Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(4):738-748. doi:10.7150/ijbs.30106 This issue Cite
Research Paper
USP17 Suppresses Tumorigenesis and Tumor Growth through Deubiquitinating AEP
Xi Chen2*, Chen Wang3*, Keman Liao1, Sunhai Zhou1, Lu Cao4, Jiayi Chen4, Cheng Xu4 ![]() , Yingying Lin1
, Yingying Lin1 ![]()
1. Department of Neurosurgery, Renji Hospital, School of Medicine, Shanghai Jiao-tong University, Shanghai, 200127, China.
2. CAS key laboratory of Tissue Microenvironment and Tumor, Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, 200025, China.
3. Shanghai Institute of Advanced Immunochemical Studies, ShanghaiTech University, Shanghai 201210, China.
4. Department of Radiation Oncology, Ruijin Hospital, School of Medicine, Shanghai Jiao-tong University, Shanghai 200025, China.
*These authors contributed equally to this work.
Received 2018-9-21; Accepted 2018-12-20; Published 2019-1-29
Abstract

Ubiquitin-specific protease 17 (USP17), a novel member of deubiquitinase, is reported to play essential roles in several solid tumors. However, the expression and function of USP17 in breast cancer tumorigenesis remains ambiguity. Here we found that the mRNA level of USP17 was lower in breast cancer tissues than normal tissues. Meanwhile, higher USP17 level was detected in normal epithelial cell MCF-10A and a less-malignant cell MCF-7 than malignant cell line MDA-MB-231. Inhibition of USP17 in MCF7 cells enhanced tumorigenesis and tumor growth while overexpression of USP17 in malignant MDA-MB-231 cells reduced its tumorigenesis and growth ability in vitro and in vivo. Further study revealed that USP17 interacted with and deubiquitinated Asparaginyl endopeptidase (AEP), resulting in decreased protein levels of AEP. Moreover, knockdown of AEP inhibited breast cancer tumorigenesis and growth in vitro and in vivo through the inactivation of ERK signaling. Taken together, our works indicate that USP17 deubiquitinates AEP, down-regulates its protein level, and inhibits breast cancer tumorigenesis through disturbing ERK signaling. Thus, our data suggests that USP17 is a potential tumor suppressor in breast cancer and AEP is a promising target in breast cancer therapy.
Keywords: Breast cancer, AEP, USP17, Deubiquitination, ERK
Introduction
Breast cancer is the second leading causes of cancer death in females all over the world and the incidence of new cases ranks first in female [1]. Therefore, it is quite urgent to uncover new tumor suppressors and therapeutic targets in breast cancer. The process of ubiquitination and de-ubiquitination have been discovered to maintain the stability and homeostasis of large amount of crucial proteins and participate in regulation of important intracellular process and signaling pathways like apoptosis [2, 3], cell cycle [4, 5] and cancer [6]. The ubiquitination process is controlled by multiple enzymes including ubiquitin activating enzyme (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin ligase (E3) [7]. After transferring by E1, E2 and E3 enzymes, the ubiquitin polypeptides are added to target proteins [8]. The activity, cellular translocation and other functions of the substrates can be altered when lysine 63 (K63) ubiquitination chains is formed [9, 10]. When K48 of the target proteins was modified with ubiquitin chains, the targets undergo degradation through the proteasome system [11]. The process of ubiquitination can be reversed by de-ubiquitinating enzymes (DUBs), a family of proteins that clear ubiquitin chains from the substrate [12].
The ubiquitin-specific protease 17 (USP17) is an immediate early gene that can be rapidly induced by cytokines like CXCL12, CXCL8, IL-4 and IL-6 to regulate cell migration and leukocytes growth and differentiation [13, 14]. However, the expression and function of USP17 in various cancers were obscure until now. It has been showed that the expression of USP17 was higher in human organs like heart, liver and pancreas than various human cancer cell lines and USP17 overexpression leads to apoptosis in cancer cells [15]. USP17 interacted with suppressor of defective silencing 3(SDS3) and negatively regulated histone deacetylase (HDAC) activity resulting in cell proliferation inhibition [16]. Moreover, bromodomain and extra-C terminal (BET) inhibitor JQ1 and HDAC inhibitor mocetinostat significantly increased the expression of USP17 in breast cancer cells and led to decreased cell viability through attenuating the RAS/MAPK pathway [17].
Interestingly, the oncogenic characteristics of USP17 have also been reported. The results indicated high level of USP17 in lung, colon, esophagus and cervix tumor samples to promote cell G1-S transition and cell proliferation [18]. Besides, USP17 was overexpressed in non-small cell lung cancer tissues and patients with high level of USP17 had reduced survival [19]. In osteosarcoma, USP17 was up-regulated in tumor tissues and cell lines and USP17 facilitated cell migration and invasion through deubiquitinating and stabilizing SMAD4 [20].
Asparaginyl endopeptidase (AEP) is a protease that cleaves asparagine residues through lysosome and the activation of AEP is depended on low pH thresholds [21, 22]. It has been reported that AEP is required for the process of kidney physiology [23], bone metabolism [24], macrophage phagocytosis [25] and antigen processing [26]. More and more studies uncovered crucial roles of AEP in tumors. AEP was demonstrated overexpressed in various tumors like breast cancer [27], leukemia [28], colorectal cancer [29]. Our previous experiments indicated that elevated AEP promoted breast cancer pulmonary metastasis through maintaining ubiquitination homeostasis of AEP by E3 ligase TRAF6 and DUB USP17, and was correlated with poor prognosis [30].
