Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(5):942-952. doi:10.7150/ijbs.30930 This issue Cite
Research Paper
Estrogen Receptor Beta (ERβ) Mediated-CyclinD1 Degradation via Autophagy Plays an Anti-Proliferation Role in Colon Cells
Yong Wei1, Can Huang2, Haoyu Wu1, Jian Huang1 ![]()
1. Hubei Key Laboratory of Cell Homeostasis, College of Life Sciences, Wuhan University, Wuhan, Hubei, P.R China
2. Wuhan Agricultural Inspection Center, Hubei, P.R China
Received 2018-10-24; Accepted 2019-2-10; Published 2019-3-10
Abstract

Dysfunction of autophagic degradation machinery causes tumorigenesis, including colorectal cancer (CRC). Overexpression of CyclinD1 in CRC has been reported. Recent evidence also suggests that ERβ deficiency is related to the pathogenesis of CRC. Very little is known, however, about the detailed molecular mechanisms underlying the relationship among ERβ, autophagy, and CyclinD1 in CRC. Here, results showed that ERβ played an anti-proliferation role in HCT116 through impairing cell cycle but not apoptosis. Additionally, CyclinD1 accumulation was increased in response to chloroquine (CQ) or in MEF Atg7 knockout cells. Further, ERβ could inhibit the mammalian target of rapamycin (mTOR) or activate Bcl-2/adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) to promote autophagy in HCT116. In summary, these results indicate that ERβ-mediated CyclinD1 degradation can inhibit colon cancer cell growth via autophagy.
Keywords: ERβ, autophagy, colorectal cancer, mTOR, BNIP3, CyclinD1
Introduction
The diagnosed morbidity and the mortality of CRC rank third and fourth worldwide, respectively [1]. The incidence rate of CRC in pre-menopausal women is 40% lower than age-matched men [2], and young women have a better overall survival prognosis than older women with menopause. Such evidence suggests that female sex hormones, especially estrogens, form the basis for this protective effect in women. The functions of estrogen are regulated by estrogen receptors (ERs) which are coded by ERα (ESR1) and ERβ (ESR2). Since the level of ERα remains low and stable in both normal and cancerous colonocytes, the protective effect of estrogen may be regulated by ERβ [3]. ERα is abundantly expressed in the human colonic epithelium, while the ERβ expression has a progressive decline with the worsening stage and grade of tumor in CRC [4]. Thus, these findings suggest ERβ may be represented as a promising therapeutic target for CRC.
Loss of cell cycle control is an obvious alteration in tumor growth which has been proposed to be obligatory in cancer development. CyclinD1 (CCND1), a key regulator for regulating G1 to S phase progression, is often overexpressed in more than half of human CRC patients, suggesting a positive correlation between CyclinD1 and colorectal tumorigenesis [5]. Although previous studies have indicated potential associations between ERβ activation and CyclinD1 decrease in human prostate cancer and breast cancer [6,7], the direct relationship between ERβ and CyclinD1 in CRC is still poorly understood. Together, despite sufficient evidence of ERβ as a regulator for CRC suppression, future analysis is required before definitive conclusions on the protective role in CRC can be drawn.
Autophagy is a conserved evolutionary and intracellular degradation pathway in eukaryotic cells, involved in the degradation of damaged organelles and long-lived proteins by lysosomes [8]. In recent years, autophagy induction has been increasingly regarded as a suppression mechanism for tumor development [9]. In mammals, defects in autophagy are associated with tumorigenesis in CRC [10-12]. In contrast, excessive autophagy is associated with cell death called autophagic cell death (ACD) [13], which often occurs in apoptosis-defective cells and acts as a novel tumor suppressor mechanism [14]. mTOR, an inhibitory modulator of autophagy, often controls multiple physiological processes such as cell-cycle progression and cell growth [15,16]. Nevertheless, mTOR is frequently hyperactivated in CRC [17]. One report shows that estrogen can promote autophagy by mTOR suppression in bovine mammary epithelial cells [18]. However, little evidence has been found associating ERβ with mTOR in CRC. Meanwhile, few studies show that ERβ exerts an anti-proliferation role in breast cancer via autophagy promotion and cell cycle arrest, suggesting a suppressive role of autophagy in the cell cycle [19]. These case studies confirm the importance of ERβ-mediated an antiproliferation role in CRC. However, whether ERβ induced-autophagy via mTOR could impair cell cycle in HCT116 remains unclear. The ubiquitin-proteasome system (UPS) is another primary degradation system of ubiquitinated proteins in eukaryotic cells [20]. Accumulating evidence shows that CyclinD1 can be degraded in a proteasome-dependent manner, leading to G1 cell cycle arrest [21-23]. MG132, a proteasome inhibitor, blocks the proteasome-regulated CyclinD1 degradation [24]. Notably, in a recent article, Wu et.al (2018) show a novel mechanism that CyclinD1 can be degraded by an autophagic process in hepatocellular carcinoma in addition to proteasome pathway [25]. However, whether autophagy participates in CyclinD1 degradation remains unknown in CRC.
In our study, we provide evidence that ERβ could inhibit HCT116 growth by inducing CyclinD1 degradation via autophagy activation. Hence, promotion of autophagy with the clearance of CyclinD1 can be an underlying strategy for ERβ to treat CRC, and the identification of crucial elements in this tactic could bring about potential targets for CRC therapy.
Materials and Methods
Cell culture and treatments
For details, please see supplementary data file.
Plasmids and siRNA transfection
pEGFP-C1-ERβ was a gift from Michael Mancini (Addgene plasmid #28237); RFP-p62 was generously provided by professor Wei Ding (Capital Medical University, Beijing, China); RFP-LC3 and Phage-puro-ERβ fusion plasmids were constructed by our lab previously. ERβ silencing siRNA, mTOR silencing siRNA and control siRNA were designed and synthesized by GenePharma (Suzhou, China). The sequences are as follows: ERβ sense, 5'-CCAGCCAUGACAUUCUAUATT-3' and antisense, 5'-UAUAGAAUGUCAUGGCUGGTT-3'; mTOR sense, 5'GCCGCAUUGUCUCUAUCAATT-3' and antisense, 5'-UUGAUAGAGACAAUGCGGCTT-3'; control siRNA sense, 5'-UUCUCCGAACGUGUCACGUTT3' and antisense, 5'-ACGUGACACGUUCGGAGAATT-3'. All plasmids or siRNA oligonucleotides were done with Lipofectamine 2000 (Invitrogen, USA) for 48 h according to the manufacturer's protocols.
RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted using Trizol extraction kits (Invitrogen, US) according to manufacturer's protocol and then treated with RNase-free DNase I (TaKaRa, Japan) to avoid potential DNA contamination. Reverse transcription was performed using the M-MLV Reverse Transcriptase (Promega, #M1701) and random primers. Approximately 1 μg of the total RNA was used as the template for reverse transcription. The cDNA was diluted to 100 ng/μL for use in qRT-PCR. The following primers were used: GAPDH sense, 5'-TGCACCACCAACTGCTTAGC-3' and antisense, 5'-GGCATGGACTGTGGTCATGAG-3'; ERβ sense, 5'-TCCATGCGCCTGGCTAAC-3' and antisense, 5'-CAGATGTTCCATGCCCTTGTTA-3'. Data analysis was performed using Bio-rad CFX manager system, using GAPDH as a reference transcript.
Western blotting
For details, please see supplementary data file.
Transmission electron microscope (TEM)
HCT116 cells were fixed at room temperature (RT) for 1 h in 2.5% glutaraldehyde supplemented with 0.1 M phosphate buffer saline (PBS), and then post-fixed in 1.0% osmium tetroxide for 3 h. Next, cells were scraped, spun down, serially dehydrated in ethanol baths, and embedded in blocks of epon Araldite. Ultrathin sections (60-80 nm) were stained with 4% aqueous uranyl acetate and lead citrate for 5 min, and then observed by TEM (Tecnai G2 20 Twin, FEI) at 200 KV.
Confocal microscope assay
For LC3 puncta imaging, HCT116 cells were transfected with RFP-LC3 plasmid using Lipofectamine 2000. After overnight culture, cells were transfected with Phage-puro or Phage-puro-ERβ plasmid for 24 h, fixed with 4% paraformaldehyde, and observed under the confocal microscope. To observe cellular acidic vesicular organelle (AVOs) development, HCT116 were incubated with acridine orange (AO) (1ug/ml) in PBS for 15 min at 37°C after treatment. For the exploration of lysosome function, cells were transfected with RFP-p62 plasmid or stained by Lyso-Tracker Red (Invitrogen). To detect the effect of ERβ overexpression on BNIP3 and ki67 (Proteintech) in HCT116, cells were fixed, permeabilized, and incubated with primary antibodies at 4°C overnight. Finally, cells were washed and then were incubated with fluorescent secondary antibodies (Invitrogen) at RT for 1 h. Samples were examined by a laser-scanning confocal microscope. Nuclei were revealed by 2-phenylindole dihydrochloride (DAPI) staining.
Flow-cytometric analysis of apoptosis
HCT116 cells were harvested and then rinsed in ice-cold PBS 48 h after transfection. Next, cells were stained with Annexin V-FITC and Propidium Iodide (PI) (4A Biotec, China) for 15 min at RT, apoptosis was measured by a flow cytometer (Beckman Coulter, USA) and analyzed by Summit 4.3 software (Beckman Coulter, USA).
Flow-cytometric analysis of cell cycle
At 48 h after transfection, HCT116 cells were permeabilized, washed with ice-cold PBS twice, and fixed in 4% paraformaldehyde overnight at 4 °C. Next, fixed cells incubated with 10% saponin and DAPI (1 mg/ml) in PBS at RT for 30 min. Cell cycle assay was performed by a flow cytometer (Beckman Coulter, USA) and analyzed by Flowjo software (Tree Star, OR).
Statistical analysis
All experiments were repeated at least three times and presented as the mean ±S.D. Data analyses were performed using GraphPad Prism curve comparisons. P values < 0.05 were considered to indicate significant differences.
Results
ERβ plays an anti-proliferation role through impairing cell cycle but not apoptosis in HCT116 cells
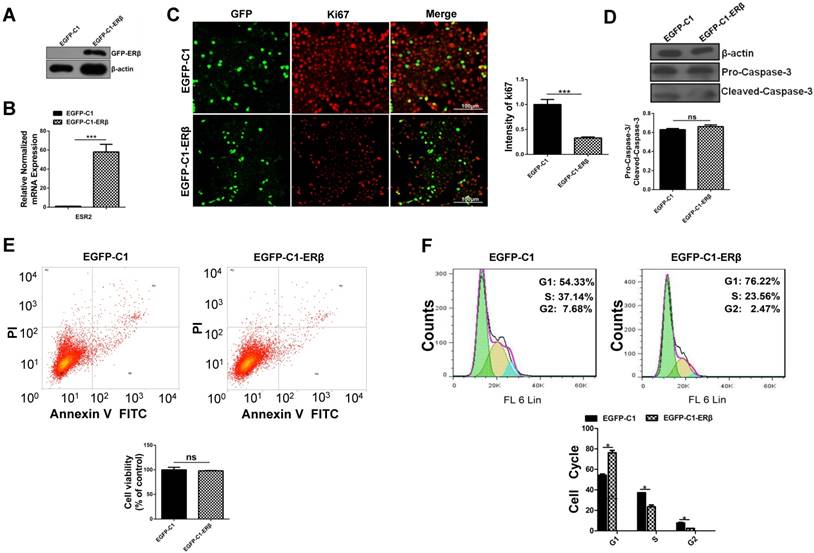
To assess the role of ERβ in tumor cell growth, HCT116 cells were transfected with EGFP-C1 and EGFP-C1-ERβ plasmids for 48 h. Western blots analysis and qRT-PCR verified that ERβ was effectively overexpressed in EGFP-C1-ERβ group (Fig. 1A and 1B). Ki67, a marker for cellular proliferation, is mainly located in the cell nucleus during the S and G2 phases and in nucleoplasm during the early G1 period [26]. To assess the role of ERβ in cell growth ability, immunofluorescence intensity of Ki67 staining was observed under the confocal microscope. Compared with the EGFP-C1 group, the percentage of Ki67-positive cells was decreased significantly in EGFP-C1-ERβ group (Fig. 1C), which indicates that ERβ could play an antiproliferative role in HCT116 cells. To examine whether ERβ-induced antiproliferation is due to apoptosis, the expression of pro-Caspase-3 and cleaved-Caspase-3 was tested after ERβ transfection in HCT116. Surprisingly, there was no statistically significant difference between the two groups in this study (Fig. 1D). Consistently, Annexin V-FITC/PI staining confirmed the same result by flow cytometry assay (Fig. 1E). Next, to further investigate whether ERβ-induced antiproliferation is due to cell cycle arrest, cell cycle distribution was analyzed by flow cytometry analysis. Results showed that ERβ treatment increased the number of cells at the G1 phase and reduced the population at the S and G2 periods in HCT116 (Fig. 1F). Collectively, these results show that ERβ brings about cell cycle arrest but not apoptosis, contributing to growth suppression in HCT116.
