Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Molecular hydrogen against organ...
Molecular hydrogen reduces the...
The biological effects and...
Effective concentration and dose...
Conclusion and perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(6):1261-1275. doi:10.7150/ijbs.30741 This issue Cite
Review
Recent Advances in Studies of Molecular Hydrogen against Sepsis
Peng Qiu1, Yang Liu2, Jin Zhang1 ![]()
1. Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang, China
2. Department of Oncology, Shengjing Hospital of China Medical University, Shenyang, China
Received 2018-11-27; Accepted 2019-1-9; Published 2019-5-11
Abstract

Sepsis is a syndrome comprised of a series of life-threatening organ dysfunctions caused by a maladjusted body response to infection with no effective treatment. Molecular hydrogen is a new type of antioxidant with strong free radical scavenging ability, which has been demonstrated to be effective for treating various diseases, such as infection, trauma, poisoning, organ ischemia-reperfusion, metabolic diseases, and tumors. Molecular hydrogen exerts multiple biological effects involving anti-inflammation, anti-oxidation, anti-apoptosis, anti-shock, and autophagy regulation, which may attenuate the organ and barrier damage caused by sepsis. However, the underlying molecular mechanisms remain elusive, but are likely related to the signaling pathways involved. This review focuses on the research progress and potential mechanisms of molecular hydrogen against sepsis to provide a theoretical basis for clinical treatment.
Keywords: molecular hydrogen, sepsis, oxidative stress, apoptosis, shock, autophagy
Introduction
Sepsis is a serious systemic inflammatory response syndrome caused by infection, which is characterized by an explosive inflammatory reaction, oxidative stress, and immune system disorder, that eventually leads to septic shock and multiple organ failure[1]. At present, there are several treatments for sepsis, such as anti-inflammation and anti-shock. However, there is no definite therapeutic effect and the mortality rate remains high.
Molecular hydrogen was first reported in Nature Medicine (2007) as a natural antioxidant and selective scavenger of oxygen free radicals to treat oxidative stress [2]. A large number of studies have subsequently used it to treat various types of diseases, including infection, trauma, metabolic diseases, organ ischemia-reperfusion injury, and tumors and have achieved satisfactory efficacy[3]. Molecular hydrogen provides a variety of advantages for treating diseases due to its unique physical and chemical properties. There are three main forms of molecular hydrogen for research: hydrogen rich saline/water (HRS/W), inhaled hydrogen and hydrogen rich medium (HRM). Molecular hydrogen is safe, non-toxic and can balance the pH of body fluids[4]. Because of its small molecular weight, molecular hydrogen spreads easily and penetrates membranes into the cytoplasm, mitochondria, and even the nucleus [4]. Molecular hydrogen is non-flammable and non-explosive at the therapeutic concentration. Its effect is moderate and its metabolites are non-toxic. Although it has antioxidant capacity, molecular hydrogen does not interfere with normal metabolism or redox reactions [5]. In addition, molecular hydrogen promotes cell detoxification, increases cell hydration, and strengthens the host immune system[5].
Current studies have indicated that molecular hydrogen exerts its biological effects in two ways, one of which is reacting with hydroxyl radicals and peroxynitrite directly, and the other is modulating specific gene expression or signaling pathways[3] (Fig. 1).
Molecular hydrogen exerts biological effects. Molecular hydrogen can be ingested in a variety of ways and exerts biological effects, including anti-oxidation, anti-inflammation, anti-apoptosis, anti-shock, and autophagy regulation through scavenging free radicals directly and regulating signal transduction and gene expression indirectly.
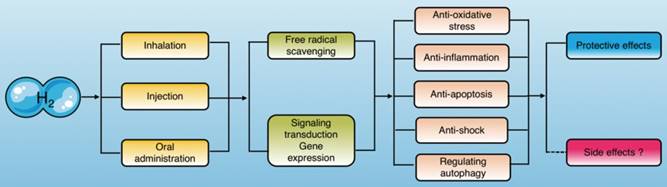
Based on these therapeutic advantages, molecular hydrogen has been widely used in studies of organ protection during sepsis in recent years, and has yielded ideal results. Molecular hydrogen attenuates the injury and dysfunction of important organs (heart, liver, lung, kidneys, and brain) and physiological barriers (epithelial cell barrier, vascular endothelial cell barrier) by suppressing oxidative stress and inflammation as well as reducing apoptosis [6-10] and regulating sepsis-induced autophagy[11, 12]. However, the underlying molecular mechanisms of these effects have not been elucidated. Therefore, understanding the research status of molecular hydrogen against sepsis and the underlying mechanisms are of great significance for treating sepsis.