In this report, we demonstrated a tumor suppressor function of USP17 in breast cancer. We showed that USP17 was down-regulated in breast cancer tissues and malignant breast cancer cell lines and USP17 overexpression significantly decreased breast cancer cell growth while USP17 knockdown dramatically enhanced breast cancer cell growth in vitro and in vivo. Furthermore, we presented evidence that AEP was a substrate for USP17 de-ubiquitination, and USP17 overexpression resulting in reduced AEP level. Meanwhile, our findings showed that AEP promoted breast cancer tumorigenesis and tumor growth, which indicated that USP17 serves as a tumor suppressor gene in breast cancer through down-regulating AEP protein level.
Materials and methods
Cell lines
Breast cancer cell lines, including MCF-7 and MDA-MB-231, and HEK-293T cell line were cultured in DMEM (HyClone, Logan,UT) medium containing 10% fetal bovine serum (HyClone, Logan, UT). The normal mammary epithelial cell line MCF-10A were cultured in the MEBM medium (CC-3150, Clonetics) with additives and 100 ng/ml cholera toxin. All the cell lines were incubated at 37℃ with 5% CO2. AEP rich medium was collected from the cell culture medium of HEK-293L cells, which secreted large amount of AEP protein.
Plasmids construction and cell line construction
AEP, Flag-tagged USP17, Flag-tagged USP17 C89S mutant, Flag-tagged TRAF6 and HA-tagged Ubiquitin plasmids were cloned into pcDNA3.1, or pCMV plasmids. To construct USP17 overexpressed MCF-7 cells, USP17 was cloned into the pMSCV-puro plasmid. shRNA sequences targeting USP17 and AEP were synthesized by Invitrogen and cloned into pLKO.1 plasmid. Both the retrovirus and lentivirus were packaged using psPAX2 and pMD2G plasmids. The stable cell line of USP17 OE MCF-7, USP17 KD MDA-MB-231 and AEP KD MDA-MB-231 cell lines were obtained by adding the virus supernatant to cell culture mediums and selected by puromycin. The sequences for USP17 overexpression, USP17 KD and AEP KD were noted below:
USP17 overexpression:
USP17-MSCV-F: 5'-CCGCTCGAGATGGAGGACGACTCACTCTACT-3'.
USP17-MSCV-R: 5'-AAGGGCGGCCGCCTGGCACACAAGCAGAGC-3'.
USP17 KD:
USP17-KD-1-F: 5'-GATCTCCCGAAGTCACCACTCTCATGTTTCAAGAGAACATGAGAGTGGTGACTTCTTTTTC-3'.
USP17-KD-1-R: 5'-TCGAGAAAAAGAAGTCACCACTCTCATGTTCTCTTGAAACATGAGAGTGGTGACTTCGGGA-3'.
USP17-KD-2-F: 5'-GATCTCCCCGACGTACTTGTGATTCATTTCAAGAGAATGAATCACAAGTACGTCGTTTTTC-3'.
USP17-KD-2-R: 5'-TCGAGAAAAACGACGTACTTGTGATTCATTCTCTTGAAATGAATCACAAGTACGTCGGGGA-3'.
AEP-KD-F: 5'-GATCTCCCCGAGATGGTGTTCTACATTGAATTTCAAGAGAATTCAATGTAGAACACCATC-3'.
AEP-KD-R: 5'-GATGGTGTTCTACATTGAATTCTCTTGAAATTCAATGTAGAACACCATCTCGGGGAGATC -3'.
Gel Filtration
Superdex 200 column (GE healthcare) were used to purify the cell lysis. We used Equilibration Buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Triton X-100) for column equilibration. Two milligram of cell lysis were applied to and eluted from the column. 400 μl elution were collected at a flow rate of 0.5 ml/min.
Cell growth curve and CCK-8 assay
For cell growth curve, 1x104 cells per well were seeded in a 6-well plate and cell numbers were counted for 6 days. For CCK-8 assay, cell number was measured using CCK-8 reagent (Beyotime) according to manufacturer's instructions.
Western Blot and Immunoprecipitation
Immunoprecipitation and Western Blot experiments were performed as previously described [30, 31]. Briefly, cells were extracted with RIPA lysis buffer containing phosphatase and protease inhibitors. Cell lysates were incubated with 1μg indicated antibodies and protein A-Sepharose (GE Healthcare). The cell lysates, antibodies and sepharose mix were incubated at 4 °C overnight. Then wash the immunocomplexes four times with lysis buffer and analyzed by Western Blot assay. Antibodies used were as follow: anti-USP17 (AP5491b, Abgent), anti-AEP (AF2199, R&D Systems), anti-TRAF6 (AF3284, R&D Systems), anti-Actin (#3700P, Cell signaling technology), anti-Flag (F3165, Sigma), anti-Ubiquitin (#3933, Cell signaling technology), anti-p-ERK (#9106, Cell signaling technology), anti-ERK (#9102, Cell signaling technology), anti-p65 (#8242, Cell signaling technology), anti-p-p65 (#3033, Cell signaling technology).
RNA extraction and quantitative Real-Time PCR
RNA extraction and qPCR were performed as previously described [30]. Briefly, total RNA was extracted using TRIzol reagent (Invitrogen). PrimeScript® RT reagent Kit (Takara) was used to obtain cDNA. Quantitative Real-Time PCRs were performed using 7500 Fast Real-Time PCR System (Applied Biosystems) and Real-Time PCR reactions were performed using 2x SYBR Green Gene Expression PCR Master Mix. Primers used were as follow (5'-3'):
USP17-F: CTGCCTCCCGACGTACTTG.
USP17-R: GTTCATGGACTCCTGATGTGTC.
AEP-F: GAAACGCAAAGCCAGTTCTC.
AEP-R: GCAAGGAGACGATCTTACGC.
18S-F: AACCCGTTGAACCCCATT.
18S-R: CCATCCAATCGGTAGTAGCG.