ERβ plays an anti-proliferation role through impairing cell cycle but not apoptosis in HCT116 cells. HCT116 cells were transiently transfected with EGFP-C1 and EGFP-C1-ERβ plasmids for 48 h, and overexpression efficiency was determined by western blotting (A) and qRT-PCR (B). (C) Immunofluorescence analysis of HCT116 cells with Ki67 (red) and GFP (green), which were visualized by the confocal microscope. Bar graph (right) indicates the percentage of ki67 positive cells. Scale bar, 100μm. (D) Immunoblot analysis of pro-caspase-3 and cleaved-caspase-3. β-actin was used as an equal loading control. Bar graph (below) shows the relative ratio of pro-caspase-3 to cleaved-caspase-3 in HCT116. (E) Apoptosis was determined by flow cytometry after stained with Annexin V-FITC and PI. Bar graph (below) indicates the survival rate of treated cells. (F) Cell cycle distribution was analyzed by flow cytometer after DAPI stained. Bar graph (below) indicates the cell cycle phase distribution in treated cells. Immunofluorescence intensity and western blot were quantified using ImageJ software. Data shown are mean ± S.D. of three independent experiments. (*, P<0.05; **, P <0.01; ***, P<0.001).
Downregulated CyclinD1 contributes to ERβ-induced anti-proliferation in HCT116 cells
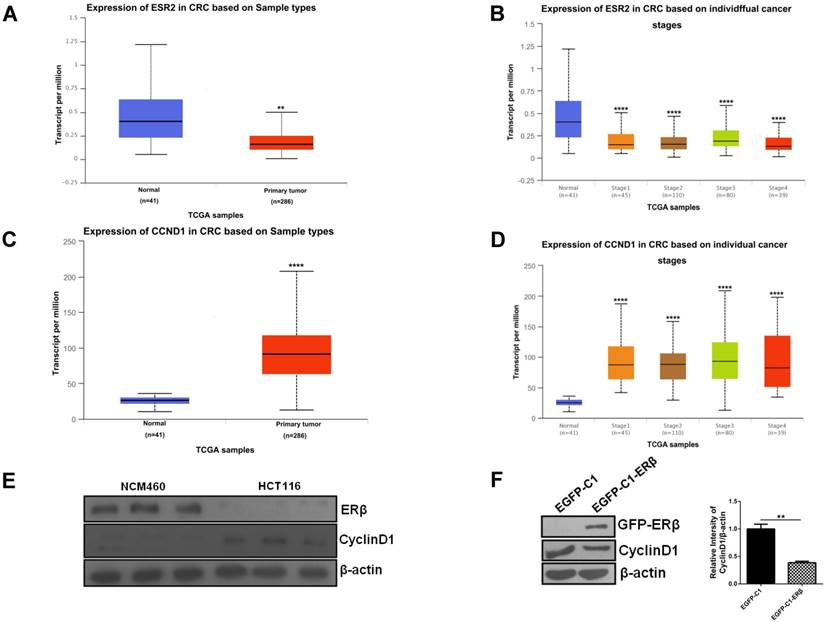
The observation of ERβ-induced G1 cell cycle arrest prompts us to evaluate how CyclinD1 changes. Firstly, we examined ESR2 and CCND1 levels in normal and CRC patients through publicly available TCGA data. Data retrieved from UALCAN web-portal [27] showed that ESR2 was downregulated in CRC patients compared with the normal colon tissue (Fig. 2A and 2B). By contrast, the expression of CCND1 was increased significantly in CRC compared with the normal colon tissue (Fig. 2C and 2D). To determine the association of ERβ and CyclinD1 in CRC, we analyzed the expression of them in NCM460 and HCT116 cells by western blotting, which confirmed the same results as UALCAN data (Fig. 2E). To further study whether ERβ reduces CyclinD1, the cell lysates from HCT116 cells transfected with vector or ERβ plasmid were collected for western blotting, and the results indicated that CyclinD1 expression was significantly decreased in contrast to the control group (Fig. 2F). Taken together, these data suggest that ERβ inhibits the proliferation of CRC cells by downregulating the expression of CyclinD1.
Downregulated Cyclin D1 contributes to ERβ-induced anti-proliferation in HCT116 cells. (A) Boxplot showing relative expression of ESR2 in TCGA samples (normal vs primary tumor). (B) Boxplot showing relative expression of ESR2 in TCGA samples. (normal vs stage 1-4 colorectal cancer patients). (C) Boxplot showing relative expression of CCND1 in TCGA samples (normal vs primary tumor). (D) Boxplot showing relative expression of CCND1 in TCGA samples. (normal vs stage 1-4 colorectal cancer patients). (E) Western blot analysis of ERβ and CyclinD1 expression in NCM460 and HCT116. (F) Cells were treated as described in (Fig. 1), then CyclinD1 was tested by immunoblotting. Bar graph (right) shows the relative ratio of CyclinD1 to β-actin in HCT116. Western blot was quantified using ImageJ software. Data shown are mean ± S.D. of three independent experiments. (*, P<0.05; **, P <0.01; ***, P<0.001; ****, P<0.0001).