Molecular hydrogen against organ injury induced by sepsis
Lipopolysaccharide (LPS) is a component of the cell wall of Gram-negative bacteria and the most important pathogenic factor in sepsis. It has been demonstrated that molecular hydrogen extenuates LPS-induced ALI in rats by reducing the release of inflammatory factors, inhibiting the aggregation of inflammatory cells, reducing oxidative stress and apoptosis[8, 11, 13]. Moreover, molecular hydrogen alleviates pulmonary edema caused by LPS through upregulating the expression of pulmonary aquaporin (AQP)[14]. These lung protective effects are thought to be associated with a reduction in LPS-induced p38 mitogen-activated protein kinase (p38 MAPK) and c-Jun N-terminal kinase (JNK) activation by molecular hydrogen[11, 14, 15]. In addition to protecting the mature lung, HRS also alleviates bronchopulmonary dysplasia (BPD) induced by LPS in neonatal mice[16]. Fibroblast growth factor receptor 4 (FGFR4) and vascular endothelial growth factor receptor 2 (VEGFR2) are important for maintaining alveolar structures and lung development[17, 18]. Oral intake of HRS ameliorates LPS-induced suppression of genes encoding FGFR4, VEGFR2, and heme oxygenase 1 (HO-1) in neonatal mice[16]. In addition, study showed[19] that LPS promotes the alveolar epithelial-mesenchymal transition (EMT) and pulmonary fibrosis by increasing the production of reactive oxygen species (ROS) and transforming growth factor-β (TGF-β). HRS alleviates oxidative stress and pulmonary fibrosis by reducing LPS-induced E-cadherin loss and α-smooth muscle actin production[20].
The liver is the most important organ for removing cytotoxic substances from the body, but may become overloaded by sepsis and exhibit injury and dysfunction[6]. A series of studies have reported that HRS reduces liver damage caused by endotoxin in rats [6, 21, 22]. Iketani et al. found that HRS Alleviates liver injury induced by oxidative stress through further increasing LPS-induced HO-1 expression and decreasing endothelin-1 (ET-1) expression [6]. Xu et al. demonstrated that HRS mitigates the pathological injury of the septic rat liver and improves survival rate by reducing the release of inflammatory cytokines and reducing hepatocyte apoptosis and oxidative stress[22]. Inhibiting signaling pathways, such as p38 MAPK, JNK, extracellular regulated protein kinase (ERK), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and reducing the second mitochondria- derived activator of caspase (Smac) level contribute to HRS-mediated liver protection[22].
Multiple studies[23-25] have demonstrated that HRS protects renal function by reducing serum creatinine and urea nitrogen levels and relieving renal structural damage caused by sepsis. Liu et al. reported that inhaling molecular hydrogen alleviates brain damage and cognitive dysfunction in septic rats by inhibiting the neuronal inflammatory response and oxidative stress, while neuronal apoptosis is reduced by increasing the expression of nuclear factor erythroid 2-related factor 2 (Nrf2) and HO-1[26]. In the septic pancreatitis rat model, Zhang et al. found that HRS exerts anti-inflammation, anti-oxidant, and antibacterial effects by inhibiting the NF-κB signaling pathway[27]. It has been demonstrated that myocarditis and insufficient energy production in the myocardium are the main causes of heart dysfunction caused by sepsis[28]. Tao et al. reported that LPS challenge increases left ventricular diameter and reduces fractional shortening and fatty acid oxidation (FAO)-related gene expression in rats, which can be attenuated by HRS[7]. However, HRS has little effect on myocardial glucose metabolism in septic rats[7]. Part of the protective mechanism of HRS on LPS-induced myocardial injury might be that HRS inhibits activation of the JNK signaling pathway, restores FAO, and increases myocardial energy production[7]. Moreover, one study showed that HRS reduces endotoxin-induced uveitis by inhibiting the aqueous humor protein[10]. However, HRS could neither effectively reduce iridocyclitis infiltration nor restore retinal function[10].
