Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(6):1177-1186. doi:10.7150/ijbs.33103 This issue Cite
Research Paper
PCBP1 inhibits the expression of oncogenic STAT3 isoform by targeting alternative splicing of STAT3 exon 23
Xiaole Wang1*, Jihua Guo1,2*, Xiaoxuan Che1, Rong Jia1 ![]()
1. The State Key Laboratory Breeding Base of Basic Science of Stomatology (Hubei-MOST) & Key Laboratory of Oral Biomedicine Ministry of Education, School & Hospital of Stomatology, Wuhan University, Wuhan, PR China.
2. Department of Endodontics, School & Hospital of Stomatology, Wuhan University, Wuhan, PR China.
*These authors contributed equally to this work.
Received 2019-1-13; Accepted 2019-3-28; Published 2019-5-7
Abstract

STAT3 plays very important roles in the initiation and development of tumors. Despite of extensive studies in repressing its activation and function via multiple ways, so far, there are few effective therapeutic methods to inhibit STAT3 in the clinic. STAT3 has two isoforms generated by alternative splicing of exon 23. STAT3α is the longer isoform and encodes the full-length oncogenic STAT3α protein. STAT3β is shorter and encodes the truncated and tumor-suppressive STAT3β protein. It remains unknown how the alternative splicing of STAT3 exon 23 is regulated. Here, we discovered that there is an exonic splicing suppressor (ESS) in exon 23. Importantly, splicing factor PCBP1 binds to this ESS. Overexpression of PCBP1 significantly reduced the proportion of STAT3α /STAT3β isoforms and the expression of STAT3α protein. Moreover, increased PCBP1 inhibited the growth of oral squamous cell carcinoma and breast cancer cells, and the expression of STAT3 target genes. Our results demonstrated that PCBP1 is the key splicing factor that promotes the switch from oncogenic isoform STAT3α to tumor-suppressive isoform STAT3β. Our results pave the way for finding new anti-STAT3 methods for cancer treatment.
Keywords: STAT3, Alternative splicing, Oncogene, head and neck cancer, PCBP1
Introduction
STAT3 (signal transducers and activators of transcription 3) is an oncogene and plays very important roles in cell proliferation, apoptosis, and metastasis [1]. Despite its important functions in cancer, so far, there are few effective therapeutic methods to inhibit STAT3 in the clinic [2]. A number of studies have shown that STAT3 is constitutively activated in many types of cancers including oral cancer by phosphorylation and dimerization [3, 4]. Many inhibitors have been developed to block the activation of STAT3. However, STAT3 can be activated by diverse receptors, which may attenuate the efficacy of STAT3 activation inhibitors. In addition, unphosphorylated STAT3 can promote cancer progression by enhancing the expression of oncogene RAS and Met [5]. Therefore, suppression of STAT3 expression may be an efficient way for cancer treatment.
STAT3 gene consists of 24 exons. Alternative splicing of exon 23 generates two isoforms, STAT3α and STAT3β. STAT3α encodes full-length oncogenic STAT3 protein (89kDa), whereas STAT3β encodes a truncated protein (80 kDa) [6]. In contrast to STAT3α, STAT3β plays negative roles in tumorigenesis [7] (Figure 1A). The lack of STAT3β contributes to enhanced inflammation and earlier tumor initiation [8]. The expression of STAT3β is strongly correlated with longer survival and less recurrence in esophageal squamous cell carcinoma (ESCC) patients. Moreover, STAT3β can suppress chemoresistance and cancer stemness of ESCC cells by blocking the transcriptional activity of STAT3α [9]. Inhibiting STAT3α expression led to tumor regression [10]. However, the mechanism of regulating alternative splicing of STAT3 exon 23 is unclear.
Screening of key motifs in STAT3 exon 23 and splicing factors which are responsible for regulating the alternative splicing of STAT3 exon 23. (A) Schematic diagram of the alternative splicing pattern of human STAT3 exon 23. The boxes represent exons, and the solid lines between the boxes represent the introns. The dashed lines above the introns indicate the direction of RNA splicing. STOP represents the terminating codons. Isoform STAT3α mRNA encodes an oncoprotein, while isoform STAT3β encodes a tumor suppressor. (B) Diagram of STAT3 minigene. Genomic sequence of STAT3 from 3′ part of exon 22 to 5′ part of exon 23 was amplified from CAL 27 cells and cloned into pEGFP-N1. To map potential regulatory motifs, exon 23 in minigene was serially mutated. The mutated bases were underlined. (C) RT-PCR analysis of alternative splicing of exon 23 in HEK293 cells transfected with wild-type (wt) or mutant (mt) minigenes. Relative α/β represents the ratio of band intensities of isoform α vs β. GAPDH served as a loading control. Diagrams on the right show the structures of STAT3 minigene and spliced products. Short line above exon 22 stands for the forward primer, and short line below GFP gene stands for the backward primer. (D) Screening analysis for splicing factors which regulates alternative splicing of STAT3 exon 23. HEK293 cells were transfected with indicated RNA splicing factors or control plasmid (pEGFP-N1). Alternative splicing of endogenous STAT3 exon 23 was analyzed by RT-PCR with primers located in exon 21 and 23. Diagrams on the right show the structures of STAT3 pre-mRNA and spliced products. Short lines above or below exons stand for primer positions. An exon 22/23 backward junction primer was used to specifically amplify short product, STAT3β. (E) Overexpression of exogenous T7 tagged PCBP1 was confirmed by western blot. LV: lentivirus. GAPDH served as a loading control. (F-G) Alternative splicing of STAT3 and the expression of PCBP1 in normal and oral cancer cells were analyzed by RT-PCR or western blot.