Immunofluorescence assays
Immunofluorescence experiments were performed as previously described [31]. Briefly, 2×105 cells were seeded on coverslips for each well of a 6-well plate. Cells were washed 3 times with PBS before fixed with 4% paraformaldehyde in PBS. Cells were blocked with PBS containing 1% goat serum for 30 min. Antibodies were incubated at 4 °C overnight. Cells were washed 6 times with PBS for totally 3 hours and incubated with secondary antibodies for 1 hour at RT. Samples were observed with a Zeiss laser-scanning confocal microscope (LSM Meta 510). Single sections are shown. Images were processed (colored and merged) with the Zeiss (LSM 510) software.
Tumor xenografts in vivo
Animal experiments were approved by the institutional biomedical research ethics committee of Shanghai Institutes for Biological Sciences (Shanghai, China). 2 x 105 MDA-MB-231 cells and 1x 106 MCF7 cells were injected in the mammary fat pat of 6-8 weeks BALB/c nude mice (female, n=5 per group) respectively. Tumor size was measured every week with a digital caliper and tumor volumes were calculated with the equation: V= (length [mm] x width2 [mm2])/2. The mice were sacrificed at 28 days post tumor cell injection.
Statistical analyses
All experiments were repeated at least three times and all data were presented as means ± SD. Statistical analyses were performed with GraphPad Prism 5.0 (GraphPad Software, Inc.). The linear regression model was used to evaluate the relationship between the expression levels of Bcl-3 and other factors. The significant differences in mean values were evaluated by the two-tailed Student's t-test. P<0.05 was considered statistically significant.
Results
USP17 expression is down-regulated in breast cancer
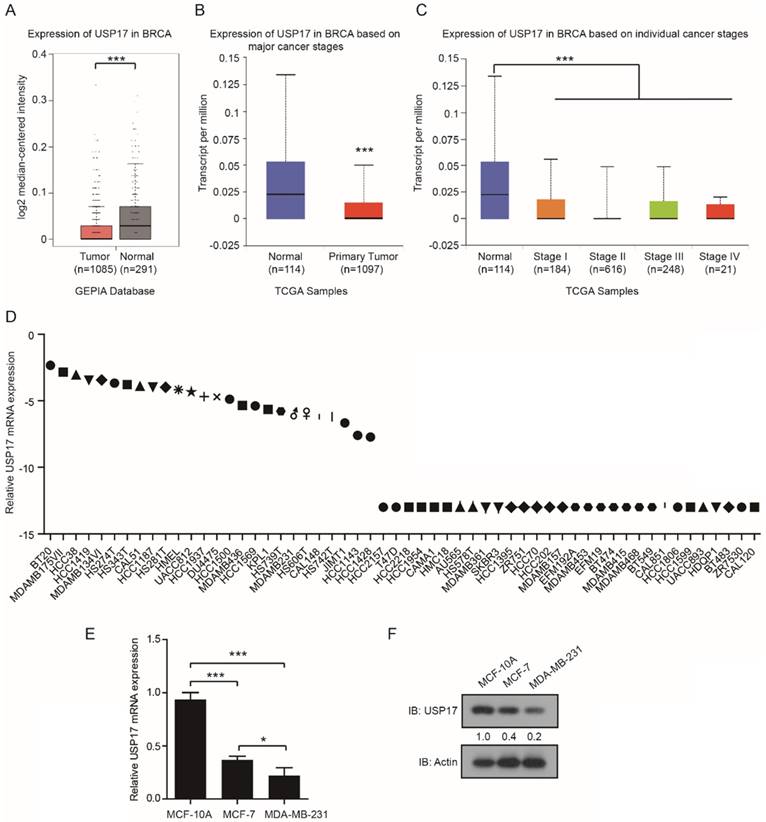
Firstly, we analyzed the expression levels of USP17 in 1085 cases of breast cancer tissues and 291 cases of normal breast tissues using the Gene Expression Profilling Interactive Analysis (GEPIA) database (http://gepia.cancer-pku.cn/). The results indicated USP17 mRNA levels were higher in normal tissues than in breast cancer tissues (Fig. 1A). Similar results were obtained using The Cancer Genome Atlas (TCGA) data of 1097 breast cancer tissues and 114 normal tissues from UALCAN (http://ualcan.path.uab.edu/index.html; [32]) (Fig. 1B). Additionally, the analysis from UALCAN showed the mRNA expression of USP17 was decreased in all stages of breast cancer tissues compared with normal tissues, which suggested that USP17 expression in breast cancer was inversely correlated with the stages of disease progression (Fig. 1C). Furthermore, we found USP17 mRNA expression was quite different in a panel of breast cancer cell lines through The Broad-Novartis Cancer Cell Line Encyclopedia (CCLE) database (https://portals.broadinstitute.org/ccle) (Fig. 1D). Next, we detected the expression of USP17 in different breast cancer cell lines with varied malignant grade. Interestingly, the mRNA (Fig. 1E) and protein levels (Fig. 1F) of USP17 was higher in MCF-10A, a normal mammary epithelial cell line and MCF-7, a less-malignant cell line, than MDA-MB-231 cells, a malignant breast cancer cell line. Together, these results suggested that USP17 played a crucial role in breast cancer tumorigenesis.
USP17 expression in breast cancer cells and patients. A-B. Expression of USP17 in human breast cancer specimens using the GEPIA Database (A) and TCGA data from UALCAN(B). C. Expression of USP17 in different stages of breast cancer versus normal tissues from TCGA data. D. USP17 expression in a panel of breast cancer cell lines using CCLE database. E. qRT-PCR analysis of USP17 mRNA expression in MCF-10A, MCF-7 and MDA-MB-231 cell lines. F. Immunoblot analyses of USP17 in MCF-10A, MCF-7 and MDA-MB-231 cell lines.
Altered USP17 expression changed breast cancer cell growth in vitro and in vivo
To investigated the function of USP17 in breast cancer, we constructed USP17 overexpressed (OE) MDA-MB-231 cell line and USP17 knockdown (KD) MCF-7 cell line. The overexpression and knockdown efficient was assessed through q-RT-PCR and Western Blot assay (Fig. 2A, B).
