Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(5):953-961. doi:10.7150/ijbs.30857 This issue Cite
Research Paper
KLF4-Mediated CDH3 Upregulation Suppresses Human Hepatoma Cell Growth and Migration via GSK-3β Signaling
Li Li1, Shijun Yu1, Qiong Wu1, Ning Dou1, Yandong Li1,2 ![]() , Yong Gao1
, Yong Gao1 ![]()
1. Department of Oncology, Shanghai East Hospital, Tongji University School of Medicine, Shanghai 200120, China;
2. Research Center for Translational Medicine, Shanghai East Hospital, Shanghai 200120, China
Received 2018-10-22; Accepted 2019-2-7; Published 2019-3-10
Abstract

P-cadherin (CDH3), a classical cell adhesion molecule involved in tissue integrity and cell localization, has been implicated in many types of cancer. However, little is known about its function and regulatory mechanism in hepatocellular carcinoma (HCC). Here we report that CDH3 was positively regulated by kr¨uppel-like transcription factor 4 (KLF4), which is a crucial tumor suppressor gene in HCC, at mRNA level in HCC cell lines. Luciferase reporter assay and chromatin immunoprecipitation assay indicated that KLF4 directly bound to CDH3 promoter and transcriptionally activated CDH3 expression. Consistently, CDH3 expression was closely related with KLF4 expression in patients' samples and both proteins exhibited a downregulated expression pattern in cancer samples. Functionally, enforced CDH3 expression suppressed and silenced CDH3 expression promoted HCC cell growth and migration in vitro. Mechanistically, we observed that GSK-3β was regulated by CDH3 and may function as a possible downstream effector of CDH3. Knockdown of GSK-3β showed a similar phenotype with CDH3 silencing. Taken together, these findings establish the KLF4/CDH3/GSK-3β axis as an important regulatory mechanism in HCC development.
Keywords: KLF4, CDH3, hepatocellular carcinoma, GSK-3β
Introduction
The classical cadherins (CDHs) are a major class superfamily of transmembrane glycoproteins, which are responsible for calcium-dependent cell-cell adhesion, and mediate specific homophilic protein interactions[1]. Many of them are involved in the regulation of tumor metastasis [2]. Two famous members are E-cadherin (CDH1) and N-cadherin (CDH2)[1]. The repression of E-cadherin and induction of N-cadherin are one of characteristics of tumor cells with metastatic potential[3]. This kind of expression alteration usually links to a transcriptional program called epithelial-to-mesenchymal transition (EMT)[4]. It has been reported that loss of E-cadherin expression correlates with a poorer prognosis in hepatocellular carcinoma (HCC)[5-8]; N-cadherin expression is increased in HCC, and the upregulation of N-cadherin is relevant to the high metastatic potential of HCC and worse surgical prognosis[7, 9]. Unlike the E- and N-cadherin, CDH3 (P-cadherin), another member of the classical cadherin family, has not been fully investigated in HCC and its regulatory mechanism remains to be well defined.
The Krüppel-like factors (KLFs) belong to a family of evolutionarily conserved transcription factors with a C-terminal three-zinc-finger DNA-binding domain in mammals [10, 11]. They play crucial roles in cell differentiation, proliferation, growth, apoptosis and development upon external stimuli [12, 13]. Among the KLF family members, KLF4 received significant attention because of the discovery that KLF4 as one of the four Yamanaka factors (Oct3/4, c-Myc, Sox2, and Klf4) in the process of stem cell reprogramming or iPS (inducible pluripotent stem cells) [14]. KLF4 is mostly expressed in post mitotic and terminally differentiated epithelial cells of the skin, lungs, and those in the gastrointestinal tract [11]. The role of KLF4 has been widely studied in multiple types of cancer and has been found to work as a tumor suppressor or an oncogene in a tissue type-specific manner. The researches to date have confirmed that KLF4 has tumor suppressive properties in colorectal cancer[15], pancreatic ductal cancer[16], nasopharyngeal cancer (NPC)[17], primary lung cancer [18], bladder cancer[19], gastric cancer[20] and liver cancer [21]. On the contrary, in oral and breast cancer KLF4 promote cancer cell growth and metastasis [22]. The first systematic report that KLF4 is a tumor suppressor in HCC was carried out in 2012, and scientists have found that the influence of the interaction of KLF4 and VDR, HNF-6, Smad7 on HCC progression until now [23-25]. So as to further supplement our understanding of the role of KLF4 in HCC, our group explored the correlation between KLF4 and CDH3, and the influence of CDH3 on the development of HCC.
In the present study, we found that CDH3 was transcriptionally regulated by KLF4 and served as a tumor suppressor in HCC consistent with the role of KLF4. Furthermore, our results revealed that GSK-3β signaling may be involved in the CDH3-mediated regulation of tumor cell growth and migration. These observations will provide new insights into molecular mechanism of HCC development and potential targets for HCC treatment.
Materials and Methods
Human Tissue Microarray (TMA) and Immunohistochemistry
A commercially available tissue microarray (TMA) purchased from AlenaBio, Xi'an, China was used, and patient information was provided by the manufacturers. The TMA consists of 64 HCC specimens. Immunohistochemical analysis was performed with anti-KLF4 (1:200, Proteintech, 11880-1-AP) and anti-CDH3 antibody (1:50, Proteintech, 13773-1-AP). Two independent researchers evaluated these immunohistochemical staining in double-blind way. The percentage of stained cells (0-100%) and the staining intensity (negative/weak, intermediate, strong) were used to assess each cancer samples. The research was approved by the Medical Ethics Committees of Shanghai East Hospital, Tongji University.