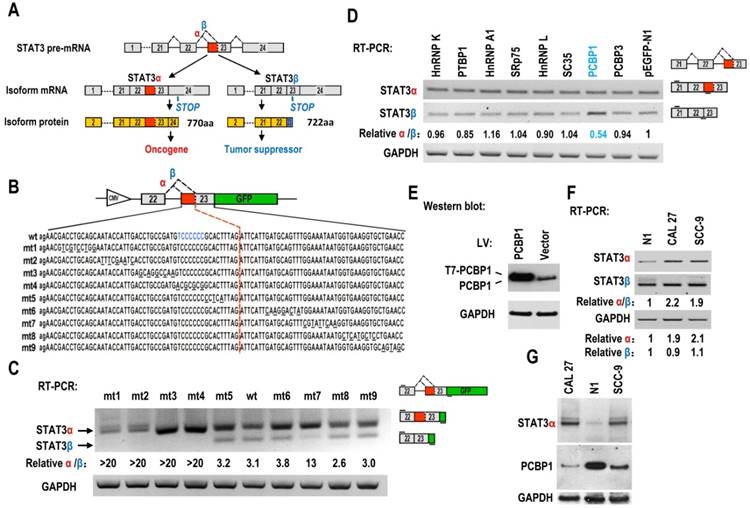
Alternative splicing of pre-mRNA is mainly regulated by splicing factor. PCBP1 [poly(rC)-binding protein 1, or hnRNP E1], is a tumor-suppressive splicing factor and regulates the process of pre-mRNA, mRNA stability, and translation {summarized in Ref. [11] [12]}. It also functions as a chaperone for the delivery of iron in cell [13]. Overexpression of PCBP1 suppresses the tumorigenicity of cancer cells, such as prostate cancer [14, 15], colon cancer [16], gastric cancer [17], ovarian cancer [18], and thyroid carcinoma cells [19]. In the present study, we discovered that PCBP1 is the key splicing factor that regulates alternative splicing of STAT3 exon 23 and promotes the switch from oncogenic isoform STAT3α to tumor-suppressive isoform STAT3β. Our results pave the way for finding new anti-STAT3 methods for cancer treatment.
Results
Regulatory motifs in STAT3 exon 23
The regulatory mechanisms of alternative splicing of STAT3 exon 23 in cancers are unknown. Oral squamous cell carcinoma (OSCC) is the most common type of head and neck squamous cell carcinoma. We constructed a human STAT3 minigene, including exon 22, intron 22 and exon 23 (Figure 1B). Then, we made serial mutations from nt 1 to nt 92 of exon 23 to screen potential regulatory motifs for controlling alternative splicing of exon 23. RT-PCR result showed that all mutants (mt1 to mt4) between proximal and distal splice sites almost totally lost the usage of distal splice site (short product), indicating that these sequences contain regulatory motifs required for the expression of STAT3β. In contrast, the mutants downstream of distal splice site have no effect on the alternative splicing of exon 23, except mutant mt7 (Figure 1C).
PCBP1 regulates the alternative splicing of STAT3 exon 23
Alternative splicing of pre-mRNA is mainly regulated by RNA splicing factors. Therefore, we overexpressed 26 splicing factors in HEK 293 cells and analyze the alternative splicing of STAT3 exon 23 (Figure 1D, Figure S1, S2). We found that PCBP1 is the one that most significantly decreases the ratio of STAT3α vs STAT3β isoform compared with control (fold change>1.5, P<0.01) (Figure 1D, 1E, Figure S1B). PCBP1 was reported as a tumor-suppressive gene [16]. Our results suggested that PCBP1 increases the level of STAT3β, which is correlated with its tumor-suppressive function.
In addition, we found that the ratio of STAT3α /β and the RNA level of STAT3α expression in normal primary cultured oral gingival epithelial cells were significantly lower than those in oral cancer cells (Figure 1F). The protein levels of PCBP1 in normal cells were significantly higher than those in the oral cancer cell lines, CAL 27 and SCC-9. On the contrary, cancer cells have significantly higher level of STAT3α protein than normal cells (Figure 1G). These results indicate that the expression of STAT3α may be inhibited by the high level of PCBP1 in normal cells.
PCBP1 interacts with an exonic splicing suppressor in exon 23
PCBP1 can interact with splicing silencing elements in alternative exons and suppresses their inclusion. We hypothesized that PCBP1 binds to an exonic splicing suppressor (ESS) in exon 23 and inhibits the usage of proximal splice site of exon 23 and the expression of STAT3α. Sequence analysis suggested that the original sequence of mt4, “UCCCCCCG”, is similar to known C-rich binding motifs of PCBP1 [20] (Figure 1B). We synthesized biotin labeled wild-type or mutant ESS RNA and performed pulldown assay with 293 total extract. As expected, PCBP1 can bind to wild-type ESS (called wt4), not mutant ESS (mt4), indicating that PCBP1 may bind to “UCCCCCCG” and promote the usage of distal 3' splice site of exon 23 and the expression of STAT3β (Figure 2A).
PCBP1 controls the alternative splicing of STAT3 exon 23. (A) RNA pulldown assay was used to analyze the interaction between PCBP1 and STAT3 RNA. Biotinylated oligo RNAs [including wt4 or mt4 based on minigene mt4, and a positive PCBP1 binding control sequence (PCBP1+)] were incubated with HEK293 total cellular extract. The total proteins binding to RNAs (Beads) were blotted with a mouse anti-PCBP1 antibody. Supernatants after pulldown: total proteins after pulldown in supernatants. (B) CAL 27 or SCC-9 cells were stably transfected with T7 tagged PCBP1 or control lentivirus and the alternative splicing of exon 23 was detected by RT-PCR. Relative α/β represents the ratio of band intensities of α vs β isoform. GAPDH served as a loading control. Diagrams on the right show the structures of STAT3 pre-mRNA and spliced products. Short lines above or below exons stand for primer positions. An exon 22/23 backward junction primer was used to specifically amplify short product, STAT3β. (C) Western blot showed the overexpression of T7 tagged PCBP1 and the expression level of cellular STAT3 and phosphorylated STAT3 (p-STAT3). GAPDH served as a loading control. (D) PCBP1 was knocked down in normal primary oral mucosal epithelial cells. Knockdown efficiency of PCBP1, and the expression of STAT3 and phosphorylated STAT3 were analyzed by western blot. The alternative splicing of exon 23 was detected by RT-PCR. GAPDH served as a loading control. (E) RT-PCR analysis showed that overexpression of PCBP1 downregulates the expression of STAT3 targets (Bcl-xl and Survivin) in both CAL 27 and SCC-9 cells.