ERβ-evoked CyclinD1 depletion and anti-proliferation are correlated with activated autophagy in HCT116 cells
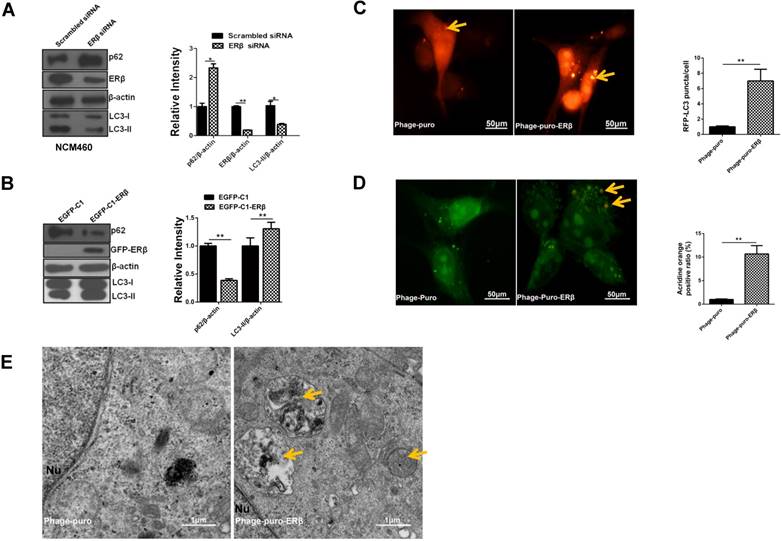
In apoptosis-defective cells, the induction of autophagy has been commonly charged with anticancer therapies in multiple kinds of cancers and shown to elicit an effect on tumor growth inhibition [9]. When autophagy occurs, the soluble LC3-I is converted to LC3-II, and LC3-II then localizes to isolated membranes and autophagosomes [28]. Autophagy cargo receptor SQSTM1/p62, a marker for autophagic flux, is continuously degraded by autophagy [29]. UALCAN analysis showed that SQSTM1 level was increased, while MAP1LC3B expression was decreased in TCGA samples (normal vs primary tumor) (Fig. S1A-1D). Such results were also confirmed in NCM460 and HCT116 cells by western blotting (Fig. S1E). Given that ERβ participates in autophagy in human seminoma [30], whether ERβ induces autophagy in HCT116 remains unclear. Next, our results certified that ERβ knockdown reduced LC3-II and increased p62 expression in NCM460, while ERβ overexpression reversed it in HCT116 (Fig. 3A and 3B). Consistently, confocal assay showed that the sub-cellular redistribution of RFP-LC3 was increased in HCT116 transfected with ERβ plasmid, indicating ERβ exhibited preferential activity for increasing the formation of autophagosomes (Fig. 3C). AO staining intensity is used to predict autophagy level [31]. In comparison with the control group, the number of AO-positive AVOs was increased obviously in the ERβ-transfected group (Fig. 3D). Furthermore, autophagy is also monitored morphologically by assessing the accumulation of autophagic vacuoles in the TEM assay. Fig. 3E illustrated that autophagosomes and autophagolysosomes were increased in the cytoplasm of an ERβ-transfected group compared to the control group. In summary, these results strongly demonstrate that the autophagic function is defective and ERβ promotes autophagy in HCT116.
ERβ-evoked Cyclin D1 depletion and anti-proliferation are correlated with activated autophagy in HCT116 cells. (A) Cells were transfected with scrambled siRNA and ERβ siRNA for 48 h, and then cell lysates were analyzed by related antibodies. Bar graph indicates the relative ratio of LC3-II, p62 and ERβ to β-actin in NCM460. (B) Cells were treated as described in (Fig. 1), then tagged proteins were analyzed by immunoblotting. Bar graph indicates the relative ratio of LC3-II and p62 to β-actin of triplicate experiments in HCT116. (C) Immunofluorescence analysis of LC3 dot in treated HCT116. Arrows indicated RFP-LC3 puncta. Bar graph indicates the quantification of cells with RFP-LC3 dots in three treated cells randomly chosen. Scale bar, 50μm. (D) Immunofluorescence analysis of AVOs in treated HCT116. Arrows indicated acidic vacuoles. Bar graph indicates the number of AVOs per cell in about three cells randomly chosen. Scale bar, 50 μm. (E) Representative electron micrographs of HCT116 cells treated with Phage-puro or Phage-puro-ERβ for 48 h. Scale bar, 1 μm. Arrows indicated autophagic vacuoles. Immunofluorescence intensity and western blot were quantified using ImageJ software. Data shown are mean ± S.D. of three independent experiments. (*, P<0.05; **, P<0.01).
ERβ-mediated autophagy exerts an anti-proliferation role through promoting CyclinD1 degradation in HCT116 cells
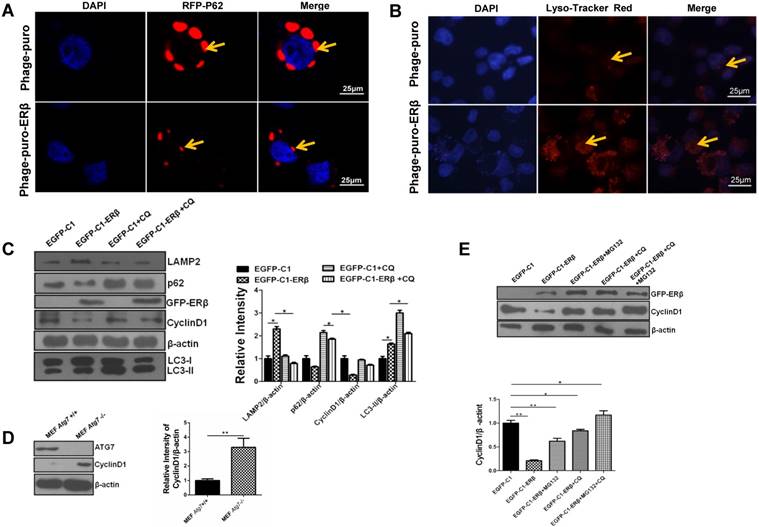
Lysosome plays a vital role in the UPS and the autophagy-lysosome system in mammals [32]. Results showed that lysosome function was reinforced under ERβ transfection, as evidenced by the decreased fluorescence intensity of RFP-p62 and increased fluorescence intensity of Lyso-Tracker Red in HCT116 (Fig. 4A and 4B). Since ERβ not only impairs cell cycle but also promotes autophagic functions, we propose that the cell cycle arrest is related to enhanced autophagy in HCT116 cells. To test whether ERβ induces CyclinD1 degradation via autophagy, CyclinD1 expression was tested in response to CQ (MCE, USA). CQ impairs autophagy function, leading to the significant accumulation of LC3-II in the cytoplasm [33]. As shown in Fig. 4C, ERβ-induced degradation of CyclinD1 protein was blocked by CQ. Autophagic flux and Lysosome-associated membrane protein type 2 (LAMP2, a marker for lysosome) were also blocked in the presence of CQ. Similarly, MEF Atg7 (-/-) cells showed higher CyclinD1 accumulation compared to wild-type (WT) MEF Atg7 (+/+) cells (Fig. 4D). Next, to determine whether proteasome participates in ERβ-regulated CyclinD1 degradation, the effect of ERβ on CyclinD1 expression was analyzed in the presence of the proteasome inhibitor MG132 (S1748, Beyotime). CyclinD1 degradation after ERβ transfection for 36 hours was blocked in the presence of MG132 as well as CQ, and this effect was enhanced when the cells received combination treatment with CQ and MG132 (Fig. 4E). These results suggest that not only proteasome but also autophagy participates in ERβ-regulated CyclinD1 protein degradation in HCT116 cells.