Tissue specimens and cell culture
37 paired samples from the HCC and adjacent normal hepatic tissues were obtained from patients at Shanghai East Hospital, Tongji University School of Medicine, China. Informed consent was obtained from all patients, and the study was approved by the Ethics Committee of the Shanghai East Hospital. The human embryonic kidney (HEK) 293T cells, the human fetal liver L02 cells and other human HCC cell lines, including Huh7, Focus, YY-8103, HCC-LM3, Hep3B, Sk-Hep-1, WRL68 and PLC/PRF/5 were taken from our laboratory stocks. All the cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM), which was supplemented with 10% fetal bovine serum and antibiotics (100µg/mL penicillin and 100 µg/mL streptomycin). Cells were grown in a 5% CO2 atmosphere at 37°C.
Construction of stable CDH3 or KLF4 knockdown Cell Lines and RNA interference
pLV3 lentiviral particles containing non-targeting shRNA and shRNA targeting CDH3 or KLF4 were purchased from Genepharma (Shanghai, China). The targeting sequences of CDH3 and KLF4 shRNA were: 5'-CCAAUAUCUGUCCCUGAAA-3' and 5'-TTGTGGATATCAGGGTATAAA-3'. The stable cell lines were established by transducing these lentiviruses into HCC cells respectively and stable pools of cells were selected with 5μg/mL puromycin for at least 1 week. For GSK-3β knockdown, two small interference RNAs were chemically synthesized by GenePharma, Shanghai, China. The sequences targeting GSK-3β were siGSK-3β-1: 5'-GGGCCUUAUAUACUCUAAA-3' and siGSK-3β-2 5'-GCCUCAAAGUAGUCCAUAU-3'.
Plasmid construction and cell transfection
The CDH3 cDNA (GeneBank Accession Number: NM_001317195.1) was cloned into pcDNA3.1 to construct overexpression plasmid pcDNA3.1-CDH3. The KLF4 overexpression plasmid pcDNA3.1-KLF4-FLAG was a gift from Dr. Keping Xie. Two fragments (-784, +116; -579, +116) including CDH3 promoter region associated with transcription initiation site were cloned into the pGL3-basic vector (Promega, USA). The two reporter plasmids were then named pGL3-CDH3-627 and pGL3-CDH3-439 as indicated in Figure 2B. Mutant constructs in KLF4 binding sequence CACCC to CAGGG were generated by site-directed mutagenesis. All above constructs were verified by DNA sequencing. Cell transfection was used Lipofectamine 3000 reagent (Invitrogen, CA, USA) according to the manufacturer's instruction.
Western Blotting
Cells seeded in 6-well plates were lysed by scraping into lysis buffer. The protein concentration of each lysate was examined using the BCA protein assay kit (TaKaRa, Japan). Western blotting was performed according to standard protocol with the following antibodies and dilutions: anti-KLF4 (Proteintech, #11880-1-AP, 1:1000), anti-CDH3 (Proteintech, #13773-1-AP, 1:500), anti-GSK-3β (Cell Signaling Technology, #12456, 1:1500), anti-Phospho-GSK-3β (Ser9) (Cell Signaling Technology, #9323, 1:1500), anti-AKT (Cell Signaling Technology, #9272, 1:1500), anti-Phospho-Akt (Ser473) (Cell Signaling Technology, #4051, 1:1500), anti-Erk1/2 (Thr202/Tyr204) (Cell Signaling Technology, #4695, 1:1500), anti-Phospho-Erk1/2 (Thr202/Tyr204) (Cell Signaling Technology, #4370, 1:1500), anti-p70S6K (Proteintech, #14485-1-AP, 1:500), anti-Phospho-p70 S6 Kinase (Cell Signaling Technology, #9204, 1:1500), anti-Smad3 (Cell Signaling Technology, #9513, 1:1500), anti-Phospho-Smad3 (Ser423/425) (Cell Signaling Technology, #9520, 1:1500), anti-β-catenin (Cell Signaling Technology, #8480, 1:1500) and anti-GAPDH (Proteintech, #60004-1-Ig, 1:10000).
Quantitative Real-time PCR (qRT-PCR)
The total RNAs were extracted from cultured cells or tissues with Trizol reagent (Invitrogen, USA). Then, the cDNAs were synthesized and used as templates for detecting the expression level of CDH3 and KLF4 by qRT-PCR. ABI 7500 Real Time PCR System and Syber green reagent (TaKaRa, Japan) were used according to the instructions from the manufacturer. The primer sequences were as follows: CDH3 (F: 5'-GTCCCTTCCCCCAGAGACTGAA-3', R: 5'- CCTCAAAATCCAAACCCTTCC-3'), KLF4 (F: 5'- AGAGGAGCCCAAGCCAAAG-3', R: 5'-CGTCCCAGTCACAGTGGTAAG-3'), β-actin (F: 5'-AGAGCCTCGCCTTTGCCGATCC-3', R: 5'- CTGGGCCTCGTCGCCCACATA-3').