Altered tumorigenesis and tumor growth of breast cancer cells by USP17 expression level. qRT-PCR analysis and Immunoblot analysis of USP17 expression in USP17 overexpressed MDA-MB-231 cells. qRT-PCR analysis and Immunoblot analysis of USP17 expression in USP17 knocked down MCF-7 cells. C-D. The cell growth curve of MDA-MB-231(C) and MCF-7 (D) cells with USP17 overexpression and USP17 knocked down. E-F. Tumor volume in mice injected with MDA-MB-231 cells (E) and MCF-7 (F) with USP17 overexpression and USP17 knocked down (n=5 for each group). All data are presented as the mean ± SD. *P < 0.05, **P <0.01.
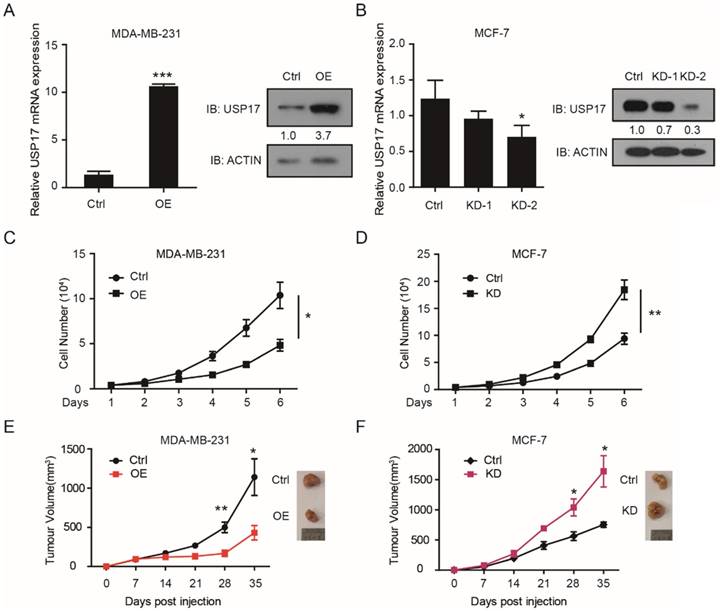
Two shRNA sequences were used to knockdown USP17 in MCF-7 cells and we chose the second shRNA sequence which play better knockdown efficiency for the experiments below. Then, we examined whether USP17 affects cell growth capability of breast cancer cells in vitro. According to the growth curve results, we found that overexpressed USP17 in MDA-MB-231 significantly inhibits cell growth compared with control cells (Fig. 2C). On the contrary, USP17 depletion dramatically enhanced cell growth when compared with control cells (Fig. 2D).
Next, we evaluated the effects of USP17 in the tumorigenicity of breast cancer cells in vivo. USP17-OE MDA-MB-231 cells, USP17-KD MCF-7 cells and their control cells were injected into the fat pad of BALB/c nude mice. After 4 weeks' evaluation, USP17-OE MDA-MB-231 cells displays reduced tumor growth capability (Fig. 2E), and MCF-7-KD cells showed significantly higher tumorigenicity (Fig. 2F). Taken together, our results uncovered the tumor suppressor function of USP17 in breast cancer cells.
USP17 interacts with and deubiquitinates AEP
We have previously reported that AEP can be ubiquitinated by TRAF6 and deubiquitinated by USP17 [30]. To further investigate the regulation of AEP by USP17, we performed co-immunoprecipitation (IP) assay to confirm the interaction of AEP and USP17. The plasmids encoding AEP, Flag tagged TRAF6 and Flag tagged USP17 were co-transfected into 293T cells. Then cell lysates were immunoprecipitated with AEP antibodies. Western Blot assay showed that both TRAF6 and USP17 were presented in the complex of AEP (Fig. 3A). Then, the interaction of AEP and USP17 were validated using size exclusion chromatography. As shown in Figure 3B, AEP, USP17 and TRAF6 were presented in the same complex. Furthermore, immunofluorescence staining assay revealed that AEP and USP17 co-localized in the cytoplasmic regions in cells (Fig. 3C). Together, these data demonstrated that AEP interacted with USP17.
USP17 deubiquitinates AEP. A. Co-IP analyses of AEP, USP17-Flag and TRAF6-Flag expression vectors in 293T cells and cell lysates with anti-AEP antibodies. B. Coelution of USP17, AEP and TRAF6 in lysates of 293T cells overexpressed USP17, AEP and TRAF6. C. Immunofluorescence staining of USP17 (red) and AEP (green) in 293T cells overexpressed USP17 and AEP. D. Immunoblot analysis of ubiquitination of AEP in MG-132 treatment 293T cells transfected with indicated plasmids.
