrial matrix, enhances the opening of mPTP through binding to the inner mitochondrial membrane under conditions of oxidative stress [22-24]. However, the regulation of mPTP-Cyp-D interaction in myocytes remains to be explored. Another pro-apoptotic Bcl2 family member, Nix, which originally modulates myocardial apoptosis [25], indirectly activates the mPTP and mitophagy in hypertrophied myocardium [26]. In recent years, multiple mPTP inhibitors targeting the pore opening have been developed in the context of MI. For instance, Cyclosporin A is an effective inhibitor of cyclophilin D, treatment with which protects heart from myocardial necrosis during reperfusion, indicating a therapeutic agent for patients with ischemic heart disease [27, 28].
Schematic diagram of the proposed molecular mechanism of mPTP upon I/R in the heart. I/R‐induced cardiac injury triggers abnormal mitochondrial oxidative stress, and thereby induces mPTP opening accompanied by high matrix Ca2+ concentration and release of cytochrome C, eventually leading to cell death. The ncRNAs regulating these key components of this process are shown in the diagram. mPTP: mitochondrial permeability transition pore; Cyp-D, cyclophilin D.
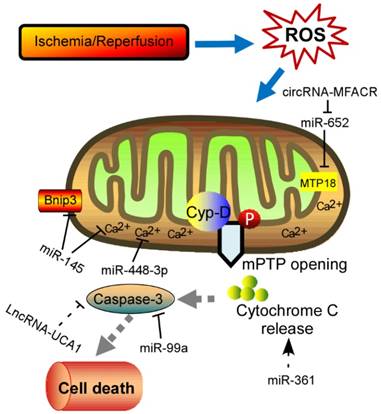
There are eleven distinct mitochondrial sites associated with substrate oxidation and ETC that release electrons to oxygen to produce superoxide or hydrogen peroxide through a series of redox carriers in respiratory chain complexes [29, 30]. During ischemia, the respiratory chain becomes reduced along with the accumulation of the citric acid cycle intermediate succinate [31-33]. A large amount of studies established that the respiratory complex I is the entry point for electrons from NADH into respiretory chain, and mainly contributes to mitochondrial superoxide production upon reperfusion [34-36]. After reperfusion, the accumulated succinate is rapidly re-oxidized, driving extensive ROS generation by reverse electron transport at mitochondrial complex I [32]. Besides, NADPH oxidases are major sources of ROS during cardiac hypertrophy. NADPH oxidase is able to produce superoxide by reducing one electron from O2, which is activated in various myocardial diseases [37-40]. There are seven NADPH oxidase isoforms that form the Nox family: Nox's 1-5 and Duox1 and Duox2 [41, 42]. These proteins share 21%-59% identity to Nox2, but differ in subcellular localization, molecular composition, and expression [43, 44]. Among them, Nox1, Nox2, Nox4 and Nox5 are expressed in specific cell types within the heart, including cardiomyocytes, cardiac fibroblasts, vascular smooth muscle cells (VSMCs), endothelial cell (ECs), and resident and infiltrating leukocytes [44, 45]. Nox2 and Nox4 are the main isoforms expressed in cardiomyocytes. Deficiency of Nox2 in vivo significantly attenuated angiotensin II (Ang II) induced cardiac hypertrophy [46]. It is reported that Nox2-mediated ROS appears to involve the activation of Erk1/2, Akt, and NF-κB pathway [47-50]. Karim et al demonstrated that Nox2-deficient mice showed significant decline in hypoxia response, oxidative stress, fibrosis, and inflammation, indicating that Nox2 is a critical mediator of I/R injury [51]. Recently, Nox2 was identified to be up-regulated under the prenatal exposure to maternal diabetes and enhanced myocardial I/R injury in the adult offspring through the VEGF-Akt-mTOR-Nox2 signaling [50]. Similarly, the expression levels of Nox4 are significantly increased upon pressure overload [52]. When generating mice with a genetic deletion of Nox4, the null mice developed exaggerated contractile dysfunction, hypertrophy and cardiac dilatation during exposure to chronic overload [53]. Investigation of underlying mechanisms revealed that Nox4 enhanced stress-induced activation of cardiomyocyte hypoxia inducible factor 1 (Hif1) and the release of vascular endothelial growth factor, resulting in increased paracrine angiogenic activity. It is noted that Sadoshima et al found that O2- production in mitochondria were abolished in cardiac-specific Nox4-/- mice in response to pressure overload, and the null mice exhibited significantly attenuated cardiac hypertrophy, interstitial fibrosis and apoptosis, and better cardiac function compared with WT mice [54]. This study suggested that Nox4 in cardiac myocytes can induce further ROS production, which may act as an amplifying mechanism. Another research pointed that mitochondrial ROS derived from Nox4 oxidize the cysteines of aconitase-2 and citrate synthase, leading to mitochondrial dysfunction and apoptosis in cardiomyocytes during heart failure [55]. Anyway, further experiments need to explore the relationship between the generation of ROS and different Nox isoforms in the setting of heart diseases in the future [56, 57].