Cell proliferation assay
Proliferation assay was performed by Cell Counting Kit-8 method. Briefly, the cells were seeded at a density of 3000 cells per well with 100 μl complete medium in 96-well cell culture plates in triplicate, and incubated at 37°C and humidified 5% CO2. At 1 to 5 day after seeding, 10 µl CCK8 reagent (Dojindo, Shanghai, China) was added and incubated for 75 min at 37 °C. Then, the absorbance values at 450 nm were measured using a Spectra Max M2 (Molecular Devices, Sunnyvale, CA, USA). Each experiment was repeated at least three times.
Colony Formation Assay
For colony formation assays, 2000 cells were seeded into a 6-well plate and cultured for about 2-3 weeks. These cultures were then stained with crystal violet (Sangon Biotech, Shanghai, China) and photographed. Each experiment was repeated at least three times.
Cell migration assays
Transwell chamber assay was conducted to detect cell migration. 50,000 cells in 400 μl DMEM without FBS were seeded onto the upper compartment of Transwell (Corning, US). The lower chamber was filled with 800 μl DMEM and 10% fetal bovine serum as chemoattractant. The cells were incubated for 24 h at 37 °C and 5% CO2 and then non-migrated cells were removed from the top of the chamber with a cotton swab. The rest cells were fixed by 5% glutaraldehyde, stained with Crystal Violet, and photographed using a microscope. All assays were repeated three times in triplicate.
Luciferase Reporter Assay
HCC cells were plated in 24-well plates. 24 h later cells were co-transfected with human pGL3-CDH3-627 or pGL3-CDH3-439 firefly luciferase reporter plasmid together with pRL-SV40 renilla luciferase construct, KLF4 or empty vector using lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturer's instruction. The luciferase activity in the cells was quantified using a dual luciferase assay system (Promega, USA) 24 h after transfection. Each experiment was performed in triplicate and repeated at least three times.
Chromatin immunoprecipitation (ChIP)
ChIP was performed in exponentially growing Huh7 cell using a ChIP assay kit (Millipore, CA, USA) according to the manufacturer's instructions. The resulting precipitated DNA samples were analyzed using PCR to amplify a 156-bp region of the CDH3 promoter with the primers 5'-GTCCAGAAGTTCAAGACCA-3' (sense) and 5'-GGCTGAAGCGATTCTCCTC-3' (antisense) and a 161-bp region of the CDH3 promoter with the primers 5'-GCTGGAGATTATGAGACTA-3' (sense) and 5'-GAGTGCTCAGGTTACAGG-3' (antisense). The PCR products were resolved electrophoretically on an agarose gel and visualized using Bio-red staining. Each experiment was repeated at least three times.
Statistical analysis
Data obtained from experiments were expressed as mean ±standard deviation (SD) from three independent experiments, and analyzed using the χ2 test or Student's t test to assess differences between experimental groups. All statistical analyses were performed with GraphPad Prism 5 software. P <0.05 were recognized as significant. “*” indicates P<0.05; “**” indicates P<0.01.
Results
CDH3 is positively regulated by KLF4 in HCC cells
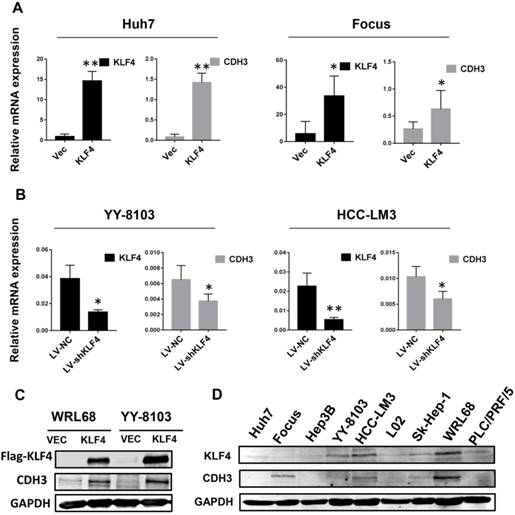
To determine whether there is a correlation between the expression of KLF4 and CDH3, we first investigated the mRNA levels of CDH3 upon KLF4 overexpression or silencing in HCC cells by qRT-PCR. The results showed that enforced expression of KLF4 in Huh7 and Focus cells led to increased CDH3 expression (Figure 1A), while in KLF4-silenced YY-8103 and HCC-LM3 cells, the expression of CDH3 was decreased (Figure 1B). Importantly, western blot analysis also indicated that CDH3 was induced by KLF4 overexpression in protein level (Figure 1C). Moreover, we obtained a consistent trend of protein expression between KLF4 and CDH3 in multiple HCC cell lines (Figure 1D). These results manifest that there is a conspicuous positive activation of CDH3 expression by KLF4 in HCC cells.
CDH3 expression is activated by KLF4 in HCC cells. (A) KLF4 overexpression by transient transfection promoted CDH3 expression by qRT-PCR. (B) The mRNA level of CDH3 detected by qRT-PCR was reduced in KLF4 stably silenced HCC cells. *P<0.05, **P<0.01. (C) Overexpression of KFL4 enhanced CDH3 expression at protein level in WRL68 and YY-8103 cells detected by western blot analyses. (D) Western blots showing the expression pattern of KLF4 and CDH3 in a series of HCC cell lines.