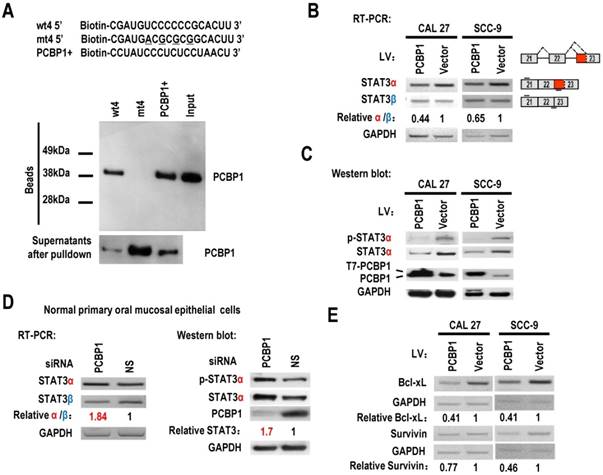
PCBP1 represses the expression of STAT3α in oral squamous cell carcinoma cells
STAT3 is involved in the tumorigenesis of OSCC. We wonder whether overexpression of PCBP1 can reduce the expression of STAT3, especially STAT3α, in OSCC cells. We established stable overexpression of PCBP1 in CAL 27 and SCC-9 cells, and found that overexpression of PCBP1 significantly reduced the ratios of STAT3α vs β and the levels of STAT3α by RT-PCR analysis (Figure 2B). Western blot also showed that the expression of STAT3α and phosphorylation of STAT3α decreased significantly upon overexpression of PCBP1 (Figure 2C). In contrast, in normal cells, knockdown of PCBP1 significantly increased the ratio of STAT3α vs β and the RNA levels of STAT3α compared with control group (Figure 2D), as well as the protein level of STAT3α and phosphorylation of STAT3α (Figure 2D). In addition, we found that mt4 abolished PCBP1's inhibitory effects on STAT3α expression (Figure S5). These results demonstrate that PCBP1 represses STAT3α expression.
STAT3 plays key roles in the initiation of tumorigenesis by promoting the expression of oncogenic targets, including Bcl-xL and survivin. Bcl-xL has central roles in suppressing apoptosis. Survivin can also block apoptosis. We found that the transcription levels of survivin, and Bcl-xL were reduced in CAL 27 and SCC-9 cells with PCBP1 overexpression (Figure 2E). These results further confirmed PCBP1 repressed the expression of STAT3α.
PCBP1 inhibits the proliferation and colony formation of OSCC cells
PCBP1 has been considered as a tumor suppressor. We found that overexpression of PCBP1 significantly inhibited the growth of CAL 27 and SCC-9 cells (Figure 3A-B). In addition, cells with PCBP1 overexpression showed significantly less colonies compared with control cells (Figure 3C-D). In line with these findings, knockdown of PCBP1 promoted cell proliferation in both CAL 27 and SCC-9 cancer cells (Figure S4C and S4D). Moreover, knockdown of PCBP1 increased the ratio of STAT3α vs STAT3β and the expression of STAT3α protein in both CAL 27 and SCC-9 cancer cells (Figure S4A and S4B). These results suggested that PCBP1 also acts as a tumor suppressor and inhibits the proliferation of oral cancer cells.
Downregulating the ratio of STAT3 α/β inhibits the cell growth of OSCC cell through overexpression of PCBP1. (A) CAL 27 or SCC-9 cells with T7-PCBP1 stable overexpression were seeded into 12 well plates on Day 0. Cell numbers were counted on Day 2 and Day 4. (B) The histograms show apparent statistically significant difference between the PCBP1 overexpression and control groups on Day 4. Data are the means ± SE, n=3. (C, D) Effects of PCBP1 overexpression on the clonogenic ability of CAL 27 and SCC-9 cells. One thousand cells were seeded in 6 cm dishes and cultured for 10 days. Representative images are shown (C). (D) The histograms summarized the number of colonies. Data are the means ± SE, n =3. (E) Overexpression of STAT3α rescued cell growth inhibition induced by PCBP1 overexpression. CAL 27 cells were transfected with T7-PCBP1 expression lentivirus, STAT3α-GFP expression lentivirus, empty control lentivirus and/or GFP expression control lentivirus. Transfected cells were divided into four groups: T7-PCBP1 + GFP, Vector + GFP, T7-PCBP1 + STAT3, and Vector + STAT3. Cell number was counted at Day 2 and Day 4. (F) The histograms summarized the numbers of cells counted on Day 4. Data are the means ± SE, n =3. LV: lentivirus. (G) Western blot displayed the overexpression of exogenous T7 tagged PCBP1 and exogenous STAT3α-GFP fusion protein. GAPDH served as a loading control. (H-I) STAT3α overexpression rescues the inhibition of PCBP1 overexpression on the clonogenic ability of CAL 27 cells. One thousand cells were seeded into 6 cm dishes and cultured for 10 days. Representative images are shown (H). (I) The histograms summarized the numbers of colonies. Data are the means ± SE, n =3. (J) Expression levels of the indicated STAT3 target genes (Bcl-xl and survivin) were analyzed by RT-PCR. GAPDH served as a loading control. (K-M) STAT3α overexpression rescues the inhibition of PCBP1 overexpression on the tumorigenesis of CAL 27 cells. Transfected CAL 27 cells in panel (G) was inoculated into nude mice (5 × 105 cells per mice). (K-L) Tumors were dissected out and weighed on day 27. (M) Growth curves of tumors in nude mice (n = 5 mice in each group). Data are presented as mean ± SE. *P < 0.05 compared with PCBP1 + GFP group.