The Framework of ROS-ncRNA-Cardiovascular Disease
In recent years, increasing evidences have supported that ncRNAs are involved in the pathogenesis of many cardiovascular diseases demonstrated by both loss-of-function and gain-of-function approaches [58]. Accumulation of ROS in cardiomyocytes results in differential expression of ncRNAs, subsequently contributing to cell apoptosis and heart pathology. Among these ncRNAs, miRNAs are most intensively studied, which inhibit protein translation or target mRNA degradation by bind to the untranslated regions (UTRs) of mRNA through complementary base pairing [59, 60]. Moreover, a single miRNA might have multiple molecular targets if only the target genes containing a sequence complementary to the “seed sequence” of the miRNA [61]. Abundant of miRNAs have been identified as a powerful modulator in the process of myocardial disorders under oxidative stress (Table 1). Intracellular Ca2+ overload can relay signals through Ca2+-regulated enzymes, such as the calcium/calmodulin dependent protein kinase II (CaMKII), which has acknowledged as a ROS-activated signaling molecule that affects adverse outcomes after MI [62, 63]. Cha et al demonstrated that miR-145 decreased Ca2+ overload and apoptosis in H2O2-treated cardiomyocytes [64]. Consistent with inhibition of CaMKII, miR-145 overexpression protects against ROS-induced cellular injury responses. Li et al also reported that miR-145 inhibited oxidative stress-associated cardiomyocyte apoptosis by regulating the mitochondrial apoptotic pathway through directly targeting Bnip3 [65]. Excessive level of ROS would also activate nuclear factor-κB (NF-κB) pathway that is a redox-sensitive pathway [66]. A positive feedback-loop consisting of NF-κB, miR-21, and programmed cell death 4 (PDCD4) was recently proposed [67]. H2O2-induced NF-κB up-regulates miR-21, and PDCD4 is a direct target for miR-21, suggesting that miR-21 contributes a critical role in ROS-mediated cardiomyocytes injury. ROS have been implicated in modulating myocardin expression during cardiac hypertrophy, and myocardin is induced in response to hypertrophic signals [68, 69]. Further experiments in vitro and in vivo examined that different magnitudes and chronicities of ROS levels trigger the differential regulation of miR-1 and that, in turn, the miR-1-dependent regulation of myocardin expression contributes to the development of hypertrophy or apoptosis [70]. As mentioned, NOX2-driven oxidative stress is involved in several cardiac dysfunctions [71]. Kyrychenko et al found drastic downregulation of miR-448-3p in hearts of a mice model of duchenne muscular dystrophy [61]. Downregulation of miR-448-3p leads to the increase in the expression of Ncf1 gene, which encodes the Nox2 regulatory subunit p47phox, as well as to the substantial increase in ROS production and enhanced Ca2+ signaling in dystrophic heart. Another pivotal regulator of redox response is Nrf2, a basic leucine zipper transcription factor, which is essential for the coordinately increased expression of multiple antioxidant enzymes [72, 73]. It is observed that miR-144 inhibitor abates high glucose-induced ROS formation, reduces apoptosis and improves cardiac function in streptozotocin (STZ)-induced diabetic mice by directly targeting Nrf2 [74]. It is currently known that H2O2 works as an exogenous ROS that leads to mitochondrial fission and cytochrome c release, and the consequent apoptotic pathway [75, 76]. Therefore, lot of research was performed to investigate the relationship between miRNAs and apoptosis in response to H2O2 treatment. For instance, Li et al reported that miR-361 and prohibitin 1 (PHB1) constitute an axis that regulates mitochondrial fission and apoptosis in response to H2O2 treatment in cardiomyocytes [76]. MiR-361 overexpression induced mitochondrial fission and myocardial infarction sizes upon myocardial ischemia injury through participating in suppression of PHB1 translation. In addition, another miR-499 is significantly increased in response to ROS stimulus and could protect cardiomyocytes from H2O2-induced apoptosis [77]. Upon to oxidative stress, the JNK pathway was activated and then induced miR-499 which acts as an upstream mediator of the mitochondrial pathway through PDCD4 and PACS2 to block apoptosis and heart infarction. Recently, Liu et al found miR-132 expression was downregulated in blood of heart failure patients [78]. Overexpression of miR-132 in H9C2 cells dramatically increased the anti-oxidant stress and anti-apoptotic ability, laying a theoretical guidance for clinical targeted drugs. Lipopolysaccharide (LPS) could significantly suppress viability and increase cell death and ROS level [79]. In addition, miR-99a was reported to be capable of promoting BCL2 and suppressing the activation of caspase 3, thereby attenuating LPS-induced oxidative injury in H9c2 cells, possibly via a Notch pathway [79]. Long et al found that miR-23a promotes cardiomyocyte apoptosis and myocardial infarction induced by I/R through directly suppressing the expression of MnSOD (Manganese superoxide dismutase), which is an important antioxidant to scavenge superoxide and protects cells from oxidative stress [80, 81]. Apart from them, many other miRNAs, such as miR-130a [82], miR-98 [83], etc, are investigated to be associated with ROS-related cardiomyocyte apoptosis. Moreover, excessive ROS is also able to cause oxidative modification of RNA, which is related to a variety of cardiac pathological events [84]. MiR-184 was isolated to be highly oxidized in response to ROS stimulation [84]. Such modification leads to the misrecognition of Bcl-xL and Bcl-w, which are not its native targets. Further experiments in mouse model of MI proved that the mismatch of oxidized miR-184 is responsible for the cells undergoing apoptosis. Together, these studies imply the interplay between ROS and miRNA, however, the mechanical details remain to be addressed.