Molecular hydrogen reduces the physiological barrier dysfunction caused by sepsis
Physiological barrier dysfunction is one of the pathological manifestations of sepsis [9, 29, 30]. Vascular endothelial cells, epithelial cells, and intercellular junctions are important components of physiological barrier[31, 32]. Adherens junctions (AJ), represented by cadherin, and tight junctions (TJ), represented by occluding, are the main cell junctions involved in formation of barriers[31, 32]. Components of the barrier become damaged during sepsis and normal intercellular junctions are destroyed, which leads to increased barrier permeability[29, 30, 33]. Subsequently, proteins or liquids leak out of the blood vessels into tissues, leading to edema and hypoproteinemia, which aggravate endotoxin shock[34].
Many studies have investigated how molecular hydrogen alleviates the physiological barrier damage caused by sepsis, including protective effects on vascular endothelial cells[30, 35, 36], epithelial cells (e.g., alveoli and gastrointestinal epithelium)[9, 11, 29, 37] and intercellular junctions[9, 30, 33, 38], which are characterized by decreased permeability, increased transmembrane resistance, and regulation of expression and distribution of intercellular junction- related proteins.
Molecular hydrogen improves LPS-induced hyperpermeability of the vascular endothelium, represented by an increase in trans-endothelial electrical resistance (TEER)[30, 38] and a decrease in fluorescein isothiocyanate-dextran (FITC-dextran) flux of endothelial cells[38]. Molecular hydrogen stabilizes the AJ between endothelial cells and reduces barrier permeability by reducing internalization of VE-cadherin[38] and increasing the expression of VE-cadherin in vivo[35] and in vitro[36, 38].
Similar to vascular endothelial cells, HRS has a protective effect on epithelial cells. Studies by Zhang et al.[11] and Liu et al.[37] have demonstrated that HRS attenuates the alveolar epithelial barrier damage caused by LPS, improves alveolar gas exchange, and reduces cell damage caused by alveolar epithelial cell apoptosis and excessive autophagy. Yang et al.[9] and Ikeda et al.[29] reported that molecular hydrogen also protects the gastrointestinal epithelial barrier during sepsis. LPS can disrupt barrier integrity by reducing trans-epithelial resistance [9, 33] and increasing FITC-dextran flux in a concentration-dependent manner[9], which can be attenuated by molecular hydrogen through elevating LPS-induced downregulated expression of occludin and E-cadherin[9].
According to current research results[9, 30, 35, 36, 38], molecular hydrogen protects physiological barriers mainly through the following mechanisms: regulating signaling of the Ras homolog gene family member A (RhoA) and its effector protein kinases involving mammalian diaphanous-related formin 1 (mDia1) and Rho-associated coiled-coil protein kinase (ROCK); and by reducing adhesion of inflammatory cells.
The Rho GTPases are a family of small signaling G proteins and a subfamily of the Ras superfamily. Members of the Rho GTPase family (Cdc42, Rac1, and RhoA) regulate many aspects of intracellular actin dynamics, which are essential for the formation of AJ and TJ[39]. It has been demonstrated that the Rho GTPase family has an important effect on stability and integrity of the physiological barrier by regulating cell junctions[40](Fig. 2). The baseline activities of Rac1, Cdc42, and RhoA are required to maintain the integrity of epithelial and endothelial barriers[30, 40]. However, additional activation of Rac1 and Cdc42 can exert a role in barrier stabilization[40], while increased stimulation of RhoA has a dual effect on barrier permeability[40-42] (Fig. 2).
The role and mechanisms of Rho GTPase in regulating physiological barriers. Cdc42 and Rac1 in the Rho GTPase family stabilize physiological barriers after activation, while RhoA has dual effects on the barrier. Moderate RhoA activation selectively activates mDia1 signaling to protect the barrier, while excessive activation leads to ROCK-mediated barrier disruption. Cdc42: cell division control protein 42 homolog; Rac1: Ras-related C3 botulinum toxin substrate 1; RhoA: Ras homolog gene family member A; mDia1: mammalian diaphanous-related formin 1; ROCK: Rho-associated coiled-coil protein kinase.