ERβ-mediated autophagy exerts an anti-proliferation role through promoting CyclinD1 degradation in HCT116 cells. (A) Compared with Phage-puro plasmid group, the fluorescence intensity of RFP-p62 was decreased dramatically in Phage-puro-ERβ transfected group using a confocal microscope. Scale bar, 25 µm. (B) Cells were treated as described in (A), and then images were captured by the confocal microscope followed by Lyso-Tracker Red probe to stain lysosome. Scale bar, 25 µm. (C) After transfection of EGFP-C1 and EGFP-C1-ERβ plasmids for 36 h, cells were treated with CQ (10 μM) for 12 h, and then examined the expression of related-proteins. Bar graph (right) indicates the relative ratio of LAMP2, p62 and CyclinD1 and LC3-II to β-actin in HCT116. (D) CyclinD1 and ATG7 protein levels were tested in MEF Atg7 +/+ and MEF Atg7 -/- cells. Bar graph (right) indicates the relative ratio of CyclinD1 to β-actin in HCT116. (E) After ERβ transfection for 36 h, cells were treated with CQ (10 μM, 12 h), MG132 (10 μM; 12 h), or MG132 plus CQ for 12 h. Bar graph indicates the relative ratio of CyclinD1 to β-actin in HCT116. Western blot was quantified using ImageJ software. Data shown are mean ± S.D. of three independent experiments. (*, P<0.05; **, P<0.01).
ERβ promotes autophagy through down-regulating mTOR or BNIP3 protein in HCT116 cells
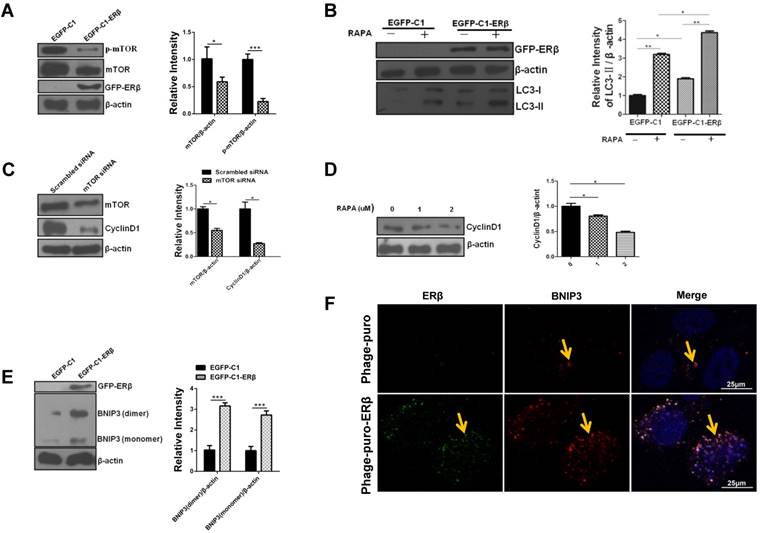
Excessive activation of mTOR is often accompanied by hyperactivity and proliferation in CRC [17]. Our data showed that the total expression of mTOR and its phosphorylation level were increased in HCT116 compared with NCM460 (Fig. S2A). As discussed above, mTOR is often accepted as a negative regulator of autophagy and can be down-regulated by estrogen in bovine mammary epithelial cells. Therefore, to further explore the mechanism of ERβ-induced HCT116 growth suppression via autophagy, mTOR expression was tested by western blotting. Results showed that ERβ reduced mTOR and p-mTOR levels in HCT116 (Fig. 5A). To better understand the mechanism of ERβ mediated-autophagy, we tested several markers of PI3K/AKT/ mTOR signaling pathways. ATG7 is an essential gene in an autophagic process [34]. In this case, the EGFP-C1-ATG7 plasmid was considered as a positive control. Overexpression of ERβ, as well as ATG7, increased the LC3-II expression in HCT116 cells. The levels of p-mTOR and p-AKT were decreased, while the PTEN, p-Beclin1 and p-ULK1 levels had a significant increase (Fig. S2B). Rapamycin (RAPA), an inhibitor of mTOR, enhanced LC3-II expression in cells transfected with EGFP-C1-ERβ plasmid than those treated with EGFP-C1 plasmid (Fig. 5B). Next, to confirm the role of mTOR in ERβ-induced CyclinD1 degradation in HCT116 cells, CyclinD1 expression was detected under mTOR siRNA or RAPA treatment in HCT116 cells. Results showed that CyclinD1 expression was decreased in response to inhibitors of mTOR (Fig. 5C and 5D). This evidence confirms that mTOR participates in ERβ-induced CyclinD1 degradation further. Considering that ERβ induces nonapoptotic cell death and regulate autophagy via mTOR in HCT116 cells, whether ERβ participates in ACD need to be explored. BNIP3 is another key regulator of ACD [35]. Studies showed that ERβ increased BNIP3 expression in HCT116 cells (Fig. 5E and 5F), suggesting ACD may participate in ERβ-induced HCT116 growth suppression. Collectively, these data indicate that autophagy can be regulated by ERβ via mTOR and BNIP3 protein in HCT116.