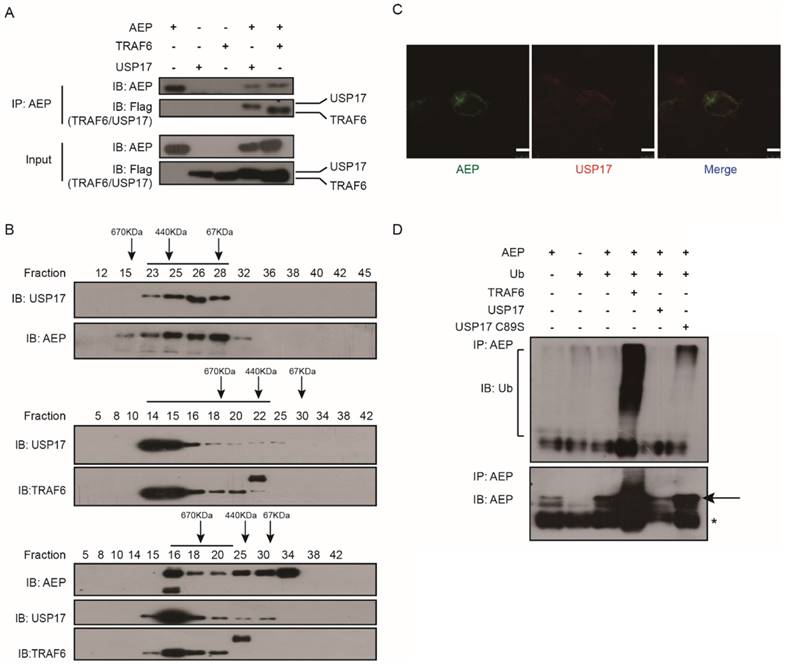
To ascertain the deubiquitination function of USP17, we co-transfected expression vectors encoding AEP, ubiquitin (Ub), TRAF6, USP17 and USP17-C89S into 293T cells. After treated with proteasome inhibitor MG132, the cell lysates were immunoprecipitated with AEP antibodies. Western Blot assay were performed to detect the ubuiqitination level of AEP in each panel. As showed in Figure 3D, TRAF6 overexpression dramatically increased AEP ubiquitination level while USP17, but not inactive mutant USP17 C89S, overexpression significantly attenuated AEP ubiquitination. Together, our results confirmed the interaction of AEP and USP17, which mediated the removal of ubiquitin chain from AEP.
USP17 down-regulates AEP protein level
As we have shown previously, TRAF6 could increase the stability and secretion of AEP [30], we want to explore whether USP17 regulated AEP stability and secretion. After transfecting plasmids encoding AEP with TRAF6, USP17 and USP17 C89S, we analyzed the expression level of AEP. Consistent with previous results, TRAF6 overexpression significantly elevated both the pro-AEP (56 KDa) and the active- AEP (36 KDa) level, while USP17 overexpression decreased the two forms of AEP expression. Interestingly, USP17 mutant plasmid C89S have no effect on reducing AEP protein level (Fig. 4A). To confirm the results, we transfected 293T cells with increasing amounts of Flag-USP17 expression plasmid and analyzed by Western Blot. The results revealed that AEP expression decreased gradually with more USP17 expressed in cells (Fig. 4C). As the mature AEP (46 KDa) can be secrete to the culture medium, to explore the regulation of USP17 on AEP secretion, we detected the AEP level in cell culture medium from USP17-OE MDA-MB-231 and control cells. The same amount of protein from cell culture medium was loaded to detect the expression of AEP. Western Blot results showed a dramatically decrease in AEP secretion when USP17 was overexpressed (Fig. 4B).
USP17 down-regulates AEP protein level. A. Immunoblot analysis of AEP and Flag-tagged TRAF6, USP17 and USP17 C89S in MDA-MB-231 cell line. B. Immunoblot analysis of AEP in conditional medium of control and USP17 overexpressed MDA-MB-231 cells. C. Immunoblot analysis of AEP in MDA-MB-231 cells transfected with USP17 plasmids. D. qRT-PCR analysis of USP17 and AEP in MDA-MB-231 cells overexpressed USP17 and USP17C89S. E. qRT-PCR analysis of USP17 and AEP in USP17 knocked down MCF-7 cells.
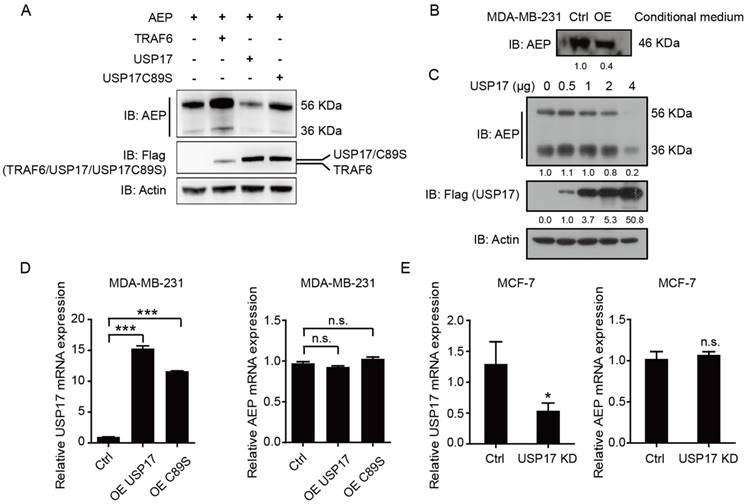
To ascertain the regulation of USP17 on AEP stability was relied on its deubiquitinase activity, we performed the q-RT-PCR assay to detected AEP mRNA expression when USP17 expression was altered. Neither overexpression of USP17 nor C89S mutant influenced AEP mRNA level (Fig. 4D). Consistently, USP17 depletion barely altered AEP mRNA level (Fig. 4E). Thus, these data indicated that USP17 down-regulated AEP protein level in post-translational pathway.
Suppression of AEP reduced tumorigenesis and tumor growth in vitro and in vivo
AEP is initially found to participate in tumor invasion and metastasis progress. Recently, Andrade reported that nucleoplasmic calcium regulates cell proliferation through AEP [33]. However, the function of AEP in tumorigenesis and tumor growth remains unclear. In order to further investigate whether USP17 inhibits breast cancer tumorigenesis through down-regulation AEP, we constructed AEP knockdown stable breast cancer cell lines in MDA-MB-231 cell line. The pro-AEP significantly decreased in AEP knockdown cells (Fig. 5A). And depletion of AEP significantly reduced cell growth in vitro (Fig. 5B). Moreover, culture medium containing different AEP protein concentration were used to treat MDA-MB-231 cells, and we found that supernatant containing higher AEP protein concentration significantly promoted MDA-MB-231 cell growth in vitro (Fig. 5C). Consistently, the cell cycles analysis showed that in cells in which AEP was silenced, there was a significant reduction in the fraction of cells in G2/M (25.2±2% of control cells, as compared to 7±1% of AEP knockdown cells; P<0.05) (Fig. 5D).