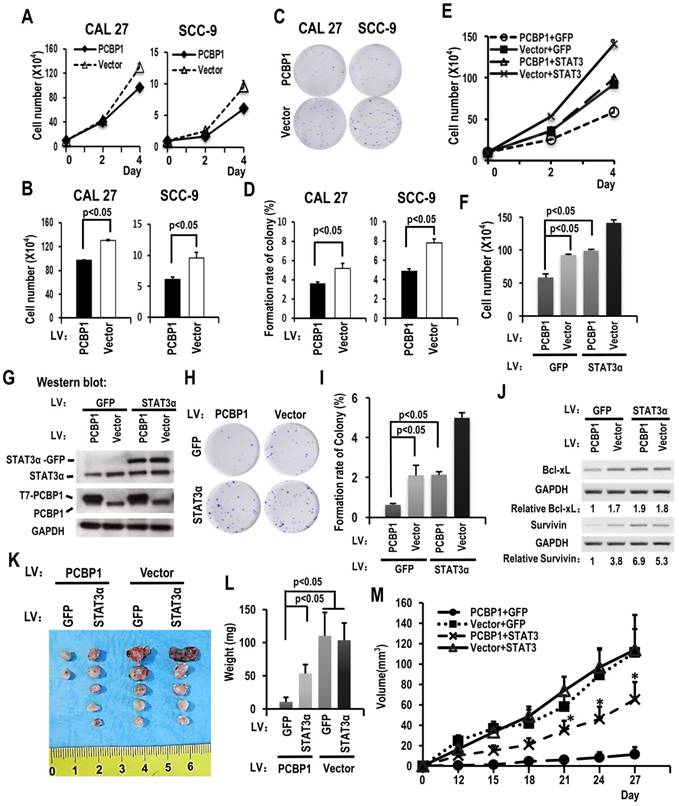
STAT3α rescues the cell proliferation and colony formation in PCBP1 overexpressed OSCC cells
To understand whether PCBP1 inhibits cancer cell proliferation through suppressing STAT3α expression, we established cell lines in CAL 27 as following: overexpression of PCBP1, overexpression of both PCBP1 and STAT3α, overexpression of STAT3α, or vector control. Western blot analysis confirmed the overexpression of T7 tagged PCBP1 and/or GFP tagged STAT3α (Figure 3G). Cell proliferation assay revealed that overexpression of STAT3α almost completely rescued cell growth and colony formation in PCBP1-overexpressed cells (Figure 3E-I). Moreover, overexpression of STAT3α can restore the expression of Bcl-xL and survivin in PCBP1-overexpressed cells (Figure 3J). Similar results were also observed in SCC-9 cells (Figure S3). These results demonstrated that PCBP1 inhibits cancer cell proliferation through inhibiting the expression of oncogene STAT3α.
STAT3α partially rescues the tumorigenesis in PCBP1 overexpressed OSCC cells
To understand whether PCBP1 inhibits the tumorigenesis of cancer cells through suppressing STAT3α expression. We also inoculated previous CAL 27 cells with overexpression of PCBP1, overexpression of both PCBP1 and STAT3α, overexpression of STAT3α, or vector control into nude mice. We found that overexpression PCBP1 significantly inhibited tumorigenesis. Co-overexpression of STAT3α can partially rescue tumor formation in PCBP1-overexpressed cells (Figure 3K-M). These results demonstrated that PCBP1 suppresses cancer cell tumorigenesis partially through inhibiting the expression of oncogene STAT3α.
PCBP1 inhibits the expression of STAT3α and cell proliferation in breast cancer cells
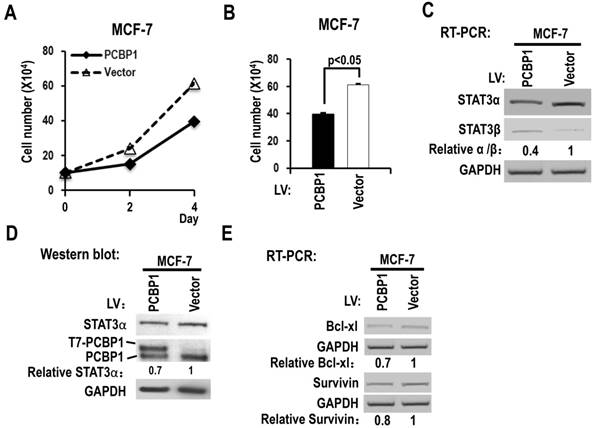
We also analyzed the functions of PCBP1 in MCF-7, a breast cancer cell line. We found that overexpression of PCBP1 significantly reduced the cell proliferation of MCF-7 cells (Figure 4A-B), decreased the ratio of STAT3α vs STAT3β isoform and the protein level of STAT3α (Figure 4C-D), and suppressed the expression of Bcl-xL and survivin compared with controls (Figure 4E). These results suggested that PCBP1-dependent STAT3α/STAT3β regulation may be a common activity in tumor cells.
Overexpression of PCBP1 reduces the ratio of STAT3α/β and suppresses the cell proliferation of a breast cancer cell line (MCF-7). (A, B) MCF-7 cells were stably transfected with T7 tagged PCBP1 or control lentivirus and seeded into 12 well plates on Day 0. Cell numbers were counted on Day 2 and Day 4 (A). (B) The histograms show significant difference between the PCBP1 overexpression and control groups on Day 4. Data are the means ± SE, n =3. (C) The alternative splicing of exon 23 was detected by RT-PCR. Relative α/β represents the ratio of band intensities of α vs β isoform. GAPDH served as a loading control. (D) Western blot showed the overexpression of T7 tagged PCBP1 and the expression level of cellular STAT3α. GAPDH served as a loading control. (E) RT-PCR analysis showed that overexpression of PCBP1 downregulates the expression of STAT3 targets (Bcl-xL and survivin) in MCF-7 cells.