ERβ promotes autophagy through down-regulating mTOR or BNIP3 protein in HCT116 cells. (A) Cells were treated as described in (Fig. 1), then mTOR and p-mTOR were tested by immunoblotting. Bar graph (right) shows the relative ratio of mTOR and p-mTOR to β-actin in HCT116. (B) HCT116 cells were transiently transfected with EGFP-C1 and EGFP-C1-ERβ plasmids, then treated with rapamycin or not. Tagged proteins were analyzed by immunoblotting. Bar graph (right) indicates the relative ratio of LC3-II to β-actin of triplicate experiments in HCT116. (C) HCT116 cells were transiently transfected with siRNA to mTOR in HCT116 for 48 h, scrambled siRNA of mTOR as a control. mTOR and CyclinD1 antibodies were used to detect their expression levels by western blot. Bar graph (right) indicates the relative ratio of mTOR and CyclinD1 to β-actin of triplicate experiments in HCT116. (D) Tagged proteins were analyzed by immunoblotting in HCT116 treated with different concentrations of rapamycin. Bar graph indicates the relative ratio of CyclinD1 to β-actin in HCT116. (E) Cells were treated as described in (Fig. 1), Then BNIP3 was tested by immunoblotting. Bar graph (right) indicates the relative ratio of BNIP3 to β-actin of triplicate experiments in HCT116. (H) Cells were transfected with Phage-puro and Phage-puro-ERβ plasmids, then the fluorescence intensity was detected by a confocal microscope. Scale bar, 25 μm. Data shown are mean ± S.D. of three independent experiments. (*, P<0.05; **, P < 0.01).
Discussion
Epidemiological data indicate a positive effect of estrogen replacement therapy (HRT) on CRC, and ERβ is necessary for the action of estrogen, suggesting that ERβ is a valid target for the treatment of CRC [36]. Therefore, using selective agonists of ERβ to treat CRC is a novel method when HRT of CRC has severe side effects in reproductive organs [37]. Considering that ERβ in HCT116 was either very low or absent compared with NCM460 (Fig. 2), using its agonists may not have apparent effects. Therefore, future targets should be focused on novel agonists with both high affinity and high selectivity. To raise the expression of ERβ obviously, transient transfection of ERβ plasmid was applied to this study. The present study determined a novel function of ERβ in regulating autophagy-dependent CyclinD1 degradation and ACD in HCT116, which contributed to HCT116 cell growth suppression.
ERβ, a nuclear transcription factor, has been considered as an anti-tumor regulator in many cancers through various mechanisms. In our study, Ki67 staining confirmed that ERβ suppressed HCT16 cell proliferation. However, there was no significant change in the relative ratio of Caspase-3/pro-Caspase-3 and apoptosis rate between vector group and ERβ transfection group. A potential mechanism by which ERβ transfection inhibits proliferation in HCT116 is by modulation of key regulators of the cell cycle. It was reported previously that ERβ induces G1 arrest by inducing CyclinE1 decrease in HCT8 cells [38]. Our results showed that ERβ can give rise to G1-S phase arrest in HCT116 (Fig.1F). For G1-S phase arrest, CyclinD1 is one of the key molecules that mediates G1 to S phase development in various cancers. High levels of CyclinD1 are observed early in the development in many cancers including CRC, which indicates that promoting CyclinD1 to down-regulation is a promising therapeutic approach for CRC [5,39]. It has been proved that the expression of CyclinD1 can be suppressed by PTEN-mediated autophagy, leading to G0-G1 phase arrest [40]. Additionally, ERβ activates autophagy through the binding of sp1 on PTEN promoter in human seminoma cell lines [30]. Meanwhile, given that ERβ exerts an anti-proliferation role in breast cancer via autophagy promotion [19], this evidence inspires us to ask whether ERβ regulates autophagy and promotes CyclinD1 decrease in HCT116.
The role of autophagy has been recognized in regulating cancer cells growth and death. Recently, defects in autophagy have been found in the promotion of tumors, such as loss of Beclin1 in lung and liver tumors [41] and heterozygous loss of Atg5 in human melanomas [42]. Similarly, Beclin1 and Atg5 are also frequently and greatly down-regulated in tissue samples from CRC patients and colon cancer cell lines [10,11]. Furthermore, UV irradiation resistance-associated gene (UVRAG), another positive regulator of autophagy, is scarce in CRC. Overexpression of UVRAG not only restores autophagy but also inhibits HCT116 cell growth [43]. UALCAN data and western blot assay showed that the defective autophagy could be one of the events involved in the development of CRC. Afterward, we transfected ERβ plasmid into HCT116 and detected autophagic flux through various methods, such as AO staining, subcellular RFP-LC3 distribution, and TEM. As anticipated, our results demonstrated that ERβ, a tumor suppressor, enhanced the autophagic process in HCT116 by promoting autophagosome maturation and autolysosome formation (Fig. 3). Together, the above results suggest that ERβ promotes autophagy in HCT116.
UPS and autophagy have been considered as complementary systems for protein degradation [20], including cell cycle proteins. CyclinA2, a key regulator of the cell cycle often accumulates from late G1 to M phase and can be degraded by UPS and autophagy in MCF-7 breast cancer cells [44]. Moreover, CyclinD1, one of the key molecules that mediates G1 to S phase development in various cancers, can be degraded by protease or autophagy pathways in ovarian liver cancer cells and hepatocellular carcinoma, separately [21,25]. Our study showed that ERβ gave rise to G1-S phase arrest rather than apoptosis in HCT116. These above results prompt us to ask whether ERβ regulated-cell cycle arrest was due to CyclinD1 clearance via autophagy or UPS in HCT116. Firstly, given that lysosome is the final stage of protein degradation, our results indicated that the lysosomal function was enhanced under ERβ overexpression. Next, CQ, an inhibitor of the autophagy-lysosome fusion process, could block the degradation of CyclinD1 in HCT116. Consistent results were also observed in MEF Atg7 knockout cells, suggesting autophagy exerts a key role in CyclinD1 degradation. mTOR is a key negative autophagy modulator, which coordinately meditates the balance between cell growth and cell death in various cancers [45]. Western blot showed that mTOR expression was suppressed by ERβ. Furthermore, our results demonstrated that siRNA of mTOR or low concentrations of rapamycin (2µM) reduced CyclinD1 expression significantly in HCT116, which is similar to the result in MDA361 breast cancer cells [46]. These above observations suggest that autophagy plays a significant role in ERβ-induced CylinD1 degradation via mTOR suppression. Besides autophagy, results showed that CylinD1 degradation was blocked in the presence of proteasome inhibitor MG132. These results indicate that CyclinD1 can be degraded through both autophagy and proteasome degradation pathways in HCT116.