KLF4 directly regulate CDH3 transcription
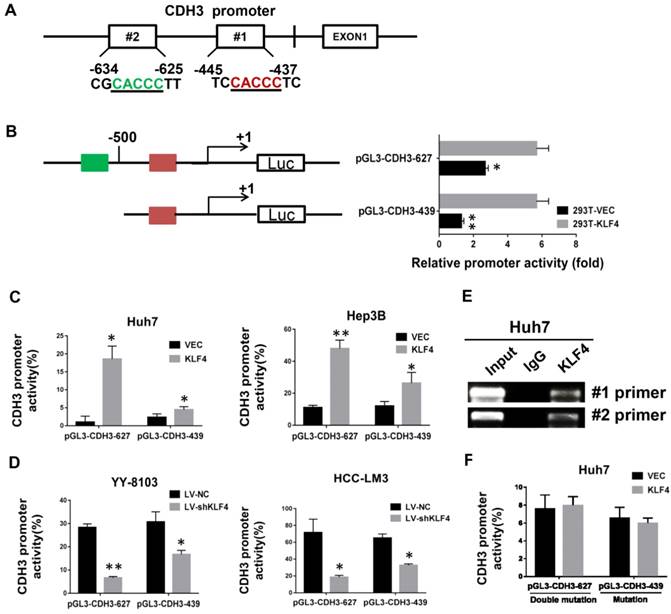
By bioinformatics analysis, we found there are two putative binding sites of KLF4 in the promoter region of CDH3 (Figure 2A). To approve whether KLF4 regulates CDH3 expression in a direct manner, we generated two promoter luciferase constructs for CDH3, the long promoter pGL3-CDH3-627 and the short promoter pGL3-CDH3-439. The subsequent luciferase reporter assay in 293T cells demonstrated that KLF4 overexpression markedly promoted the activity of CDH3 promoter (Figure 2B). The results were then confirmed in Huh7 and Hep3B cells as indicated in Figure 2C. In contrast, the relative luciferase activity of CDH3 promoter was decreased with KLF4 knockdown in YY-8103 and HCC-LM3 cells (Figure 2D). In addition, a chromatin immunoprecipitation assay was performed in Huh7 cells. The PCR data indicated that KLF4 protein could bind to the endogenous CDH3 promoter regions (Figure 2E). When KLF4 binding sequence was mutated in pGL3-CDH3-627 and pGL3-CDH3-439 constructs, the effects of KLF4 on CDH3 promoter activities were significantly abolished (Figure 2F). Collectively, these data strongly suggest that KLF4 can positively regulate CDH3 expression in HCC cell lines by directly binding to the promoter region of CDH3.
Direct transcriptional activation of CDH3 by KLF4 in HCC cells. (A) The two potential binding sites for KLF4 in the promoter of CDH3. (B) CDH3 promoter reporters (pGL3-CDH3-627 and pGL3-CDH3-439) were transfected into 239T cells in triplicate with KLF4 expression plasmids or control vectors for 24 h. The CDH3 promoter activity was then examined using a dual luciferase assay kit. (C) The CDH3 promoter reporter activities were determined in KLF4 overexpressed Huh7 and Hep3B cells. (D) The CDH3 promoter reporter activities were determined in KLF4 silenced YY-8103 and HCC-LM3 cells. The experiments were performed independently three times. *P<0.05, **P<0.01. (E) Chromatin immunoprecipitation assays were performed with a specific anti-KLF4 antibody or IgG and oligonucleotides flanking the KLF4 binding sites were amplified by PCR. (F) The activities of mutant CDH3 promoters were determined by dual luciferase assays in KLF4 overexpressed Huh7 cells.
CDH3 expression correlates with KLF4 level in HCC specimens
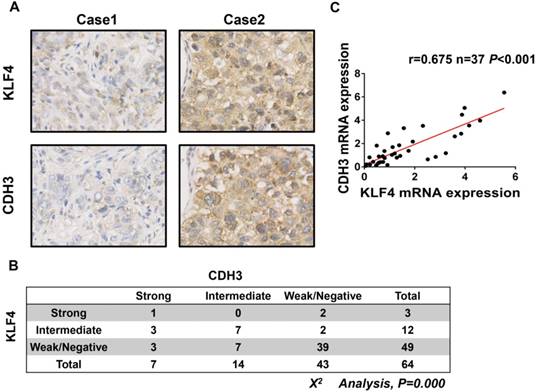
To further address the relationship between CDH3 and KLF4, we performed immunohistochemical staining of both proteins in a HCC tissue microarray. As shown in Figure 3A and 3B, weak/negative staining of CDH3 was detected in 67.2% (43 of 64) of HCC samples, whereas 76.6% (49 of 64) of HCC samples exhibited low expression of KLF4. A positive correlation between CDH3 and KLF4 protein levels was observed in these HCC samples (Figure 3B). Furthermore, qRT-PCR was employed to analyze the mRNA levels of both genes in additional 37 paired of HCC samples and corresponding normal tissues. The statistical analysis revealed that the expression of KLF4 was positively correlated with that of CDH3 in mRNA level (Figure 3C). Together, these data suggest that CDH3 expression is associated with KLF4 level in clinical samples.