AEP promotes breast cancer tumorigenesis. A. Immunoblot analysis of AEP in AEP knocked down MDA-MB-231 cells. B. The cell growth curve of MDA-MB-231 cells with AEP knocked down. C. MTT assay to detect cell number after AEP protein treatment in MDA-MB-231 cells. D. Cell cycle distribution in control and AEP knocked down MDA-MB-231 cells. E. Control and AEP knockdown MDA-MB-231 cells stably expressed GFP were injected into mammary fat pad of female nude mice. Representative tumors are shown by the fluorescent intensity. F. Quantification of the fluorescent intensity in E. G. Representative hematoxylin and eosin sections of tumors from E. H-I. Tumor volumes in mice injected with control, AEP knockdown, USP17 overexpression and AEP USP17 double overexpressed MDA-MB-231 cells.
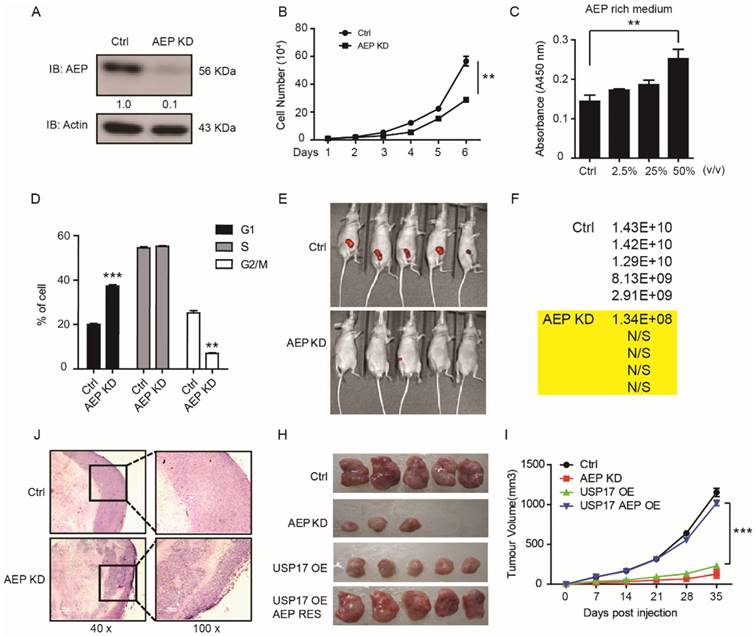
Furthermore, we inoculated AEP KD and control MDA-MB-231 cells which contained green fluorescent protein (GFP) into the fat pad of nude mice to investigate the function of AEP in orthotopic breast cancer model. The tumor formation rate and tumor volume were much lower in mice inoculated with AEP knockdown cells compared with control group which indicated by fluorescent intensity (Fig. 5E, F). Then lower tumor grade was found in xenografts generated from AEP knockdown cell compared with control xenografts through HE assays (Fig. 5J).
In addition, we performed rescue experiments to confirm the function of USP17 and AEP in breast cancer tumorigenesis in vivo. The significantly reduced tumor growth caused by USP17 overexpression can be rescued by AEP overexpression (Fig. 5H, I). Taken together, our results indicate that AEP plays important role in tumorigenesis and tumor growth, and USP17 inhibits breast cancer proliferation through down-regulation of AEP level.
USP17 inhibits AEP mediated activation of ERK signaling
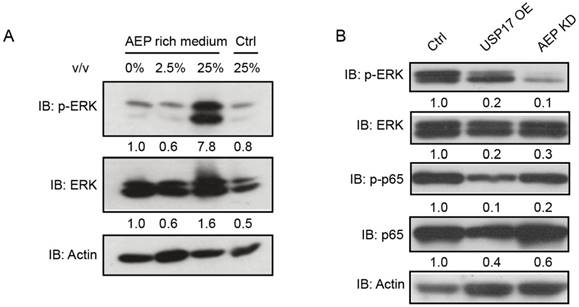
To characterize how USP17 and AEP regulate breast cancer proliferation, we examined several important signaling pathways in AEP protein treated MDA-MB-231 cells. Western Blot assay indicated that p-ERK, but not total ERK, dramatically increased after AEP stimulation (Fig. 6A). Meanwhile, both USP17 overexpression and AEP knockdown caused reduced p-ERK level (Fig. 6B). Thus, our results showed that AEP promotes breast cancer through the activation of ERK signaling and USP17 inhibits breast cancer proliferation by down-regulating AEP protein level.
USP17 inhibits AEP mediated activation of ERK signaling. A. Immunoblot analysis of p-ERK and ERK in MDA-MB-231 cells after treated with different concentration of AEP. B. Immunoblot analysis of p-ERK, ERK, p-p65 and p65 in control, USP17 overexpression and USP17 knockdown MDA-MB-231 cells.
In summary, we have characterized the tumor suppressor function of USP17 and tumor promoting function of AEP in breast cancer cells. USP17 inhibits breast cancer proliferation through deubiquitinating and enhancing AEP turnover. Moreover, AEP promotes breast cancer cell proliferation partially dependent on activation of ERK signaling and USP17 functions to disrupt this process.
Discussion
The ubiquitin proteasome system (USP) is one of the components of the DUB family which reverse ubiquitination process to regulate protein levels and protein functions. Recent years, USPs has been reported play essential roles in cancer biology and therapy [34]. In this study, we uncovered the function of deubiquitinase USP17 in breast cancer and demonstrated that USP17 inhibited breast cancer cell proliferation in vivo and in vitro through regulating AEP protein level. Furthermore, we discovered that AEP promoted breast cancer cell proliferation in vivo and in vitro via activating ERK signaling.