Summary of ncRNAs linked to ROS and their actions in cardiovascular diseases (CVDs)
| ncRNAs | Targets | Promotional or Antagonistic | CVDs | Refs |
|---|---|---|---|---|
| miR-145 | CaMKII | Anta- | Cardiomyocytes apoptosis | [64] |
| miR-21 | PDCD4 | Pro- | Cardiomyocytes apoptosis | [67] |
| miR-208a | PTPN4 | Anta- | Myocardial infarction | [115] |
| miR-130a | Smad4 | Pro- | Myocardial infarction | [82] |
| miR-98 | Fas | Anta- | Myocardial infarction | [83] |
| miR-1 | myocardin | Anta- | Cardiac hypertrophy | [69, 70] |
| miR-448- 3p | Ncf1 | Anta- | Dystrophic cardiomyopathy | [61] |
| miR-144 | Nrf2 | Anta- | Diabetic cardiomyopathy | [74] |
| miR-361 | PHB1 | Pro- | Cardiomyocytes apoptosis | [76] |
| miR-499 | PACS2 | Anta- | Myocardial infarction | [77] |
| miR-132 | Smad3 | Anta- | Heart Failure | [78] |
| miR-99a | Capase 3 | Anta- | LPS-induced oxidative injury | [79] |
| miR-23a | MnSOD | Pro- | Myocardial infarction | [80, 81] |
| miR-184 | Bcl-xL/ Bcl-w | Pro- | Myocardial infarction | [84] |
| lncRNA- APF | miR-188-3p/ATG7 | Pro- | Myocardial infarction | [92] |
| lncRNA- UCA1 | p27 | Pro- | Myocardial infarction | [93] |
| lncRNA- ROR | Nox2 | Pro- | Myocardial infarction | [94] |
| lncRNA- CARL | miR-539/Phb1 | Anta- | Myocardial infarction | [116] |
| lncRNA- FTX | miR-29b-1-5p/ Bcl2l2 | Anta- | Cardiomyocytes apoptosis | [117] |
| circRNA- MFACR | miR-652-3p/ MTP18 | Anta- | Cardiomyocytes apoptosis | [99] |
Besides miRNAs, lncRNAs are recently described as new regulators of redox pathway in cardiovascular diseases. LncRNAs are a set of RNAs lacking protein-coding potential, which are located throughout the whole genome [85]. It displays multiple cellular functions, such as capturing miRNAs, guiding transcription factors (TFs), and affecting the three-dimensional structure of chromatin [86-88]. In recent years, many studies have revealed that I/R-induced oxidative stress could enhance cardiomyocyte autophagy, which is related to a variety of pathological disorders [89]. It is well known that ATG7 is a key autophagy-promoting gene that functions in the regulation of cell death and survial [90, 91]. Consistent with these reports, miR-188-3p was defined to inhibit autophagic program and myocardial infarction through suppressing the translation of ATG7 [92]. Anoxia/reoxygenation (A/R) have been reported to induce autophagy and a significant increase in the APF expression levels. Moreover, lncRNA-APF affected the activity of miR-188-3p and its downstream target ATG7 by sponging to miR-188-3p, and subsequently participated in regulating the signal for autophagy and cell death in the heart. Another example is that lncRNA-UCA1 is involved in I/R or H2O2-induced cell apoptosis and ROS production in the rat model [93]. The tumor suppressor gene p27 was frequently upregulated in the ischemic heart associated with enhanced ROS production, and triggered cell apoptosis by activating cleaved-caspase 3. Follow-up experiments demonstrated that UCA1 was decreased in I/R-induced heart injury and contributed to the apoptosis of cardiomyocytes mediated by caspase3 through targeting p27. Recent study unveiled that lncRNA-ROR is highly expressed in serum of myocardial I/R injury patients, and overexpression of lncRNA-ROR could aggravate myocardial I/R damage along with increased cardiomyocyte apoptosis [94]. In this context, lncRNA-ROR also modulates the p38/MAPK pathway, which is closely related to oxidative stress. When expressing lncRNA-ROR in H9c2 cells, the production of intracellular ROS and mitochondrial ROS was enhanced by activating the NADPH oxidase. Recent research found that lncRNA Plscr4 was upregulated in hypertrophic mice hearts, and overexpression of lncRNA-Plscr4 reserved the increased ROS production following Ang II treatment by acting as an endogenous sponge of miR-214 [95]. Furthermore, the interaction between Plscr4 and miR-214 attenuated the inhibitory effects of miR-214 on Mfn2, which was involved in the mitochondrial fusion and fission in the hypertrophic heart [95]. Recently, another lncRNA- TINCR is downregulated in diabetic cardiomyopathy and lncRNA-TINCR overexpression inhibited apoptosis of cardiomyocytes under high glucose treatment [96]. Further experiments indicated that lncRNA-TINCR expression level can be used