CDH3 expression is associated with KLF4 level in HCC specimens. (A) TMA specimens including 64 HCC tissues were prepared for immunostaining with specific antibodies against CDH3 and KLF4. Representative images of CDH3 and KLF4 expression in HCC specimens were shown. (B) Data showed that the CDH3 expression levels directly correlated with the KLF4 expression levels in HCC. (C) A positive correlation between KLF4 and CDH3 mRNA level in additional 37 paired HCC tissues was measured by linear regression (r=0.675, P<0.001).
Downregulation of CDH3 promotes HCC cell proliferation and migration in vitro
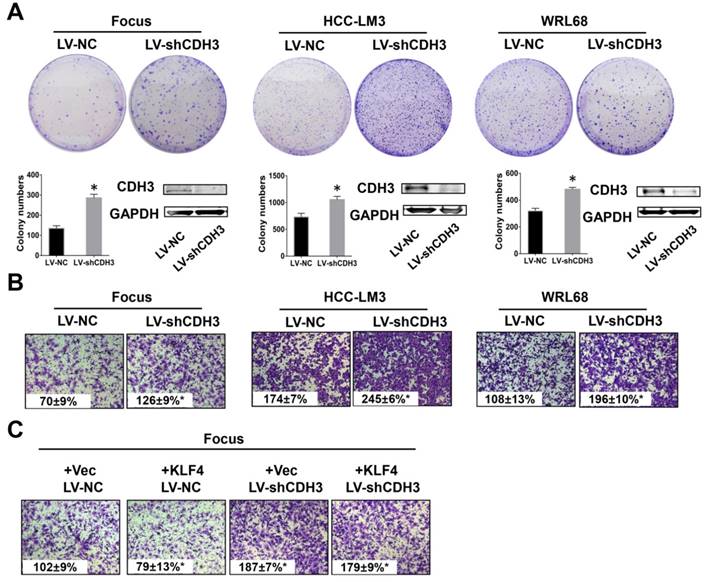
To test the possible function of CDH3 in HCC as mentioned above, we examined cell growth and migration in CDH3 stably silenced HCC cells. The expression level of CDH3 was verified by western blot as shown in Figure 4A. Colony formation assays demonstrated that CDH3 knockdown accelerated HCC cells growth compared with corresponding control cells (Figure 4A). Meanwhile, transwell chamber assays revealed that CDH3 knockdown cells had stronger migratory ability than that of control cells (Figure 4B). These findings implicated that CDH3 downregulation facilitates the tumorigenic properties of HCC cells in vitro. To further elucidate the functional relevance between CDH3 and KLF4, KLF4-overexpressing plasmid was transfected into CDH3 knockdown cells and then these cells were conducted with transwell chamber assays. The final results indicated that CDH3 reduction attenuated the suppression of KLF4 on cell migration (Figure 4C), suggesting CDH3 may be one of downstream effectors of KLF4.
Downregulation of CDH3 induced cell proliferation and migration. (A) Colony formation assays were performed to detect the growth of LV-shCDH3-infected Focus, HCC-LM3 and WRL68 cells and control cells, respectively. Colonies were counted and captured. The levels of CDH3 expression were examined via western blot. *P<0.05. (B) Transwell assays were performed to detect changes in migratory abilities of Focus, HCC-LM3 and WRL68 cells treated with LV-shCDH3 or LV-shNC control. (C) KLF4 expression plasmid was transfected into CDH3 silenced HCC cells and control cells, respectively. Transwell chamber assays were then performed to detect cell migration of these cells.
Overexpression of CDH3 suppresses growth and migration of HCC cells
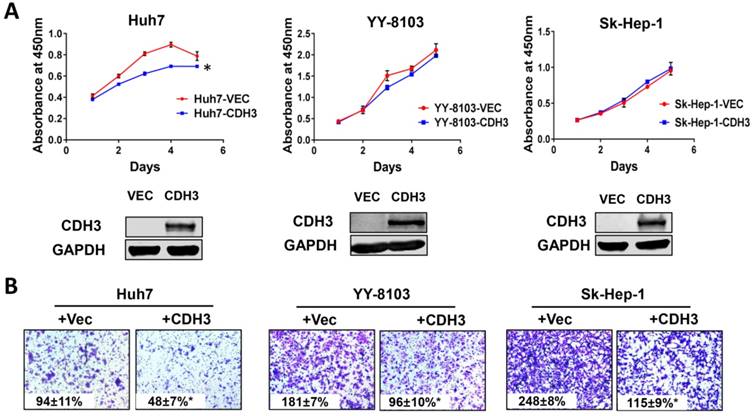
We next examined the effects of CDH3 overexpression in cultured HCC cells. Enhanced CDH3 expression by transient transfection decreased cell growth rate in Huh7 cells, but not in YY-8103 or Sk-Hep-1 cells (Figure 5A), suggesting the suppressive roles induced by CDH3 may be cell-type specific. However, transwell assay showed that CDH3 overexpression retarded cell migration in all three HCC cell lines (Figure 5B). The overexpression efficiency was verified by western blotting (Figure 5A). Our outcomes revalidated that CDH3 is a tumor suppressor that can inhibit tumor growth and migration of HCC cells, yet the impact on the latter is more significant.
The effect of CDH3 overexpression on growth and migration of HCC cells. (A) Cell proliferation of Huh7, YY-8103 and Sk-Hep-1 cells transfected with CDH3 plasmid or control empty vector was evaluated using CCK8 assay. And the level of CDH3 expression was examined using western blot. *P<0.05. (B) Transwell assays were conducted to evaluate migration of CDH3 overexpressed Huh7, YY-8103 and Sk-Hep-1 cells.