Previous reports found that USP17 expressed in several tumor biopsies and indicated that USP17 might be a biomarker in tumors [18]. More studies demonstrated that USP17 inhibition reduced cell proliferation by disturbing G1-S phase cell cycle progression [18]. Moreover, the up-regulated USP17 in osteosarcoma promoted cell proliferation, epithelial-mesenchymal transition through deubiquitinating and stabilizing SMAD4 [20]. The expression differences of USP17 in various cancers may cause by the inflammatory signals in tumor microenvironments and in breast cancer cells the expression of USP17 predicts the malignant degree of different cell lines. In our study, we uncovered the tumor suppressor function of USP17 in breast cancer. We found that the inhibitory role of USP17 in breast cancer proliferation was based on the homeostasis of AEP protein level. USP17 interacted with and deubiquitinated AEP, then sustained cellular AEP protein level.
Our previous data have shown that AEP promotes breast cancer invasion and metastasis via ubiquitination by TRAF6 [30]. Recent years, AEP has been found overexpressed in a variety of tumors, like breast cancer [27, 30], colorectal cancer [29] and ovarian cancer [35]. And the AEP expression correlated with poor prognosis in multiple cancer types, such as breast cancer [30], colorectal cancer [29], gastric cancer [36], and ovarian cancer [35]. Although AEP has been reported to promote metastasis through activation of matrix metalloproteinase 2 (MMP2) and cathepsins [37, 38], the mechanisms of AEP promoting breast cancer proliferation remain unclear.
In this study, we confirmed the relationship of AEP and USP17 in breast cancer cells and identified USP17 as the debuiquitinase that regulate the ubiquitination level and protein stability of AEP. In general, deubiquitination of proteins by DUB enzymes generates the more expression level of proteins prohibiting them from degradation by the 26S proteasome. In our results, we found that the protein level of AEP is decreased with USP17 overexpression instead. The mechanism of how USP17 regulate AEP protein level remains to be seen and we speculated that there may be other protein molecular, such as heat shock protein (HSP), participate in this process.
Moreover, AEP is required for the G1/S transition, as depletion of AEP caused cell cycle arrested at G1 phase. AEP knockdown also suppressed the expression of p-ERK, leading to the inhibition of ERK signaling pathway. Thus, AEP promoted breast cancer cell proliferation via activating ERK signaling, while USP17 suppressed breast cancer tumorigenesis by down-regulating AEP protein level. Both USP17 overexpression and AEP knockdown significantly inhibited breast cancer cell growth. Because of the tumor-promoting function of AEP in breast cancer, an inhibitor of AEP or AEP antibody might be used in animal model to evaluate its function of tumorigenesis. Besides, a significant decrease of p65 and p-p65 were observed when USP17 was overexpressed. But the knockdown of AEP did not influence the expression of p65 and p-p65. These data suggested that USP17 has other substrates that promote the tumorigenesis of breast cancer through regulation of p65 and p-p65.
In summary, our data demonstrated that USP17 was down-regulated in breast cancer samples and cell lines. USP17 inhibited breast cancer cell growth and proliferation in vivo and in vitro through deubiquitinated AEP and sustain AEP protein level. In normal cells, AEP protein level is maintained in low grade because of high level of USP17. However, AEP is overexpressed in breast cancer cells, following USP17 is down-regulated.
Abbreviations
DUBs: Deubiquitinating enzymes; USP: Ubiquitin proteasome system; USP17: Ubiquitin-specific protease 17; SDS3: Suppressor of defective silencing 3; HDAC: Histone deacetylase; BET: Bromodomain and extra-C terminal; MAPK: Mitogen-activated protein kinase; AEP: Asparaginyl endopeptidase; DMEM: Dulbecco's modified eagle medium; CCK-8: Cell counting kit-8; ERK: extracellular regulated protein kinases; TRAF6: TNF receptor associated factor 6; MMP2: matrix metalloproteinase 2.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81671203, 81402042, 81772654, 81702950), Shanghai science and technology (14140903400, 14YF1402600, 16140902900) and China Postdoctoral Science Foundation (2017M611633).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
2. Grimm LM, Goldberg AL, Poirier GG, Schwartz LM, Osborne BA. Proteasomes play an essential role in thymocyte apoptosis. EMBO J. 1996;15:3835-44
3. Kurokawa M, Kim J, Geradts J, Matsuura K, Liu L, Ran X. et al. A network of substrates of the E3 ubiquitin ligases MDM2 and HUWE1 control apoptosis independently of p53. Sci Signal. 2013;6:ra32
4. Teixeira LK, Reed SI. Ubiquitin ligases and cell cycle control. Annu Rev Biochem. 2013;82:387-414
5. Craney A, Rape M. Dynamic regulation of ubiquitin-dependent cell cycle control. Curr Opin Cell Biol. 2013;25:704-10
6. Kirkin V, Dikic I. Ubiquitin networks in cancer. Curr Opin Genet Dev. 2011;21:21-8
7. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373-428
8. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503-33
9. Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275-86
10. Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610-6
11. Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J. et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133-45
12. Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363-97