to effectively diagnose diabetic cardiomyopathy. These above studies may help in the development of new strategies for the treatment of heart diseases. However, further challenges lie in exploring the detail influence of lncRNAs in cardiomyocytes dysfunction upon oxidative stress. Another class of ncRNA, circRNAs, which form covalently closed continuous loop with its 3' and 5' ends joined together, are emerged as key regulators in multiple cellular processes by acting as competing endogenous RNAs to miRNAs [97, 98]. So far, only one circRNA (mitochondrial fission and apoptosis-related circRNA, MFACR) is illustrated during oxygen deprivation induced by MI [99]. As is known, anoxia/ reoxygenation to cardiomyocyte causes an absolute deficit in oxygen supply and triggers the accumulation of fragmented mitochondria, which consequently leads to apoptotic cell death and cardiac dysfunction. In this study, circRNA-MFACR regulates myocardial apoptosis in the heart by directly targeting and downregulating miR-652-3p; this in turn blocks mitochondrial fission and cardiomyocyte cell death by suppressing MTP18 translation. Accordingly, circRNAs may serve as a potential therapeutic avenue for cardiovascular diseases in the future.
Future Considerations
For decades, ROS acts as signaling molecules in the pathogenesis of cardiovascular diseases related to hypoxia, cardiotoxicity and ischemia-reperfusion. Here, we describe the participation of ncRNAs in the regulation of mitochondrial-mediated cardiomyocytes death and consequent heart diseases. Taken together with their emerging role in redox signaling, the ncRNAs would offer novel and powerful approaches to restore or reverse the effects of pathological heart conditions. Due to the existence in the extracellular environment, the extracellular miRNAs are defined as circulating miRNAs who have been detected in various biological fluids including sera, plasma, urine, tears, saliva, and so on [100]. Up to now, several circulating miRNAs have been reported to be potential biomarkers of ROS-related cardiac diseases including myocardial infarction, myocardial hypertrophy, cardiac fibroblasts and heart failure [101-103]. For instance, miR-499 is a representative biomarker in the early diagnosis of myocardial infarction and heart failure, which is detected earlier than other traditional biomarkers (such as cTnT, CK-MB) [101, 104]. It has been shown that Nox-derived ROS contributes to the redox signaling during the development of HF [44]. Tijsen and colleagues demonstrated that miR-423-5p exhibits a strong ability to diagnose HF (area under the curve, AUC of 0.91) [105]. Besides, many other potential biomarkers of ROS-related cardiac diseases have been demonstrated, such as miR-21, miR-378, miR-208, and miR-940 etc [102, 106]. Up to now, a mitochondrial long noncoding RNA (LIPCAR) was strongly associated with cardiovascular mortality in patients with HF, which might participate in regulating mitochondrial oxidative phosphorylation [107].
In recent years, some antioxidant-based strategies against various cardiovascular diseases have been developed, of which, ncRNAs own new and powerful therapeutic intervention through inducing or repressing miRNA expression. NcRNA mimics or inhibitors can be easily synthesized and operate in vivo, for example, inhibiting miR-208a improved survival and cardiac function during HF progression in rat model [108]. Intracardiac injection of mimics of miR-21 along with miR-1 and miR-24 reduced infarct size in a rat model [109]. However, one of the major obstacles in the development of ncRNA-based therapy is the low bioavailability, which prevents small RNA molecules from delivering into circulatory system [110]. To address this problem, several strategies have been applied, such as liposomes, exosomes, and polymers [111]. Currently, there have been many clinical trials using RNA-based drugs [112, 113], for example, a study of Santaris Pharma's LNA-modified anti-miR-122 for the treatment of hepatitis C virus infection has completed phase 1 and phase 2 clinical trials without causing viral resistance [114]. Together above observations, many challenges remain to be solved in clarifying the potential of ncRNAs as exciting new therapeutic tools for ROS-related cardiovascular disorders in the future.
Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No: 31701733, 81470522, 31430041), Applied Basic Research Programs of Qingdao, China (Grant No: 17-1-1-46-jch), and Shandong Provincial Natural Science Foundation, China (Grant No: ZR2016CQ31).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kroese LJ, Scheffer PG. 8-hydroxy-2'-deoxyguanosine and cardiovascular disease: a systematic review. Curr Atheroscler Rep. 2014;16:452
2. Bartz RR, Suliman HB, Piantadosi CA. Redox mechanisms of cardiomyocyte mitochondrial protection. Front Physiol. 2015;6:291
3. Cortes-Rojo C, Rodriguez-Orozco AR. Importance of oxidative damage on the electron transport chain for the rational use of mitochondria-targeted antioxidants. Mini Rev Med Chem. 2011;11:625-32
4. Fruehauf JP, Meyskens FL Jr. Reactive oxygen species: a breath of life or death? Clin Cancer Res. 2007;13:789-94
5. Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214-20
6. Melo Z, Ishida C, Goldaraz MP, Rojo R, Echavarria R. Novel Roles of Non-Coding RNAs in Opioid Signaling and Cardioprotection. Noncoding RNA. 2018:4
7. Di Mauro V, Barandalla-Sobrados M, Catalucci D. The noncoding-RNA landscape in cardiovascular health and disease. Noncoding RNA Res. 2018;3:12-9
8. Dong Y, Liu C, Zhao Y, Ponnusamy M, Li P, Wang K. Role of noncoding RNAs in regulation of cardiac cell death and cardiovascular diseases. Cell Mol Life Sci. 2018;75:291-300
9. Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16:71-84
10. Mikhed Y, Gorlach A, Knaus UG, Daiber A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015;5:275-89
11. Makhdoumi P, Roohbakhsh A, Karimi G. MicroRNAs regulate mitochondrial apoptotic pathway in myocardial ischemia-reperfusion-injury. Biomed Pharmacother. 2016;84:1635-44
12. Abdellatif M. Differential expression of microRNAs in different disease states. Circ Res. 2012;110:638-50
13. Magenta A, Greco S, Gaetano C, Martelli F. Oxidative stress and microRNAs in vascular diseases. Int J Mol Sci. 2013;14:17319-46
14. Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29-38
15. Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res. 2015;116:531-49
16. Koopman WJ, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA. et al. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal. 2010;12:1431-70
17. Stowe DF, Camara AK. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid Redox Signal. 2009;11:1373-414
18. Di Lisa F, Bernardi P. Modulation of Mitochondrial Permeability Transition in Ischemia-Reperfusion Injury of the Heart. Advantages and Limitations. Curr Med Chem. 2015;22:2480-7
19. Braunersreuther V, Jaquet V. Reactive oxygen species in myocardial reperfusion injury: from physiopathology to therapeutic approaches. Curr Pharm Biotechnol. 2012;13:97-114
20. Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307( Pt 1):93-8
21. Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol. 2015;78:129-41
22. Connern CP, Halestrap AP. Chaotropic agents and increased matrix volume enhance binding of mitochondrial cyclophilin to the inner mitochondrial membrane and sensitize the mitochondrial permeability transition to [Ca2+]. Biochemistry. 1996;35:8172-80
23. Connern CP, Halestrap AP. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J. 1994;302( Pt 2):321-4
24. Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M. et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887-92
25. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC. et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. P Natl Acad Sci USA. 2007;104:19500-5
26. Dorn GW 2nd. Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. 2010;3:374-83
27. Hausenloy DJ, Boston-Griffiths EA, Yellon DM. Cyclosporin A and cardioprotection: from investigative tool to therapeutic agent. Brit J Pharmacol. 2012;165:1235-45
28. Kornfeld OS, Hwang S, Disatnik MH, Chen CH, Qvit N, Mochly-Rosen D. Mitochondrial Reactive Oxygen Species at the Heart of the Matter New Therapeutic Approaches for Cardiovascular Diseases. Circulation Research. 2015;116:1783-99
29. Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radical Bio Med. 2016;100:14-31
30. Rich PR. Mitochondrial cytochrome c oxidase: catalysis, coupling and controversies. Biochem Soc T. 2017;45:813-29
31. Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C. et al. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016;23:254-63
32. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL. et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431-5
33. Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C. et al. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016;23:254-63
34. Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417:1-13
35. Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA. et al. The Oxygen Free Radicals Originating from Mitochondrial Complex I Contribute to Oxidative Brain Injury Following Hypoxia-Ischemia in Neonatal Mice. J Neurosci. 2012;32:3235-44
36. Kohlhaas M, Nickel AG, Maack C. Mitochondrial energetics and calcium coupling in the heart. J Physiol. 2017;595:3753-63
37. Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464-76
38. Zhang M, Kho AL, Anilkumar N, Chibber R, Pagano PJ, Shah AM. et al. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. 2006;113:1235-43
39. Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension. 2003;42:206-12
40. Jha JC, Watson AMD, Mathew G, de Vos LC, Jandeleit-Dahm K. The emerging role of NADPH oxidase NOX5 in vascular disease. Clin Sci (Lond). 2017;131:981-90
41. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245-313
42. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181-9
43. Geiszt M. NADPH oxidases: new kids on the block. Cardiovasc Res. 2006;71:289-99
44. Zhang M, Perino A, Ghigo A, Hirsch E, Shah AM. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxid Redox Signal. 2013;18:1024-41
45. Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic Biol Med. 2018;117:76-89
46. Peng T, Lu X, Feng Q. Pivotal role of gp91phox-containing NADH oxidase in lipopolysaccharide-induced tumor necrosis factor-alpha expression and myocardial depression. Circulation. 2005;111:1637-44
47. Hingtgen SD, Tian X, Yang J, Dunlay SM, Peek AS, Wu Y. et al. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics. 2006;26:180-91
48. Hirotani S, Otsu K, Nishida K, Higuchi Y, Morita T, Nakayama H. et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation. 2002;105:509-15
49. Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003;35:851-9
50. Zhang L, Wang X, Wu Y, Lu X, Chidiac P, Wang G. et al. Maternal diabetes up-regulates NOX2 and enhances myocardial ischaemia/reperfusion injury in adult offspring. J Cell Mol Med. 2018;22:2200-9
51. Karim AS, Reese SR, Wilson NA, Jacobson LM, Zhong W, Djamali A. Nox2 is a Mediator of Ischemia Reperfusion Injury. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2015;15:2888-99
52. Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD. et al. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93:802-5
53. Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M. et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121-6
54. Kuroda J, Ago T, Matsushima S, Zhai PY, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. P Natl Acad Sci USA. 2010;107:15565-70
55. Ago T, Kuroda J, Pain J, Fu CX, Li H, Sadoshima J. Upregulation of Nox4 by Hypertrophic Stimuli Promotes Apoptosis and Mitochondrial Dysfunction in Cardiac Myocytes. Circulation Research. 2010;106:1253-U183
56. Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY. et al. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860-6
57. Zhang M, Perino A, Ghigo A, Hirsch E, Shah AM. NADPH Oxidases in Heart Failure: Poachers or Gamekeepers? Antioxid Redox Sign. 2013;18:1024-41
58. Latronico MVG, Condorelli G. MicroRNAs and cardiac pathology. Nature Reviews Cardiology. 2009;6:418-29
59. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102-14
60. Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Current Opinion in Cell Biology. 2009;21:452-60
61. Kyrychenko S, Kyrychenko V, Badr MA, Ikeda Y, Sadoshima J, Shirokova N. Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy. Cardiovascular Research. 2015;108:324-34
62. Yang YB, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y. et al. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol-Heart C. 2006;291:H3065-H75
63. Yoo B, Lemaire A, Mangmool S, Wolf MJ, Curcio A, Mao L. et al. Beta1-adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;297:H1377-86
64. Cha MJ, Jang JK, Ham O, Song BW, Lee SY, Lee CY. et al. MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIdelta. Biochem Biophys Res Commun. 2013;435:720-6