CDH3 expression affects GSK-3β level
To determine the molecular mechanism by which CDH3 suppresses tumor cell proliferation and migration, western blot analysis was used to measure the expression alteration of a series of signaling transduction factors including GSK-3β, p-GSK-3β, AKT, p-AKT, Erk, p-Erk, P70s6k, p-P70s6k, SMAD3, p-SMAD3 and β-catenin in CDH3-silenced Focus cells. As shown in Figure 6A, the total level of GSK-3β was reduced, whereas other factors had no significant change in CDH3 knockdown cells. To attain a trustworthy conclusion, we further elucidated the protein level of GSK-3β in CDH3-overexpressing cells via western blot assay. Consistent with the results above, CDH3 overexpression boosted GSK-3β expression in YY8103 and HCC-LM3 whether treated with insulin or not (Figure 6B).
CDH3 affects GSK-3β expression in HCC cells. (A) Western blot analysis was used to measure the expressions of GSK-3β, p-GSK-3β, AKT, p-AKT, Erk, p-Erk, P70S6K, p-P70S6K, SMAD3, p-SMAD and β-catenin in CDH3 silenced Focus cells. (B) The levels of GSK-3β and p-GSK-3β were examined in CDH3 overexpressed YY8103 and HCC-LM3 cells using western blot assay. The cells were treated with or without insulin for 10 min. (C) TCF/LEF luciferase activity was measured by dual luciferase reporter kit and the results illustrated that CDH3 expression had no effect on the transcription activity of Wnt/β-catenin signaling. (D) The GSK-3β knockdown cell line was constructed by transfecting siGSK-3β into Focus and YY-8103 cells and detected by western blotting. (E) The migration assay was conducted in Focus and YY-8103 cells after transfection with siGSK-3β (or control).
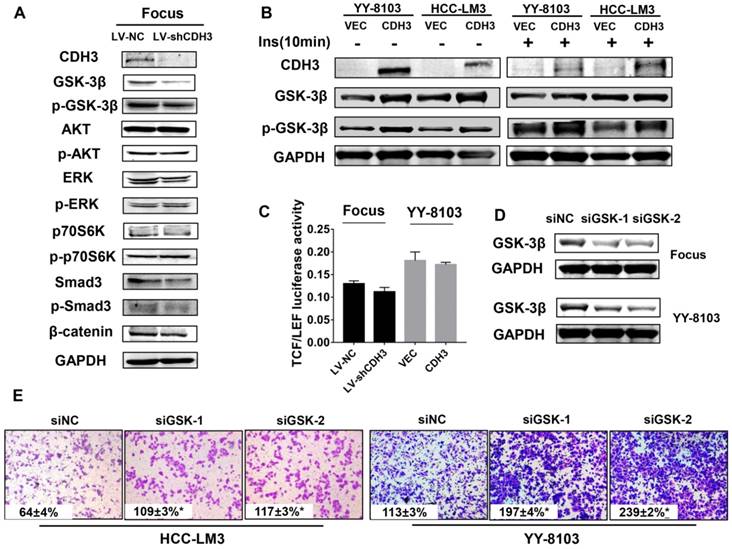
As known, Wnt/β-catenin signaling plays a crucial role in HCC[26]. GSK-3β is identified as a critical negative regulator of this pathway, for inactivation of GSK-3β results in the translocation and accumulation of β-catenin and subsequently affects downstream gene transcription [27]. However, as our data shown in Figure 6A, β-catenin level was not altered with the downregulation of CDH3. Thus, we supposed that CDH3 may not be involved in the regulation of Wnt/β-catenin pathway. For further confirmation, we designed a luciferase reporter assay to measure TCF/LEF luciferase activity for judging whether CDH3 affects the downstream gene expression of Wnt/β-catenin pathway. As expected, TCF/LEF luciferase activity was not changed significantly when CDH3 was overexpressed or silenced (Figure 6C). To verify the role of GSK-3β, transwell chamber assays were accomplished in the HCC cell lines with GSK-3β knockdown by two small interfering RNAs (Figure 6D). The results showed that the migration ability of HCC cells was elevated with GSK-3β downregulation (Figure 6E), in accordance with those phenotypes of CDH3 knockdown. Taken together, these results suggest that GSK-3β may act as downstream factor of CDH3 and mediate the tumor suppressive function of CDH3 in HCC.
Discussion
Hepatocellular carcinoma (HCC) is the prime leading cause of cancer death in the world [28]. Its incidence has gradually increased in recent years [29]. Thus, it is still urgent to explore the molecular pathogenesis of HCC for new drug target development and therapy approach improvement. In the present study, we identified CDH3 as a tumor suppressor in HCC and the deregulation of KLF4/CDH3/GSK-3β axis might contribute to this disease.