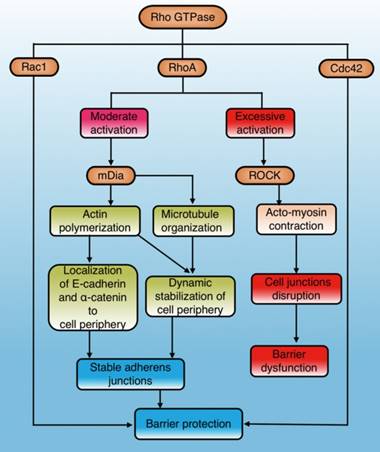
RhoA is the most studied molecule in the Rho family and its main effectors are ROCK and mDia1. ROCK promotes the contraction of myosin and aggregation of actin, while mDia1 promotes aggregation of actin and organization of microtubules [41, 43]. Actin aggregation is important for correct localization of AJ components, and microtubule organization stabilizes the cell periphery[41]. RhoA/ROCK regulates cell adhesion, migration, proliferation, and apoptosis by controlling the arrangement of the actin skeleton and cell contraction[44]. RhoA/mDia plays an important role in the localization of E-cadherin at the cell junction and the formation of AJ[45, 46]. Gavard et al. reported that both intensity and space distribution of active RhoA impact the downstream signaling options [42]. Moderate activation of RhoA selectively activates mDia signaling to stabilize cell junctions and the periphery, whereas excessive activation tends to cause ROCK-dependent myosin contraction and disrupt cell junctions[40, 41] (Fig. 2).
Studies have demonstrated that LPS leads to endothelial hyperpermeability by decreasing VE- cadherin expression[47] and increasing phosphorylation of myosin light chain [48-50] by activating the RhoA/ROCK signaling pathway. In contrast, HRM has been demonstrated to reduce LPS-induced neutrophil/polymorphonuclear cell (PMN) adhesion to endothelial cells, increase the expression of vascular cell adhesion molecule-1 (VCAM-1) and E-selectin, and improve the decreased TEER and VE-cadherin expression in endothelial cells by inhibiting the Rho/ROCK signaling pathway[30]. In addition, molecular hydrogen improves the decreased expression of mDia induced by LPS in epithelial cells[9]. Inhibiting RhoA or knockdown of mDia eliminates the molecular hydrogen-induced benefits of occludin, E-cadherin, and AJ[9], indicating that molecular hydrogen exerts a protective effect on the epithelial barrier through RhoA or mDia signaling. As strong proof, researchers have revealed that molecular hydrogen reduces LPS-induced excessive activation of RhoA to a moderate level and increases the expression of mDia; thus, preventing destruction of TJ and AJ and alleviating intestinal epithelial barrier dysfunction[9].
Excessive numbers of inflammatory cells during sepsis activate and adhere to endothelial cells through adhesion molecules, which subsequently leads to endothelial cell apoptosis, disruption of cell junctions, cell remodeling, and other damage[30]. Studies have shown that LPS increases the production of multiple adhesion molecules, including VCAM-1, intercellular adhesion molecule-1 (ICAM-1) and E-selectin, which increase adhesion and aggregation of monocytes to vascular endothelial cells and induce decomposition of TJ or redistribution of VE-cadherin[30, 36, 38]. Instead, HRM reduces the damage of endothelial cells and AJ by downregulating the expression of adhesion molecules; thus, reducing permeability of the endothelial barrier[30, 36, 38].
Notably, the inhibition of adhesion molecules by molecular hydrogen is associated with the RhoA/ mDia/ROCK signaling pathway. RhoA mediates LPS-induced ICAM-1 expression in endothelial cells by activating p38 and NF-κB[51]. The absence of mDia results in more adhesion of neutrophils to endothelial cells in sepsis[52], and the ROCK inhibitors Y-27632 or fasudi reduce the expression of adhesion molecules[30]. In addition, some researchers believe that HRS may reduce the production of adhesion molecules by activating the Nrf2-mediated HO-1 signaling pathway[36].