13. de la Vega M, Kelvin AA, Dunican DJ, McFarlane C, Burrows JF, Jaworski J. et al. The deubiquitinating enzyme USP17 is essential for GTPase subcellular localization and cell motility. Nat Commun. 2011;2:259
14. Burrows JF, McGrattan MJ, Rascle A, Humbert M, Baek KH, Johnston JA. DUB-3, a cytokine-inducible deubiquitinating enzyme that blocks proliferation. J Biol Chem. 2004;279:13993-4000
15. Shin JM, Yoo KJ, Kim MS, Kim D, Baek KH. Hyaluronan- and RNA-binding deubiquitinating enzymes of USP17 family members associated with cell viability. BMC Genomics. 2006;7:292
16. Ramakrishna S, Suresh B, Bae SM, Ahn WS, Lim KH, Baek KH. Hyaluronan binding motifs of USP17 and SDS3 exhibit anti-tumor activity. PLoS One. 2012;7:e37772
17. Borbely G, Haldosen LA, Dahlman-Wright K, Zhao C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget. 2015;6:33623-35
18. McFarlane C, Kelvin AA, de la Vega M, Govender U, Scott CJ, Burrows JF. et al. The deubiquitinating enzyme USP17 is highly expressed in tumor biopsies, is cell cycle regulated, and is required for G1-S progression. Cancer Res. 2010;70:3329-39
19. McFarlane C, McFarlane S, Paul I, Arthur K, Scheaff M, Kerr K. et al. The deubiquitinating enzyme USP17 is associated with non-small cell lung cancer (NSCLC) recurrence and metastasis. Oncotarget. 2013;4:1836-43
20. Song C, Liu W, Li J. USP17 is upregulated in osteosarcoma and promotes cell proliferation, metastasis, and epithelial-mesenchymal transition through stabilizing SMAD4. Tumour Biol. 2017;39:1010428317717138
21. Chen JM, Dando PM, Rawlings ND, Brown MA, Young NE, Stevens RA. et al. Cloning, isolation, and characterization of mammalian legumain, an asparaginyl endopeptidase. J Biol Chem. 1997;272:8090-8
22. Chen JM, Dando PM, Stevens RA, Fortunato M, Barrett AJ. Cloning and expression of mouse legumain, a lysosomal endopeptidase. Biochem J. 1998;335( Pt 1):111-7
23. Miller G, Matthews SP, Reinheckel T, Fleming S, Watts C. Asparagine endopeptidase is required for normal kidney physiology and homeostasis. FASEB J. 2011;25:1606-17
24. Choi SJ, Reddy SV, Devlin RD, Menaa C, Chung H, Boyce BF. et al. Identification of human asparaginyl endopeptidase (legumain) as an inhibitor of osteoclast formation and bone resorption. J Biol Chem. 1999;274:27747-53
25. Descamps D, Le Gars M, Balloy V, Barbier D, Maschalidi S, Tohme M. et al. Toll-like receptor 5 (TLR5), IL-1beta secretion, and asparagine endopeptidase are critical factors for alveolar macrophage phagocytosis and bacterial killing. Proc Natl Acad Sci U S A. 2012;109:1619-24
26. Manoury B, Mazzeo D, Li DN, Billson J, Loak K, Benaroch P. et al. Asparagine endopeptidase can initiate the removal of the MHC class II invariant chain chaperone. Immunity. 2003;18:489-98
27. Gawenda J, Traub F, Luck HJ, Kreipe H, von Wasielewski R. Legumain expression as a prognostic factor in breast cancer patients. Breast Cancer Res Treat. 2007;102:1-6
28. Holland M, Castro FV, Alexander S, Smith D, Liu J, Walker M. et al. RAC2, AEP, and ICAM1 expression are associated with CNS disease in a mouse model of pre-B childhood acute lymphoblastic leukemia. Blood. 2011;118:638-49
29. Murthy RV, Arbman G, Gao J, Roodman GD, Sun XF. Legumain expression in relation to clinicopathologic and biological variables in colorectal cancer. Clin Cancer Res. 2005;11:2293-9
30. Lin Y, Qiu Y, Xu C, Liu Q, Peng B, Kaufmann GF. et al. Functional role of asparaginyl endopeptidase ubiquitination by TRAF6 in tumor invasion and metastasis. J Natl Cancer Inst. 2014;106:dju012
31. Chen X, Cao X, Sun X, Lei R, Chen P, Zhao Y. et al. Bcl-3 regulates TGFbeta signaling by stabilizing Smad3 during breast cancer pulmonary metastasis. Cell Death Dis. 2016;7:e2508
32. Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B. et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017;19:649-58
33. Andrade V, Guerra M, Jardim C, Melo F, Silva W, Ortega JM. et al. Nucleoplasmic calcium regulates cell proliferation through legumain. J Hepatol. 2011;55:626-35
34. Weathington NM, Mallampalli RK. Emerging therapies targeting the ubiquitin proteasome system in cancer. The Journal of clinical investigation. 2014;124:6-12
35. Wang L, Chen S, Zhang M, Li N, Chen Y, Su W. et al. Legumain: a biomarker for diagnosis and prognosis of human ovarian cancer. Journal of cellular biochemistry. 2012;113:2679-86
36. Li N, Liu Q, Su Q, Wei C, Lan B, Wang J. et al. Effects of legumain as a potential prognostic factor on gastric cancers. Medical oncology. 2013;30:621
37. Brix K, McInnes J, Al-Hashimi A, Rehders M, Tamhane T, Haugen MH. Proteolysis mediated by cysteine cathepsins and legumain-recent advances and cell biological challenges. Protoplasma. 2015;252:755-74
38. Ohno Y, Nakashima J, Izumi M, Ohori M, Hashimoto T, Tachibana M. Association of legumain expression pattern with prostate cancer invasiveness and aggressiveness. World journal of urology. 2013;31:359-64
Author contact
![]() Corresponding authors: Dr Yingying Lin, Department of Neurosurgery, Renji Hospital, School of Medicine, Shanghai Jiao-tong University, Shanghai, 200127, China. E-mail: yylinac.cn (Y.L.). Dr Cheng Xu, Department of Radiation Oncology, Ruijin Hospital, School of Medicine, Shanghai Jiao-tong University, Shanghai 200025, CHINA. E-mail: xucheng60com (C.X.)
Corresponding authors: Dr Yingying Lin, Department of Neurosurgery, Renji Hospital, School of Medicine, Shanghai Jiao-tong University, Shanghai, 200127, China. E-mail: yylinac.cn (Y.L.). Dr Cheng Xu, Department of Radiation Oncology, Ruijin Hospital, School of Medicine, Shanghai Jiao-tong University, Shanghai 200025, CHINA. E-mail: xucheng60com (C.X.)