Previous studies have reported that CDH3 is aberrantly expressed in various tumor tissues, and processes oncogenic or tumor suppressor properties. On one hand, in gastric, colorectal, pancreatic and breast cancers[30, 31], CDH3 promote growth, survival, angiogenesis, migration, and invasion of tumor cells; On the other hand, CDH3 inhibits tumor growth and invasion in melanoma cells, bladder cancer and non-small cell lung cancers [2, 32]. As for HCC, CDH3 was found to be downregulated in tumor tissues and its downregulation could induce tumor cell growth [33]. In our case, we provided evidence that CDH3 inhibits HCC cell proliferation and migration via colony formation assays and transwell chamber assays. Interestingly, one recent report implicated that CDH3 exists in serum and may act as a potential serum marker for monitoring colorectal cancer progression [34]. In another transcriptomic study, CDH3 abundance was found to be decreased in urine extracellular vesicles from prostate cancer patients [35]. It is obvious that CDH3 can be secreted outside the cell and exerts functions for some unknown mechanism. These above studies imply the roles of CDH3 in tumor are versatile and controversial. Therefore, it is important to investigate the upstream and downstream signaling of CDH3 for better understanding its involvement in tumor development.
By searching KLF4 consensus sequence 5'-CACCC-3' in the CDH3 promoter region, we identified two putative KLF4 binding sites on it. Following studies proved that KLF4 transcriptionally activates CDH3 expression in HCC cells. As the same time, the expression of CDH3 and KLF4 has consistent trend in clinical cancer samples. Functional analysis revealed that CDH3 serves as downstream effector of KLF4 by transwell assays. Our present data suggest that KLF4 is a key regulator of CDH3, and both proteins may play suppressive roles in HCC development.
Of note, when we explored the downstream factors of CDH3 by western blot, we observed that GSK-3β is modulated by CDH3. The functional phenotype of GSK-3β knockdown is similar with that of CDH3 downregulation as well. Glycogen synthase kinase-3 (GSK-3) belongs to a family of conserved serine/threonine kinase, comprising two highly conserved isoforms, GSK-3α and GSK-3β [36]. GSK-3 influences many important cellular processes, including cell differentiation, growth, motility and apoptosis [27]. As one of important substrates of GSK-3β, β-catenin mediates cell motility and metastasis in several cancers. Our western blot results showed that no change is observed in the expression of AKT, p70S6K, ERK, SMAD3 and β-catenin in CDH3 knockdown cell line, suggesting that other unknown possible effectors mediated GSK-3β downregulation-induced phenotype of HCC cells.
To sum up, our findings identified CDH3 as a KLF4-regulated tumor suppressor, and GSK-3β may mediate the suppressive roles of CDH3 in HCC. We highlight a new regulatory mechanism in HCC development. The KLF4/CDH3/GSk-3β axis may be considered as a new therapeutic strategy for drug target development in HCC.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (No. 81772567 and No. 81772568) and Key Disciplines Group Construction Project of Pudong Health Bureau of Shanghai (No. PWZxq2017-13).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harbor perspectives in biology. 2009;1:a003129
2. Vieira AF, Paredes J. P-cadherin and the journey to cancer metastasis. Molecular cancer. 2015;14:178
3. Hazan RB, Qiao R, Keren R, Badano I, Suyama K. Cadherin switch in tumor progression. Annals of the New York Academy of Sciences. 2004;1014:155-63
4. Gheldof A, Berx G. Cadherins and epithelial-to-mesenchymal transition. Progress in molecular biology and translational science. 2013;116:317-36
5. Gan WJ, Wang JR, Zhu XL, He XS, Guo PD, Zhang S. et al. RARgamma-induced E-cadherin downregulation promotes hepatocellular carcinoma invasion and metastasis. Journal of experimental & clinical cancer research: CR. 2016;35:164
6. Hashiguchi M, Ueno S, Sakoda M, Iino S, Hiwatashi K, Minami K. et al. Clinical implication of ZEB-1 and E-cadherin expression in hepatocellular carcinoma (HCC). BMC cancer. 2013;13:572
7. Liu YA, Liang BY, Guan Y, You J, Zhu L, Chen XP. et al. Loss of N-cadherin is associated with loss of E-cadherin expression and poor outcomes of liver resection in hepatocellular carcinoma. The Journal of surgical research. 2015;194:167-76
8. Paredes J, Figueiredo J, Albergaria A, Oliveira P, Carvalho J, Ribeiro AS. et al. Epithelial E- and P-cadherins: role and clinical significance in cancer. Biochimica et biophysica acta. 2012;1826:297-311
9. Zhan DQ, Wei S, Liu C, Liang BY, Ji GB, Chen XP. et al. Reduced N-cadherin expression is associated with metastatic potential and poor surgical outcomes of hepatocellular carcinoma. Journal of gastroenterology and hepatology. 2012;27:173-80
10. Guo K, Cui J, Quan M, Xie D, Jia Z, Wei D. et al. The Novel KLF4/MSI2 Signaling Pathway Regulates Growth and Metastasis of Pancreatic Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:687-96
11. Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J. et al. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:4370-80