The biological effects and mechanisms of molecular hydrogen against sepsis
Anti-oxidative stress
Studies have shown that excessive production or insufficient elimination of free radicals, such as ROS and reactive nitrogen species (RNS), is one of the most important pathogenic mechanisms of sepsis[53]. ROS mainly include superoxide anions (·O2-), hydroxyl radicals (·OH), and hydrogen peroxide (H2O2), and RNS include nitric oxide (·NO), nitrogen dioxide (·NO2), and peroxynitrite anion (·ONOO-).
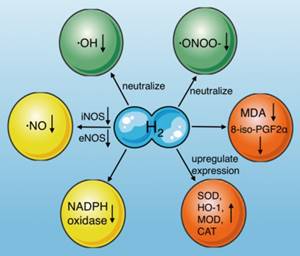
Molecular hydrogen is a natural antioxidant that antagonizes oxidative stress in several ways (Fig. 3): (1) by neutralizing ·OH[54], (2) reducing ·ONOO- and its gene expression directly[55], and inhibiting the production of nitro-tyrosine indirectly, which is an indicator of ·ONOO- generation[13], (3) by inducing antioxidant gene expression and increasing antioxidant enzyme activity, including superoxide dismutase (SOD), HO-1, catalase (CAT), and myeloperoxidase (MPO) [56], (4) by reducing the levels of oxidative stress indicators, such as 8-iso-prostaglandin F2α [57], and the lipid peroxidation marker malondialdehyde (MDA)[13], (5) by reducing ·NO production through inhibiting inducible nitric oxide synthase (iNOS)[29, 58] and endothelial nitric oxide synthase (eNOS)[59], and (6) by inhibiting NADPH oxidase activity, which is the main source of free radicals in sepsis[59-61].
Effects of molecular hydrogen on oxidative stress. Molecular hydrogen antagonizes oxidative stress through multiple pathways, including neutralizing ·OH and ·ONOO-, reducing ·NO production, upregulating antioxidant gene expression (SOD, HO-1, CAT, and MOD), suppressing NADPH oxidase activity, and reducing 8-iso-PGF2α and MDA. ·OH: hydroxyl radicals; ·ONOO-: peroxynitrite anions; SOD: superoxide dismutase; CAT: catalase, MOD: myeloperoxidase; MDA: malondialdehyde; iNOS: inducible nitric oxide synthase; eNOS: endothelial nitric oxide synthase; NADPH oxidase: nicotinamide adenine dinucleotide phosphate oxidase.
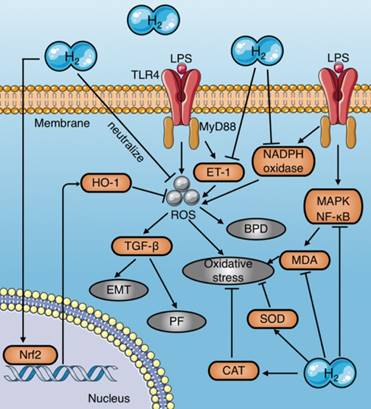
Numerous studies have demonstrated that molecular hydrogen attenuates LPS-induced tissue damage by reducing ROS, increasing antioxidant enzyme activities, and inhibiting pro-oxidant enzyme activities (Fig. 4) (Table 1). HRS reduces oxidative stress in LPS-induced BPD neonatal mice by reducing ROS production in alveolar epithelial cells[16]. In addition, molecular hydrogen reduces the LPS- induced alveolar EMT and pulmonary fibrosis by inhibiting ROS-mediated TGF-β production[20]. Iketani et al. reported that HRS pretreatment further increases the expression of HO-1 induced by LPS, while reducing the expression of ET-1[6], which is a potential endogenous vasoconstrictor that stimulates ROS production dominated by superoxide anions and aggravates oxidative stress associated with lipid peroxidation[62]. Similarly, Chen et al. found that molecular hydrogen increases Nrf2-mediated HO-1 expression, and reduces the endothelial cell injury caused by sepsis[36]. Moreover, studies have shown that HRS inhibits activation of the MAPK and NF-κB signaling pathways, thereby significantly reducing MDA levels in liver tissues of septic rats[22] and reducing oxidative stress in rats with septic peritonitis [27].