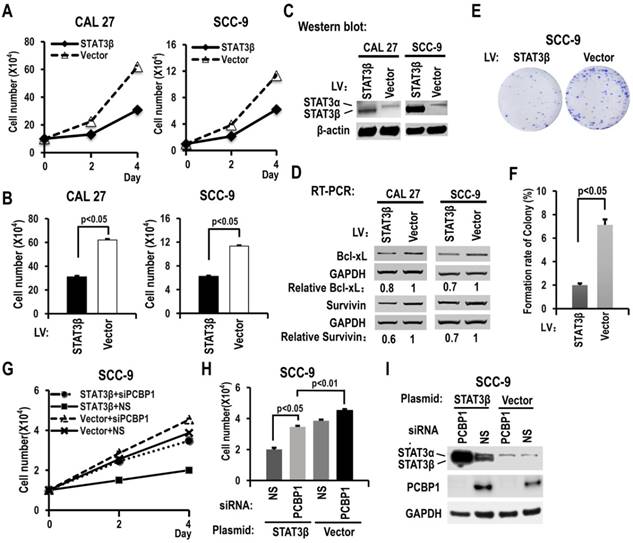
STAT3β inhibits cell proliferation and colony formation in OSCC cells, and the expression of Bcl-xL and survivin
To further understand the functions of STAT3β in OSCC, we overexpressed STAT3β in CAL 27 or SCC-9 cells. Overexpression of STAT3β significantly reduced cell proliferation and colony formation compared with controls (Figure 5A-C, 5E-F). The increased cell proliferation induced by knockdown of PCBP1 could be overcome by overexpression of STAT3β (Figure 5G-I). In addition, in contrast to STAT3α, we discovered that overexpression of STAT3β suppressed the expression of Bcl-xL and survivin (Figure 5D). These results suggested that STAT3β functions as a tumor suppressor in OSCC.
STAT3β inhibits the proliferation of OSCC cells and the expression of Bcl-xL and survivin. (A) CAL 27 or SCC-9 cells were stably transfected with STAT3β or control lentivirus. Cells were seeded into 12 well plates on Day 0. Cell numbers were counted on Day 2 and Day 4. (B) The histograms show statistically significant difference between the STAT3β overexpression and control groups on Day 4. Data are the means ± SE, n =3. (C) Western blot showed the overexpression of STAT3β. β-actin served as a loading control. (D) RT-PCR analysis showed that overexpression of STAT3β downregulates the expression of STAT3 targets (Bcl-xl and survivin) in both CAL 27 and SCC-9 cells. (E, F) Effect of STAT3β overexpression on the clonogenic ability of SCC-9 cells. One thousand cells were seeded in 6 cm dishes and cultured for 10 days. Representative images are shown (E). (F) The histograms summarized the numbers of colonies. Data are the means ± SE, n =3. (G) Overexpression of STAT3β decreased cell proliferation induced by PCBP1 knockdown in SCC-9 cells. SCC-9 cells were transfected with STAT3β, empty control plasmid (Vector), PCBP1 specific siRNA and/or non-specific (NS) siRNA. Transfected cells were divided to four groups: STAT3β + siPCBP1, STAT3β + NS, Vector + siPCBP1, and Vector + NS. Cell number was counted at Day 2 and Day 4. (H) The histograms summarized the numbers of cells counted on Day 4. Data are the means ± SE, n =3. (I) Western blot displayed the overexpression of STAT3β and knockdown efficiency of PCBP1. GAPDH served as a loading control.
Discussion
A number of inhibitors have been developed to block the oncogenic activation of STAT3 [2]. STAT3β shared coiled-coil domain (CCD), DNA-binding domain (DBD) and SH2 domain with STAT3α. Therefore, small molecule inhibitors targeting these domains will block both STAT3α's and STAT3β's functions. Although STAT3β is missing the transactivation domain and generally considered as a dominant negative form, it can play unique functions and rescue the embryonic lethality of a STAT3-null mutation mice [21]. So, the inhibitors targeting STAT3 may cause unexpected side effects due to repressing the function of STAT3β. STAT3β has been found to be a tumor suppressor [7, 8]. In line with previous studies, we also found that STAT3β represses the cell proliferation and colony formation of OSCC cells. To reduce side effects and enhance anti-tumor capability, it may be useful to develop novel methods to promote the shift from STAT3α to STAT3β by modulating the alternative splicing of exon 23 in cancer cells. Zammarchi et al. have shown that antisense oligonucleotide against proximal splice site of exon 23 can drive the shift from STAT3α to STAT3β and suppress tumor formation [10]. Our results demonstrated that increasing the expression of PCBP1 also promotes the shift from STAT3α to STAT3β and inhibits cell proliferation in both oral cancer cells and breast cancer cells, and may be a novel anti-tumor method.
In the screening experiment (Figure 1B-C), we discovered that all mutants between proximal and distal splice sites abolished the expression of STAT3β. In addition, mt7, a downstream mutant, also reduced the expression of STAT3β. By using SFmap online program, we found that splicing factor QKI might bind to the wild-type sequence of mt7 through an AAUAAU motif. Interestingly, QKI was considered as a tumor suppressor [22]. Therefore, QKI may increase the expression of STAT3β by binding to AAUAAU motif. However, we found that overexpression of QKI-6 couldn't increase STAT3β expression (Figure S1). QKI has three isoforms (QKI-5, QKI-6, and QKI-7). Further experiments are required to clarify the functions of QKI isoforms in the alternative splicing of STAT3 exon 23.
Head and neck squamous cell carcinoma is the sixth most common cancer in the world. Oral squamous cell carcinoma (OSCC) is the most common type of head and neck squamous cell carcinoma. STAT3 is significantly upregulated and constitutively activated in head and neck squamous cell carcinoma [23]. Constitutive activation of STAT3 was also found to be an early event in head and neck carcinogenesis [24]. A p-STAT3 inhibitor significantly delayed tumorigenesis in a spontaneous head and neck squamous cell carcinoma mouse model [23], suggesting STAT3 can be a therapeutic target for OSCC. However, like other cancers, there are few effective therapeutic methods to inhibit STAT3 in the treatment of OSCC. In this study, we found that overexpression of PCBP1 inhibited the expression of oncogenic STAT3α and repressed the cell proliferation of OCSS cells. Therefore, upregulation of PCBP1 may be a promising novel method to inhibit STAT3 in OSCC. In addition, we found that overexpression of PCBP1 also significantly repressed cell proliferation and decreased the expression of oncogenic STAT3α in breast cancer cell line, MCF-7. STAT3 is constitutively activated in more than 40% of breast cancer and plays key roles in cell proliferation, angiogenesis, apoptosis, and metastasis in breast cancer [25]. Our results may pave the way to novel options for the treatment of breast cancer or other types of cancer by upregulation of PCBP1.