65. Li R, Yan G, Li Q, Sun H, Hu Y, Sun J. et al. MicroRNA-145 protects cardiomyocytes against hydrogen peroxide (H(2)O(2))-induced apoptosis through targeting the mitochondria apoptotic pathway. PLoS One. 2012;7:e44907
66. Gaspar-Pereira S, Fullard N, Townsend PA, Banks PS, Ellis EL, Fox C. et al. The NF-kappaB subunit c-Rel stimulates cardiac hypertrophy and fibrosis. Am J Pathol. 2012;180:929-39
67. Wei C, Li L, Kim IK, Sun P, Gupta S. NF-kappaB mediated miR-21 regulation in cardiomyocytes apoptosis under oxidative stress. Free Radic Res. 2014;48:282-91
68. Chiu CZ, Wang BW, Shyu KG. Use of atorvastatin to inhibit hypoxia-induced myocardin expression. Eur J Clin Invest. 2012;42:564-71
69. Li M, He HP, Gong HQ, Zhang J, Ma WJ, Zhou H. et al. NFATc4 and myocardin synergistically up-regulate the expression of LTCC alpha1C in ET-1-induced cardiomyocyte hypertrophy. Life Sci. 2016;155:11-20
70. Lee S, Lim S, Ham O, Lee SY, Lee CY, Park JH. et al. ROS-mediated bidirectional regulation of miRNA results in distinct pathologic heart conditions. Biochem Biophys Res Commun. 2015;465:349-55
71. Zhao YY, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM. et al. Nox2 NADPH Oxidase Promotes Pathologic Cardiac Remodeling Associated with Doxorubicin Chemotherapy. Cancer Res. 2010;70:9287-97
72. Kensler TW, Wakabayash N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol. 2007;47:89-116
73. Li J, Johnson D, Calkins M, Wright L, Svendsen C, Johnson J. Stabilization of nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol Sci. 2005;83:313-28
74. Yu M, Liu Y, Zhang B, Shi Y, Cui L, Zhao X. Inhibiting microRNA-144 abates oxidative stress and reduces apoptosis in hearts of streptozotocin-induced diabetic mice. Cardiovasc Pathol. 2015;24:375-81
75. Wu YY, Wang DJ, Wang XD, Wang YY, Ren FL, Chang D. et al. Caspase 3 is Activated through Caspase 8 instead of Caspase 9 during H2O2-induced Apoptosis in HeLa Cells. Cell Physiol Biochem. 2011;27:539-46
76. Wang K, Liu CY, Zhang XJ, Feng C, Zhou LY, Zhao Y. et al. miR-361-regulated prohibitin inhibits mitochondrial fission and apoptosis and protects heart from ischemia injury. Cell Death Differ. 2015;22:1058-68
77. Wang JJ, Jia ZQ, Zhang CG, Sun M, Wang WP, Chen P. et al. miR-499 protects cardiomyocytes from H2O2-induced apoptosis via its effects on Pdcd4 and Pacs2. Rna Biol. 2014;11:339-50
78. Liu XL, Tong Z, Chen KY, Hu XF, Jin HX, Hou MX. The Role of miRNA-132 against Apoptosis and Oxidative Stress in Heart Failure. Biomed Res Int. 2018
79. Jing R, Zhou ZM, Kuang F, Huang L, Li CC. microRNA-99a Reduces Lipopolysaccharide-Induced Oxidative Injury by Activating Notch Pathway in H9c2 Cells. Int Heart J. 2017;58:422-7
80. Long B, Gan TY, Zhang RC, Zhang YH. miR-23a Regulates Cardiomyocyte Apoptosis by Targeting Manganese Superoxide Dismutase. Mol Cells. 2017;40:542-9
81. Cramer-Morales K, Heer CD, Mapuskar KA, Domann FE. SOD2 targeted gene editing by CRISPR/Cas9 yields Human cells devoid of MnSOD. Free Radical Bio Med. 2015;89:379-86
82. Li Y, Du Y, Cao J, Gao Q, Li H, Chen Y. et al. MiR-130a inhibition protects rat cardiac myocytes from hypoxia-triggered apoptosis by targeting Smad4. Kardiol Pol. 2018;76:993-1001
83. Sun C, Liu H, Guo J, Yu Y, Yang D, He F. et al. MicroRNA-98 negatively regulates myocardial infarction-induced apoptosis by down-regulating Fas and caspase-3. Sci Rep. 2017;7:7460
84. Wang JX, Gao J, Ding SL, Wang K, Jiao JQ, Wang Y. et al. Oxidative Modification of miR-184 Enables It to Target Bcl-xL and Bcl-w. Mol Cell. 2015;59:50-61
85. Papait R, Kunderfranco P, Stirparo GG, Latronico MVG, Condorelli G. Long Noncoding RNA: a New Player of Heart Failure? J Cardiovasc Transl. 2013;6:876-83
86. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904-14
87. Piccoli MT, Gupta SK, Thum T. Noncoding RNAs as regulators of cardiomyocyte proliferation and death. J Mol Cell Cardiol. 2015;89:59-67
88. Dong YH, Liu CY, Zhao YF, Ponnusamy M, Li PF, Wang K. Role of noncoding RNAs in regulation of cardiac cell death and cardiovascular diseases. Cellular and Molecular Life Sciences. 2018;75:291-300
89. Essick EE, Sam F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid Med Cell Longev. 2010;3:168-77
90. Ghavami S, Gupta S, Ambrose E, Hnatowich M, Freed DH, Dixon IMC. Autophagy and Heart Disease: Implications for Cardiac Ischemia-Reperfusion Damage. Curr Mol Med. 2014;14:616-29
91. Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109:151-60