Studies of molecular hydrogen against sepsis through an anti-oxidative stress effect
| Drug | Authors | Animal/cell | Disease model | Administration | Effects | Signaling pathways |
|---|---|---|---|---|---|---|
| HRW | Iketani et. al 2017[6] | C57BL/6 mice | Septic liver injury (LPS) | HRW po. × 3d | 4-HNE↓, 8-OHdG ↓ | HO-1 ↑ |
| 2% H2 | Qiu et. al 2011[13] | C57BL/6 mice | Septic lung injury (LPS) | 2% H2 inhalation× 2h | MDA ↓, SOD ↑ | Nitro tyrosine (NT) ↓ |
| HRW | Muramatsu et. al 2016[16] | Sprague-Dawley rats, A549 | Bronchopulmonary Dysplasia (LPS) | HRW po. × 10d | 8-OHdG ↓, ROS ↓ | HO-1 ↑ |
| HRS | Dong et. al 2017[20] | ICR mice | Pulmonary Fibrosis (LPS) | HRS 2.5, 5 or 10 ml/kg/d× 8d i.p. | MDA ↓, T-AOC ↑, SOD ↑, CAT ↑ | N/A |
| HRS | Xu et. al 2013[22] | Sprague-Dawley rats | Septic liver injury (LPS) | HRS 8 ml/kg/h i.v.× 6h | MDA ↓, MPO ↓ | NF-κB ↓, Smac ↓, MAPK(JNK/P38) ↓ |
| HRS | Li et. al 2013[23] | Sprague-Dawley rats | Sepsis (CLP) | HRS 5 ml/kg/h i.p. (0, 6, 18h after CLP) | MDA ↓, SOD ↑ | N/A |
| 2% H2 | Xie et. al 2010[24] | C57BL/6 mice | Sepsis (CLP) | 2% H2 inhalation (4L/min) | SOD ↑, CAT↑ | N/A |
| 2% H2 | Liu et. al 2014[25] | Wistar rats | Septic shock (LPS) | 2% H2 inhalation× 4h | MDA ↓, MPO ↓, SOD ↑ | N/A |
| HRW | Zhang et. al 2014[27] | Sprague-Dawley rats | Septic peritonitis injury (LPS/CLP) | HRW 6 ml/kg/d po. × 10d | MDA ↓, MPO ↓ | NF-κB ↓ |
HRS/W/M: hydrogen rich saline/water/media, LPS: Lipopolysaccharide, CLP: cecal ligation and puncture, MDA: malondialdehyde, SOD: superoxide dismutase, MPO: myeloperoxidase, CAT: catalase, 4-NHE: 4-hydroxy-2-nonenal, 8-OHdG: 8-hydroxy-2'-deoxyguanosine, ROS: reactive oxygen species, i.p.: Intraperitoneal injection, i.v.: Intravenous injection, po.: oral administration.
Mechanisms of molecular hydrogen against oxidative stress in sepsis. (a) Molecular hydrogen eliminates LPS-induced ROS by direct neutralization, promoting HO-1 expression mediated by Nrf2, and inhibiting LPS-induced ET-1 activation. (b) Molecular hydrogen reduces MDA by inhibiting activation of MAPK and NF-κB signaling induced by LPS. (c) Molecular hydrogen increases antioxidant enzyme activities, such as CAT and SOD. (d) Molecular hydrogen suppresses NADPH oxidase activity. EMT: epithelial-mesenchymal transition; PF: pulmonary fibrosis; BPD: bronchopulmonary dysplasia; Nrf2: nuclear factor erythroid 2-related factor 2; TLR4: toll-like receptor 4; MyD88: myeloid differentiation primary response 88; MDA: malondialdehyde; ROS: reactive oxygen species; HO-1: heme oxygenase 1; CAT: catalase; SOD: superoxide dismutase
NADPH oxidase is an important pro-oxidative enzyme that catalyzes the production of superoxide free radicals by transferring electrons from NADPH to oxygen[63]. NADPH oxidase is dormant under physiological conditions but can be rapidly activated by bacterial products and cytokines during sepsis and become the main source of free radicals[63]. Studies have demonstrated that HRS reduces ROS production and attenuates mitochondrial dysfunction by inhibiting NADPH oxidase activity in rat cardiomyocytes [59, 61]. In addition, researchers have found that molecular hydrogen reduces the levels of the p40 phox, p47 phox, and p67 phox subunits of NADPH oxidase in the cell membrane, while increasing their levels in the cytoplasm, suggesting that molecular hydrogen reduces NADPH oxidase activity by limiting the translocation of these molecules to the cell membrane[60].