Activation of STAT3 suppresses the expression of proapoptotic genes in cancer cells [26]. STAT3 also promotes the expression of anti-apoptotic genes, such as Bcl-xL and survivin. Bcl-xL is a member of the Bcl-2 family proteins, and plays central roles in suppressing apoptosis [27]. Survivin is a member of the inhibitor of apoptosis protein family, and could block apoptosis through suppressing activation of caspases [28]. It has been reported that overexpression of PCBP1 induced apoptosis by downregulating Bcl-2 [29]. Our results discovered that PCBP1 is also able to inhibit the expression of Bcl-xL and survivin induced by STAT3, suggesting a novel mechanism, by which PCBP1 induces apoptosis.
In conclusion, PCBP1 is the key splicing factor that regulates alternative splicing of STAT3 exon 23 and promotes the switch from oncogenic isoform STAT3α to tumor-suppressive isoform STAT3β. Our results may initiate new methods for anti-STAT3 treatment in cancers.
Materials and Methods
Cell culture
Normal gingival epithelial cells were derived from gingival tissues of healthy donors and cultured in keratinocyte growth medium (KGM, Lonza, Switzerland) as described in our previous study [30]. HEK-293 cell and OSCC cell line CAL 27 cell were grown in Dulbecco's modified Eagle medium (DMEM; HyClone, USA) with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic (Gibco, USA). OSCC cell line SCC-9 cell was cultured in DMEM/F12 with 10% FBS and 1% antibiotic-antimycotic (Gibco, USA) and 1‰ hydrocortisone. MCF-7 cell, a breast cancer cell line, was grown in Eagle's Minimum Essential Medium (EMEM) (HyClone, USA) with 10% FBS and 1% antibiotic-antimycotic. Cell culture is maintained at 37°C in 5% CO2. All experimental protocols were approved by the Ethics Committee at the School of Stomatology in Wuhan University. The methods were performed in accordance with the approved guidelines.
Plasmids
Genomic DNA sequence from exon 22 to exon 23 of human STAT3 was amplified from the CAL 27 genome by primer 5' CGCTGCCCCATACCTGAAGAC 3' and 5' GGTTCAGCACCTTCACCATTATTTC 3'. The PCR product was further amplified by 5' GAATTCACCATGGCTGCCCCATACCTGAAGAC 3' and 5' GGATCCGGTTCAGCACCTTCACCATTATTTC 3', and cloned into pEGFP-N1 at BamHI and EcoRI sites. The resulting minigene plasmid carried genomic sequence of STAT3 from exon 22 to exon 23. To map potential regulatory motifs, the sequence from nt 5 to 88 of exon 23 in minigene was serially mutated every 10 bases. The mutation was made by substituting a base by its complementary base every other base (Figure 1B). For STAT3α overexpression, the open reading frame of human STAT3α isoform was cloned into pLVX-IRES-PURO vector at NotI and SpeI sites. For STAT3β overexpression, the open reading frame of human STAT3β isoform was cloned into pLVX-IRES-PURO vector at NotI and SphI sites. The open reading frame of human PCPB1 was obtained from CAL 27 cells and fused with T7 tag, and then cloned into pLVX-IRES-Zsgreen vector at NotI and EcoRI sites. Plasmids expressing Flag tagged HuB, HuC, HuD, and QKI-6 were gifts from Dr. Thomas Tuschl (Addgene plasmid #65755, #65756, #65757, and #19891) [31]. Plasmid expressing Flag tagged FUS was a gift from Dr. Patrick Calsou (Addgene plasmid #60362) [32]. Plasmid expressing Flag tagged hnRNP F and hnRNP H1 were gifts from Dr. Mariano Garcia-Blanco (Addgene plasmid #21926 and #21925) [33]. Plasmid expressing Flag tagged hnRNP R was a gift from Dr. Markus Landthaler (Addgene plasmid #38067) [4]. Plasmid expressing Flag tagged SRm160 was a gift from Dr. Phillip Sharp (Addgene plasmid #11305) [34]. Plasmid expressing Flag tagged SF3B1 was a gift from Dr. Manoj Pillai (Addgene plasmid #82576) [35]. Plasmid expressing GFP tagged FOX1, NOVA1 and FOX2 were gifts from Dr. Nicolas Charlet-Berguerand (Addgene plasmid #63085, #61275 and #63086). Plasmid expressing GFP tagged RBM3 was a gift from Dr. Maria Alvarado-Kristensson (Addgene plasmid #87860) [36]. Plasmid expressing GFP tagged FLKSRP was a gift from Dr. Douglas Black (Addgene plasmid #23001) [37]. Plasmid expressing Myc tagged YBX1 was a gift from Dr. Thomas Tuschl (Addgene plasmid #19878) [38]. Plasmid expressing Myc tagged PSF was a gift from Dr. Benjamin Blencowe (Addgene plasmid #35183) [39]. Plasmid expressing Flag tagged SC35 was a gift from Dr. Zhi-Ming Zheng. T7 tagged PTBP1, hnRNP A1, and hnRNP L expressing plasmids were made in our previous publications [30, 40, 41]. HnRNP K gene was amplified from CAL 27 cells by 5' TAATAACTGATTGGTGTGCCCG 3' and 5' AAACTTTCCAGAATACTGCTTCACAC and fused with T7 tag, and then cloned into pLVX-IRES-PURO vector at NotI and SpeI sites.
RNAi and transfection
The sequence of human PCBP1 siRNA is 5' GAACCAGGUGGCAAGACAA 3', and was synthesized by Genepharma (Shanghai, China). Primary normal oral mucosal epithelial cells were transfected with 20 nM siRNA in the presence of Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer's instructions. After 48 hours, cells were transfected again.
For plasmid transfection, 293 cells were transfected with 1 µg T7 tagged PCBP1 expression or empty vector plasmid in the presence of Lipofectamine 3000 24 hours later after the cells adhered to the wall. Total protein and RNA were collected 48 hours later.
CAL 27 cell and SCC-9 cell were stably transfected with T7-tagged PCBP1 expression lentivirus, STAT3-GFP expression lentivirus, empty control lentivirus and/or GFP expression control lentivirus in the presence of Lipofectamine 3000.