The PTEN-PI3K/Akt/mTOR is often considered as a classical autophagic signaling pathway in various cancers [47]. PTEN is a pivotal downstream regulator of the PI3K/Akt and mTOR pathway, acting as a tumor suppressor gene in many kinds of human cancers [48]. Western blot showed that mTOR expression was suppressed by ERβ (Fig. 5A). Our results also confirmed that ERβ could up-regulate PTEN and then suppress PI3K/AKT/mTOR signaling pathways in HCT116 (Fig. S2). Based on our experimental data, we propose a model in which ERβ promotes autophagy through PTEN-PI3K/AKT/mTOR signals and its downstream regulators (Fig. 6). Furthermore, considering that mTOR downregulation and ULK1 activation are related to autophagic cell death (ACD) and cell cycle arrest [49], ERβ may participate in ACD in HCT116. Accumulating evidence suggests that autophagy inducers activate ACD in cancer cells that are resistant to apoptosis [50]. Our results showed that ERβ had no effect on apoptosis. In addition, apoptosis-defective cancer cells are often accompanied by ACD via BNIP3 protein [35]. Our results showed that BNIP3 was a positive regulator of ERβ-regulated ACD. Overall, these above results indicate that ERβ may induce ACD in HCT116, suggesting a novel mechanism of ERβ-regulated tumor growth suppression. Additional research is needed in order to establish the relationship between ERβ and ACD in CRC. However, it has been reported that autophagy has a dual role in promoting both cell survival and cell death [51]. Therefore, defining the role of autophagy in various cases is vital in cancer therapy.
Proposed mechanism for ERβ in regulating autophagy and anti-proliferation of colon cells. Schematic model depicting the over-expression ERβ induces autophagy through downregulating PI3K/AKT/mTOR. The up-regulated PTEN, p-ULK1 and p-Beclin1 indicate the autophagy induction. CQ could block the ERβ-induced CyclinD1 degradation, then causing antiproliferation in HCT116. BNIP3 was also involved in ERβ-regulated autophagic cell death. See the text for the detailed discussion on the roles of these signaling pathways and their interplay.
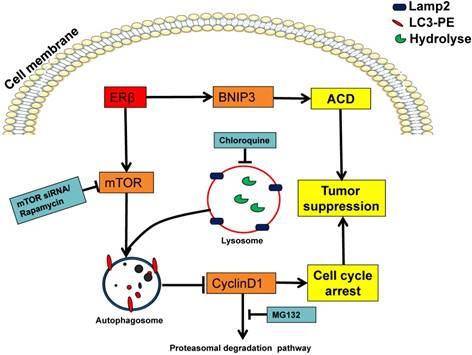
In conclusion, we investigated the role of ERβ-induced antiproliferation in HCT116. The mechanism is mainly involved in ERβ induced-CylinD1 degradation via mTOR-dependent autophagy. Additionally, ERβ induced-ACD via BNIP3 has a suppressive role in HCT116 growth. The schematic diagram of the mechanism can be seen in Fig. 6. Thus, we propose that ERβ is a favorable target for cancer therapy since it not only induces autophagy but also triggers cell cycle arrest. To our knowledge, these results provide novel insights into the underlying mechanism of the anti-proliferative properties of ERβ and generate new opportunities for the future treatment of CRC.
Supplementary Material
Supplementary figures and tables.
Abbreviations
ERβ: Estrogen receptor beta; CRC: Colorectal cancer; mTOR: mammalian target of rapamycin; CQ: Chloroquine; UPS: ubiquitin-proteasome system; AVOs: acidic vesicular organelle; LAMP2: Lysosome-associated membrane protein type 2; UVRAG: UV irradiation resistance-associated gene; BNIP3: Bcl-2/ adenovirus E1B 19-kDa-interacting protein 3; ACD: autophagic cell death; LC3: microtubule-associated protein 1 light chain 3; PTEN: phosphatase and tensin homolog deleted from chromosome 10; HRT: estrogen replacement therapy.
Acknowledgements
We are grateful to Professor Michael Mancini, Professor LongPing Wen, Professor Xiaochun Bai and Professor Wei Ding for generously providing plasmids. We also thank Dr. Du and Dr. Li for providing the support of TEM (The Core Facility and Technical Support, Wuhan Institute of Virology). This study was supported by the National Natural Science Foundation of China (NO.31870786 and 31371331) and The National Basic Research Program of China (973 Program, 2012CB720600). We thank Mr. Ryan Tames and Dr. Yujun Hou (National Institutes of Health) for critical reading of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Levi Z, Kark JD, Katz LH. et al. Adolescent body mass index and risk of colon and rectal cancer in a cohort of 1.79 million Israeli men and women: A population-based study. Cancer. 2017;123:4022-30
2. Chlebowski RT, Wactawski-Wende J, Ritenbaugh C. et al. Estrogen plus progestin and colorectal cancer in postmenopausal women. New England Journal of Medicine. 2004;350:991-1004
3. Campbell-Thompson M, Lynch IJ, Bhardwaj B. Expression of Estrogen Receptor (ER) Subtypes and ERβ Isoforms in Colon Cancer. Cancer Research. 2001;61:632-40
4. Williams C, DiLeo A, Niv Y. et al. Estrogen receptor beta as target for colorectal cancer prevention. Cancer letters. 2016;372:48-56
5. Arber N, Hibshoosh H, Moss SF. et al. Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology. 1996;110:669-74
6. Nakamura Y, Felizola SJA, Kurotaki Y. et al. Cyclin D1 (CCND1) expression is involved in estrogen receptor beta (ERβ) in human prostate cancer. The Prostate. 2013;73:590-5
7. Paruthiyil S, Parmar H, Kerekatte V. et al. Estrogen Receptor β Inhibits Human Breast Cancer Cell Proliferation and Tumor Formation by Causing a G<sub>2</sub> Cell Cycle Arrest. Cancer Research. 2004;64:423-8
8. Jin Y, Bai Y, Ni H. et al. Activation of autophagy through calcium-dependent AMPK/mTOR and PKCθ pathway causes activation of rat hepatic stellate cells under hypoxic stress. FEBS Letters. 2016;590:672-82
9. Mathew R, Kongara S, Beaudoin B. et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367-81
10. Koukourakis MI, Giatromanolaki A, Sivridis E. et al. Beclin 1 over- and underexpression in colorectal cancer: distinct patterns relate to prognosis and tumour hypoxia. British Journal Of Cancer. 2010;103:1209