92. Wang K, Liu CY, Zhou LY, Wang JX, Wang M, Zhao B. et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nature Communications. 2015:6
93. Liu YB, Zhou DL, Li GN, Ming X, Tu YF, Tian JW. et al. Long Non Coding RNA-UCA1 Contributes to Cardiomyocyte Apoptosis by Suppression of p27 Expression. Cell Physiol Biochem. 2015;35:1986-98
94. Zhang WW, Li Y, Wang P. Long non-coding RNA-ROR aggravates myocardial ischemia/reperfusion injury. Braz J Med Biol Res. 2018:51
95. Lv L, Li T, Li X, Xu C, Liu Q, Jiang H. et al. The lncRNA Plscr4 Controls Cardiac Hypertrophy by Regulating miR-214. Mol Ther Nucleic Acids. 2018;10:387-97
96. Chen Y, Tan S, Liu M, Li J. LncRNA TINCR is downregulated in diabetic cardiomyopathy and relates to cardiomyocyte apoptosis. Scandinavian Cardiovascular Journal. 2018:1-17
97. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333-8
98. Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK. et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384-8
99. Wang K, Gan TY, Li N, Liu CY, Zhou LY, Gao JN. et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death and Differentiation. 2017;24:1111-20
100. Ghai V, Wang K. Recent progress toward the use of circulating microRNAs as clinical biomarkers. Archives of toxicology. 2016
101. Xin Y, Yang C, Han Z. Circulating miR-499 as a potential biomarker for acute myocardial infarction. Annals of translational medicine. 2016;4:135
102. Xu T, Zhou Q, Che L, Das S, Wang L, Jiang J. et al. Circulating miR-21, miR-378, and miR-940 increase in response to an acute exhaustive exercise in chronic heart failure patients. Oncotarget. 2016;7:12414-25
103. Roncarati R, Viviani Anselmi C, Losi MA, Papa L, Cavarretta E, Da Costa Martins P. et al. Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2014;63:920-7
104. Liu X, Fan Z, Zhao T, Cao W, Zhang L, Li H. et al. Plasma miR-1, miR-208, miR-499 as potential predictive biomarkers for acute myocardial infarction: An independent study of Han population. Experimental gerontology. 2015;72:230-8
105. Tijsen AJ, Creemers EE, Moerland PD, de Windt LJ, van der Wal AC, Kok WE. et al. MiR423-5p as a circulating biomarker for heart failure. Circ Res. 2010;106:1035-9
106. Han Z, Zhang L, Yuan L, Liu X, Chen X, Ye X. et al. Change of plasma microRNA-208 level in acute myocardial infarction patients and its clinical significance. Ann Transl Med. 2015;3:307
107. Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A. et al. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res. 2014;114:1569-75
108. Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM. et al. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation. 2011;124:1537-47
109. Schulte C, Zeller T. microRNA-based diagnostics and therapy in cardiovascular disease-Summing up the facts. Cardiovasc Diagn Ther. 2015;5:17-36
110. Farias JG, Molina VM, Carrasco RA, Zepeda AB, Figueroa E, Letelier P. et al. Antioxidant Therapeutic Strategies for Cardiovascular Conditions Associated with Oxidative Stress. Nutrients. 2017:9
111. Baumann V, Winkler J. miRNA-based therapies: strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med Chem. 2014;6:1967-84
112. Kaczmarek JC, Kowalski PS, Anderson DG. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017;9:60
113. Christopher AF, Kaur RP, Kaur G, Kaur A, Gupta V, Bansal P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect Clin Res. 2016;7:68-74
114. Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K. et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685-94
115. Liu A, Sun Y, Yu B. MicroRNA-208a Correlates Apoptosis and Oxidative Stress Induced by H2O2 through Protein Tyrosine Kinase/Phosphatase Balance in Cardiomyocytes. Int Heart J. 2018
116. Wang K, Long B, Zhou LY, Liu F, Zhou QY, Liu CY. et al. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat Commun. 2014;5:3596
117. Long B, Li N, Xu XX, Li XX, Xu XJ, Guo D. et al. Long noncoding RNA FTX regulates cardiomyocyte apoptosis by targeting miR-29b-1-5p and Bcl2l2. Biochem Biophys Res Commun. 2018;495:312-8
Author contact
![]() Corresponding authors: Yanhan Dong, Email: yanhandongedu.cn; Peifeng Li, Email: peifliedu.cn; Kun Wang, Email: wangk696edu.cn; Tel: 86-532-82991791
Corresponding authors: Yanhan Dong, Email: yanhandongedu.cn; Peifeng Li, Email: peifliedu.cn; Kun Wang, Email: wangk696edu.cn; Tel: 86-532-82991791