Western blot
Total protein samples were lysed with 2 × SDS sample buffer, and then denatured proteins were loaded onto 10% SDS-PAGE gel, transferred to a nitrocellulose membrane, and blocked by 5% non-fat milk dissolved in TBST (Tris-buffered saline, 0.1% Tween 20). The protein samples were analyzed by the following antibodies: mouse anti-PCBP1 (Origene, USA), mouse anti-STAT3 (no. 610189; BD Biosciences, USA), mouse anti-phospho-specific STAT3 (pY705, Santa Cruz Biotechnology, USA), mouse anti-GAPDH (Santa Cruz Biotechnology), mouse anti-GFP (Santa Cruz Biotechnology), mouse anti-Flag (Sigma-Aldrich, USA), mouse anti-Myc (Yeasen, Shanghai, China) or mouse anti-T7 (Novagen, USA).
Reverse transcriptase PCR (RT-PCR)
Total RNA was extracted using the Total RNA Miniprep Kit (AxyPrep, USA) according to the manufacturer's protocol. Isolated RNA was reversely transcribed by using random hexamers (Promega, USA) and Moloney Murine Leukemia Virus Reverse Transcriptase (MMLV, Promega, USA), and amplified with Taq DNA polymerase (Takara, Japan) as described by the manufacturer. The primer pairs were as follows: 5' CGCTGCCCCATACCTGAAGAC 3' and 5′ GCTCCTCGCCCTTGCTCACCA 3′ for exogenous exon 23 alternative splicing detection in the minigene; 5' ATCGGCAGGTCAATGGTATTGC 3′ and 5' AAGGAGGAGGCATTCGGAAAGTA 3′ for endogenous STAT3α; 5' CCAAACTGCATCAATGAATGGTGT 3′ and for endogenous STAT3β; 5' CAGTGCGGCTCCCTGATTG 3′ and 5' CCTCTGGAGAGCTGGAGTCAATTC 3′ for PCBP1; 5′ GTGAATTTTTGAAACTGGACAG 3′ and 5′ CCTTTCCTAAGACATTGCTAAG 3′ for survivin; 5′ ACCCCAGGGACAGCATATCA 3′ and 5′ TGCGATCCGACTCACCAATA 3′ for Bcl-xL; 5′ GTCATCAATGGAAATCCCATCACC 3′ and 5′ TGAGTCCTTCCACGATACCAAA 3′ for GAPDH.
RNA pull-down assay
Biotin-labeled STAT3 RNA oligonucleotides wt-4 (5′ Biotin-CGAUGUCCCCCCGCACUUTT 3′ wild type), mt-4 (5′Biotin-CGAUGACGCGCGGCACUUTT 3' mutant, mutation was underlined), and PCBP1+ (5′ Biotin- CCUAUCCCUCUCCUAACUTT 3′, positive PCPB1 binding motif [42]) were synthesized by Sangon Biotech (China). The total HEK 293 cell extract was prepared by using radioimmunoprecipitation assay (RIPA) buffer (Thermo scientific, USA). Five microliter biotin-labeled RNA oligonucleotide (40μM) was immobilized onto 100μl of NeutrAvidin beads (Pierce, USA). The bead-RNA complex was then incubated with total cell extract of HEK 293 cells in a 1× binding buffer (20mM Tris, 200mM NaCl, 6mM EDTA, 5mM potassium fluoride, 5mM β-glycerophosphate, 2μg/ml aprotinin, pH 7.5) at 4°C for 2h. The proteins associated with RNA were eluted by sodium dodecyl sulfate sample buffer and analyzed by using western blot.
Colony formation assay
One thousand cells were seeded into 60-mm cell culture dishes and cultured in complete medium for 10 days at 37°C. Then, cells were fixed with 4% paraformaldehyde and stained with crystal violet. The number of colonies (at least 50 cells/colony) was counted.
Tumor induction in nude mice
CAL 27 cells stably transfected with T7-PCBP1 expression lentivirus, STAT3-GFP expression lentivirus, empty control lentivirus and/or GFP expression control lentivirus were injected subcutaneously into nude mice (5 × 105 cells per mice, 5 mice per group). Tumor sizes were monitored every 3 days. Tumors were allowed to grow for 27 days and then the mice were sacrificed and tumors were dissected out and weighed. All methods of the animal studies were conducted in accordance with the relevant institutional guidelines.
Statistical analysis
All two-group statistical comparisons of means were calculated with student's t-test (Excel, Microsoft).