11. Cho DH, Jo YK, Kim SC. et al. Down-regulated expression of ATG5 in colorectal cancer. Anticancer Res. 2012;32:4091-6
12. Choi JH, Cho YS, Ko YH. et al. Absence of autophagy-related proteins expression is associated with poor prognosis in patients with colorectal adenocarcinoma. Gastroenterology research and practice. 2014;2014:179586
13. Shimizu S, Yoshida T, Tsujioka M. et al. Autophagic cell death and cancer. International journal of molecular sciences. 2014;15:3145-53
14. Xiong HY, Guo XL, Bu XX. et al. Autophagic cell death induced by 5-FU in Bax or PUMA deficient human colon cancer cell. Cancer letters. 2010;288:68
15. Fingar DC, Richardson CJ, Tee AR. et al. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Molecular & Cellular Biology. 2004;24:200-16
16. Chatterjee A, Mukhopadhyay S, Tung K. et al. Rapamycin-induced G1 cell cycle arrest employs both TGF-β and Rb pathways. Cancer letters. 2015;360:134-40
17. Wang X-W, Zhang Y-J. Targeting mTOR network in colorectal cancer therapy. World Journal of Gastroenterology: WJG. 2014;20:4178
18. Sobolewska A, Gajewska M, Zarzyńska J. et al. IGF-I, EGF, and sex steroids regulate autophagy in bovine mammary epithelial cells via the mTOR pathway. Eur J Cell Biol. 2009;88:117-30
19. Ruddy SC, Lau R, Cabrita MA. et al. Preferential estrogen receptor beta ligands reduce Bcl-2 expression in hormone-resistant breast cancer cells to increase autophagy. Mol Cancer Ther. 2014;13:1882-93
20. Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393-8
21. Masamha CP, Benbrook DM. Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res. 2009;69:6565-72
22. Raven JF, Baltzis D, Wang S. et al. PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. J Biol Chem. 2008;283:3097-108
23. Ichikawa M, Sowa Y, Iizumi Y. et al. Resibufogenin Induces G1-Phase Arrest through the Proteasomal Degradation of Cyclin D1 in Human Malignant Tumor Cells. PLoS One. 2015;10:e0129851
24. Alao JP, Stavropoulou AV, Lam EW. et al. Histone deacetylase inhibitor, trichostatin A induces ubiquitin-dependent cyclin D1 degradation in MCF-7 breast cancer cells. Mol Cancer. 2006;5:8
25. Wu SY, Lan SH, Wu SR. et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology. 2018;68:141-54
26. Juríková M, Danihel Ľ, Polák Š. et al. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta histochemica. 2016;118:544-52
27. Chandrashekar DS, Bashel B, Balasubramanya SAH. et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017;19:649-58
28. Kabeya Y, Mizushima N, Ueno T. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720-8
29. Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Letters. 2010;584:1374-8
30. Guido C, Panza S, Santoro M. et al. Estrogen receptor beta (ERβ) produces autophagy and necroptosis in human seminoma cell line through the binding of the Sp1 on the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) promoter gene. Cell Cycle. 2012;11:2911-21
31. Paglin S, Hollister T, Delohery T. et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Research. 2001;61:439
32. Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Letters. 2010;584:1393-8
33. Klionsky DJ, Abdelmohsen K, Abe A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12:1-222
34. Komatsu M, Wang QJ, Holstein GR. et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489-94
35. Kanzawa T, Zhang L, Xiao L. et al. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene. 2004;24:980
36. Caiazza F, Ryan EJ, Doherty G. et al. Estrogen receptors and their implications in colorectal carcinogenesis. Frontiers in Oncology. 2015;5:19
37. Labrie F. Drug insight: breast cancer prevention and tissue-targeted hormone replacement therapy. Nat Clin Pract Endocrinol Metab. 2007;3:584-93
38. Martineti V, Picariello L, Tognarini I. et al. ERβ is a potent inhibitor of cell proliferation in the HCT8 human colon cancer cell line through regulation of cell cycle components. Endocrine-Related Cancer. 2005;12:455-69
39. Song HM, Park GH, Eo HJ. et al. Anti-proliferative effect of naringenin through p38-dependent downregulation of cyclin D1 in human colorectal cancer cells. Biomolecules & therapeutics. 2015;23:339
40. Chung J-H, Ostrowski MC, Romigh T. et al. The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Human molecular genetics. 2006;15:2553-9
41. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913
42. García-Fernández M, Karras P, Checinska A. et al. Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy. 2016;12:1776-90
43. Liang C, Feng P, Ku B. et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nature Cell Biology. 2006;8:688-99
44. Loukil A, Zonca M, Rebouissou C. et al. High-resolution live-cell imaging reveals novel cyclin A2 degradation foci involving autophagy. J Cell Sci. 2014;127:2145-50
45. Paquette M, El-Houjeiri L, Pause A. mTOR Pathways in Cancer and Autophagy. Cancers (Basel). 2018:10
46. Shor B, Gibbons JJ, Abraham RT. et al. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8:3831-7
47. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424
48. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014;4:64
49. Aryal P, Kim K, Park PH. et al. Baicalein induces autophagic cell death through AMPK/ULK1 activation and downregulation of mTORC1 complex components in human cancer cells. FEBS J. 2014;281:4644-58
50. Shimizu S, Kanaseki T, Mizushima N. et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221-8
51. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528-42
Author contact
![]() Corresponding author: Jian Huang, Ph.D., Room 5105, College of Life Sciences, Wuhan University, Wuhan, Hubei 430072, P.R China. Fax: +86 27 68753582; Tel: +86 27 68753582; E-mail: jianhuangedu.cn
Corresponding author: Jian Huang, Ph.D., Room 5105, College of Life Sciences, Wuhan University, Wuhan, Hubei 430072, P.R China. Fax: +86 27 68753582; Tel: +86 27 68753582; E-mail: jianhuangedu.cn