12. Hsieh MH, Chen YT, Chen YT, Lee YH, Lu J, Chien CL. et al. PARP1 controls KLF4-mediated telomerase expression in stem cells and cancer cells. Nucleic acids research. 2017;45:10492-503
13. Hu D, Gur M, Zhou Z, Gamper A, Hung MC, Fujita N. et al. Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nature communications. 2015;6:8419
14. Wei D, Wang L, Yan Y, Jia Z, Gagea M, Li Z. et al. KLF4 Is Essential for Induction of Cellular Identity Change and Acinar-to-Ductal Reprogramming during Early Pancreatic Carcinogenesis. Cancer cell. 2016;29:324-38
15. Hu R, Zuo Y, Zuo L, Liu C, Zhang S, Wu Q. et al. KLF4 Expression Correlates with the Degree of Differentiation in Colorectal Cancer. Gut and liver. 2011;5:154-9
16. Zammarchi F, Morelli M, Menicagli M, Di Cristofano C, Zavaglia K, Paolucci A. et al. KLF4 is a novel candidate tumor suppressor gene in pancreatic ductal carcinoma. The American journal of pathology. 2011;178:361-72
17. Liu Z, Yang H, Luo W, Jiang Q, Mai C, Chen Y. et al. Loss of cytoplasmic KLF4 expression is correlated with the progression and poor prognosis of nasopharyngeal carcinoma. Histopathology. 2013;63:362-70
18. Hu W, Hofstetter WL, Li H, Zhou Y, He Y, Pataer A. et al. Putative tumor-suppressive function of Kruppel-like factor 4 in primary lung carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:5688-95
19. Ohnishi S, Ohnami S, Laub F, Aoki K, Suzuki K, Kanai Y. et al. Downregulation and growth inhibitory effect of epithelial-type Kruppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochemical and biophysical research communications. 2003;308:251-6
20. Zhang N, Zhang J, Shuai L, Zha L, He M, Huang Z. et al. Kruppel-like factor 4 negatively regulates beta-catenin expression and inhibits the proliferation, invasion and metastasis of gastric cancer. International journal of oncology. 2012;40:2038-48
21. Hsu HT, Wu PR, Chen CJ, Hsu LS, Yeh CM, Hsing MT. et al. High cytoplasmic expression of Kruppel-like factor 4 is an independent prognostic factor of better survival in hepatocellular carcinoma. International journal of molecular sciences. 2014;15:9894-906
22. Yu F, Shi Y, Wang J, Li J, Fan D, Ai W. Deficiency of Kruppel-like factor KLF4 in mammary tumor cells inhibits tumor growth and pulmonary metastasis and is accompanied by compromised recruitment of myeloid-derived suppressor cells. International journal of cancer. 2013;133:2872-83
23. Sun H, Peng Z, Tang H, Xie D, Jia Z, Zhong L. et al. Loss of KLF4 and consequential downregulation of Smad7 exacerbate oncogenic TGF-beta signaling in and promote progression of hepatocellular carcinoma. Oncogene. 2017;36:2957-68
24. Sun H, Tang H, Xie D, Jia Z, Ma Z, Wei D. et al. Kruppel-like Factor 4 Blocks Hepatocellular Carcinoma Dedifferentiation and Progression through Activation of Hepatocyte Nuclear Factor-6. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:502-12
25. Li Q, Gao Y, Jia Z, Mishra L, Guo K, Li Z. et al. Dysregulated Kruppel-like factor 4 and vitamin D receptor signaling contribute to progression of hepatocellular carcinoma. Gastroenterology. 2012;143:799-810 e2
26. Monga SP. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology. 2015;148:1294-310
27. Liu B, Yang H, Pilarsky C, Weber GF. The Effect of GPRC5a on the Proliferation, Migration Ability, Chemotherapy Resistance, and Phosphorylation of GSK-3beta in Pancreatic Cancer. International journal of molecular sciences. 2018:19
28. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
29. Rao CV, Asch AS, Yamada HY. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis. 2017;38:2-11
30. Imai K, Hirata S, Irie A, Senju S, Ikuta Y, Yokomine K. et al. Identification of a novel tumor-associated antigen, cadherin 3/P-cadherin, as a possible target for immunotherapy of pancreatic, gastric, and colorectal cancers. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:6487-95
31. Taniuchi K, Nakagawa H, Hosokawa M, Nakamura T, Eguchi H, Ohigashi H. et al. Overexpressed P-cadherin/CDH3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating rho-family GTPases. Cancer research. 2005;65:3092-9
32. Van Marck V, Stove C, Van Den Bossche K, Stove V, Paredes J, Vander Haeghen Y. et al. P-cadherin promotes cell-cell adhesion and counteracts invasion in human melanoma. Cancer research. 2005;65:8774-83
33. Bauer R, Valletta D, Bauer K, Thasler WE, Hartmann A, Muller M. et al. Downregulation of P-cadherin expression in hepatocellular carcinoma induces tumorigenicity. International journal of clinical and experimental pathology. 2014;7:6125-32
34. Kumara H, Bellini GA, Caballero OL, Herath SAC, Su T, Ahmed A. et al. P-Cadherin (CDH3) is overexpressed in colorectal tumors and has potential as a serum marker for colorectal cancer monitoring. Oncoscience. 2017;4:139-47
35. Royo F, Zuniga-Garcia P, Torrano V, Loizaga A, Sanchez-Mosquera P, Ugalde-Olano A. et al. Transcriptomic profiling of urine extracellular vesicles reveals alterations of CDH3 in prostate cancer. Oncotarget. 2016;7:6835-46
36. Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochemical research. 2007;32:577-95
Author contact
![]() Corresponding authors: Yandong Li, e-mail: yandongli2017edu.cn and Yong Gao, e-mail: drgaoyongedu.cn
Corresponding authors: Yandong Li, e-mail: yandongli2017edu.cn and Yong Gao, e-mail: drgaoyongedu.cn