Abbreviations
STAT3: Signal transducers and activators of transcription 3; OSCC: Oral squamous cell carcinoma; ESS: exonic splicing suppressor.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by supported by Grant 81470741 and 81571024 from the National Natural Science Foundation of China. This work was also supported by Grant WJ2019Z014 from Health Commission of Hubei Province and Grant 2016CFA067 from Hubei Provincial Natural Science Foundation of China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yu H, Lee H, Herrmann A. et al. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736-46
2. Beebe JD, Liu JY, Zhang JT. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol Ther. 2018;191:74-91
3. Mali SB. Review of STAT3 (Signal Transducers and Activators of Transcription) in head and neck cancer. Oral Oncol. 2015;51(6):565-9
4. Baltz AG, Munschauer M, Schwanhausser B. et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46(5):674-90
5. Yang J, Liao X, Agarwal MK. et al. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21(11):1396-408
6. Ng IH, Ng DC, Jans DA. et al. Selective STAT3-alpha or -beta expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochem J. 2012;447(1):125-36
7. Niu G, Shain KH, Huang M. et al. Overexpression of a dominant-negative signal transducer and activator of transcription 3 variant in tumor cells leads to production of soluble factors that induce apoptosis and cell cycle arrest. Cancer Res. 2001;61(8):3276-80
8. Marino F, Orecchia V, Regis G. et al. STAT3beta controls inflammatory responses and early tumor onset in skin and colon experimental cancer models. Am J Cancer Res. 2014;4(5):484-94
9. Zhang HF, Chen Y, Wu C. et al. The opposing function of STAT3 as an oncoprotein and tumor suppressor is dictated by the expression status of STAT3beta in esophageal squamous cell carcinoma. Clin Cancer Res. 2016;22(3):691-703
10. Zammarchi F, de Stanchina E, Bournazou E. et al. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc Natl Acad Sci U S A. 2011;108(43):17779-84
11. Chaudhury A, Chander P, Howe PH. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1's multifunctional regulatory roles. RNA. 2010;16(8):1449-62
12. Guo J, Jia R. Splicing factor poly(rC)-binding protein 1 is a novel and distinctive tumor suppressor. J Cell Physiol. 2018;234(1):33-41
13. Shi H, Bencze KZ, Stemmler TL. et al. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008;320(5880):1207-10
14. Chen Q, Cai ZK, Chen YB. et al. Poly r(C) binding protein-1 is central to maintenance of cancer stem cells in prostate cancer cells. Cell Physiol Biochem. 2015;35(3):1052-61
15. Zhang Y, Meng L, Xiao L. et al. The RNA-Binding protein PCBP1 functions as a tumor suppressor in prostate cancer by inhibiting mitogen activated protein kinase 1. Cell Physiol Biochem. 2018;48(4):1747-54
16. Wang H, Vardy LA, Tan CP. et al. PCBP1 suppresses the translation of metastasis-associated PRL-3 phosphatase. Cancer Cell. 2010;18(1):52-62
17. Zhang ZZ, Shen ZY, Shen YY. et al. HOTAIR long noncoding RNA promotes gastric cancer metastasis through suppression of poly r(C)-Binding protein (PCBP) 1. Mol Cancer Ther. 2015;14(5):1162-70
18. Shi H, Li H, Yuan R. et al. PCBP1 depletion promotes tumorigenesis through attenuation of p27(Kip1) mRNA stability and translation. J Exp Clin Cancer Res. 2018;37(1):187
19. Zhang M, Wang X, Tan J. et al. Poly r(C) binding protein (PCBP) 1 is a negative regulator of thyroid carcinoma. Am J Transl Res. 2016;8(8):3567-73
20. Ji X, Park JW, Bahrami-Samani E. et al. AlphaCP binding to a cytosine-rich subset of polypyrimidine tracts drives a novel pathway of cassette exon splicing in the mammalian transcriptome. Nucleic Acids Res. 2016;44(5):2283-97
21. Maritano D, Sugrue ML, Tininini S. et al. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol. 2004;5(4):401-9
22. Mo HY, Jo YS, Yoo NJ. et al. Frameshift mutation of candidate tumor suppressor genes QK1 and TMEFF2 in gastric and colorectal cancers. Cancer Biomark. 2019;24(1):1-6
23. Bu LL, Zhao ZL, Liu JF. et al. STAT3 blockade enhances the efficacy of conventional chemotherapeutic agents by eradicating head neck stemloid cancer cell. Oncotarget. 2015;6(39):41944-58
24. Grandis JR, Drenning SD, Zeng Q. et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A. 2000;97(8):4227-32
25. Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: A review. Int J Cancer. 2016;138(11):2570-8
26. Timofeeva OA, Tarasova NI, Zhang X. et al. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc Natl Acad Sci U S A. 2013;110(4):1267-72
27. Catlett-Falcone R, Landowski TH, Oshiro MM. et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105-15
28. Gritsko T, Williams A, Turkson J. et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res. 2006;12(1):11-9
29. Zhang W, Shi H, Zhang M. et al. Poly C binding protein 1 represses autophagy through downregulation of LC3B to promote tumor cell apoptosis in starvation. Int J Biochem Cell Biol. 2016;73:127-36
30. Guo J, Jia J, Jia R. PTBP1 and PTBP2 impaired autoregulation of SRSF3 in cancer cells. Sci Rep. 2015;5:14548
31. Landthaler M, Gaidatzis D, Rothballer A. et al. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA. 2008;14(12):2580-96
32. Britton S, Dernoncourt E, Delteil C. et al. DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 2014;42(14):9047-62
33. Mauger DM, Lin C, Garcia-Blanco MA. HnRNP H and hnRNP F complex with Fox2 to silence fibroblast growth factor receptor 2 exon IIIc. Mol Cell Biol. 2008;28(17):5403-19
34. Cheng C, Sharp PA. Regulation of CD44 alternative splicing by SRm160 and its potential role in tumor cell invasion. Mol Cell Biol. 2006;26(1):362-70
35. Kesarwani AK, Ramirez O, Gupta AK. et al. Cancer-associated SF3B1 mutants recognize otherwise inaccessible cryptic 3' splice sites within RNA secondary structures. Oncogene. 2017;36(8):1123-33
36. Rossello CA, Lindstrom L, Glindre J. et al. Gamma-tubulin coordinates nuclear envelope assembly around chromatin. Heliyon. 2016;2(9):e166
37. Hall MP, Huang S, Black DL. Differentiation-induced colocalization of the KH-type splicing regulatory protein with polypyrimidine tract binding protein and the c-src pre-mRNA. Mol Biol Cell. 2004;15(2):774-86
38. Landthaler M, Gaidatzis D, Rothballer A. et al. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA. 2008;14(12):2580-96
39. Rosonina E, Ip JY, Calarco JA. et al. Role for PSF in mediating transcriptional activator-dependent stimulation of pre-mRNA processing in vivo. Mol Cell Biol. 2005;25(15):6734-46
40. Yu C, Guo J, Liu Y. et al. Oral squamous cancer cell exploits hnRNP A1 to regulate cell cycle and proliferation. J Cell Physiol. 2015;230(9):2252-61
41. Jia R, Zhang S, Liu M. et al. HnRNP L is important for the expression of oncogene SRSF3 and oncogenic potential of oral squamous cell carcinoma cells. Sci Rep. 2016;6:35976
42. Cho SJ, Jung YS, Chen X. Poly (C)-binding protein 1 regulates p63 expression through mRNA stability. Plos One. 2013;8(8):e71724
Author contact
![]() Corresponding author: Rong Jia, 237 Luoyu Road, Wuhan City, 430079, PR China. E-mail address: jiarongedu.cn Phone number: 86-27-87686268
Corresponding author: Rong Jia, 237 Luoyu Road, Wuhan City, 430079, PR China. E-mail address: jiarongedu.cn Phone number: 86-27-87686268