Anti-inflammation
An excessive inflammatory response is the most significant pathological process occurring in sepsis[1]. Numerous studies have shown that molecular hydrogen effectively alleviates the inflammatory response and plays a protective role in animal models of sepsis[8, 10, 11, 13, 14, 16, 21-24, 27, 36, 38, 64] (Table 2).
Studies of molecular hydrogen against sepsis through an anti-inflammation effect
| Drug | Authors | Animal/cell | Disease model | Administration | Effects | Signaling pathways |
|---|---|---|---|---|---|---|
| HRS 2% H2 | Xie et. al 2012[8] | C57BL/6 mice | Septic lung injury (LPS) | 2% H2 inhalation/HRS 10 ml/kg i.p. | PMNs↓, TNF-α↓, IL-6↓ HMGB1↓, IL-1β↓, IL-10↑ | NF-κB ↓ |
| HRS | Yan et. al 2017[10] | Sprague-Dawley rats | Endotoxin-induced uveitis (LPS) | HRS 10 ml/kg i.p. | Inflammatory cells infiltration ↓ | AqH ↓ |
| HRS | Zhang et. al 2015[11] | Sprague-Dawley rats | Septic lung injury (LPS) | HRS 8 ml/kg/h i.v.× 6h | TNF-α↓, IL-6↓ | P38 MAPK ↓ |
| 2% H2 | Qiu et. al 2011[13] | C57BL/6 mice | Septic lung injury (LPS) | 2% H2 inhalation× 2h | TNF-α↓, IL-6↓ IL-1β↓, MPO↓ | JNK ↓ |
| HRS | Tao et. al 2016[14] | Sprague-Dawley rats | Septic lung injury (LPS) | HRS 10 ml/kg i.p. (1h,4h after LPS) | PMNs ↓ | P38 MAPK ↓ JNK ↓ |
| HRW | Muramatsu et. al 2016[16] | Sprague-Dawley rats, A549 | Bronchopulmonary Dysplasia (LPS) | HRW po. × 10d | TNF-α↓, IL-6↓ | HO-1 ↑ |
| HRS | Sun et. al 2011[21] | C57BL/6 mice | Septic liver injury (LPS/GaIN) | HRS 8ml/kg/3h × 3 i.p. | TNF-α↓, IL-6↓ | JNK ↓ |
| HRS | Xu et. al 2013[22] | Sprague-Dawley rats | Septic liver injury (LPS) | HRS 8 ml/kg/h i.v.× 6h | TNF-α↓, IL-6↓ | NF-κB ↓ P38 MAPK ↓, JNK ↓ |
| HRS | Li et. al 2013[23] | Sprague-Dawley rats | Sepsis (CLP) | HRS 5 ml/kg i.p. (0,6,18h after CLP) | MPO↓, IL-6↓ HMGB1↓ | N/A |
| 2% H2 | Xie et. al 2010[24] | C57BL/6 mice | Sepsis (CLP) | 2% H2 inhalation (4L/min) | HMGB1↓ | N/A |
| 2% H2 | Liu et. al 2014[25] | Wistar rats | Septic shock (LPS) | 2% H2 inhalation× 4h | TNF-α↓, IL-6↓ IL-10↑ | N/A |
| HRW | Zhang et. al 2014[27] | Sprague-Dawley rats | Septic peritonitis injury (LPS/CLP) | HRW 6 ml/kg/d po. × 10d | WBCs↓, endotoxin↓ TNF-α↓, IL-6↓ | NF-κB ↓ |
| HRS HRM | Chen et. al 2015[36] | ICR mice, HUVECs-12 | Sepsis (CLP) HUVECs (LPS) | HRS 5 ml/kg i.p. HRM 0.6 mmol/L | TNF-α↓, IL-1β↓ HMGB1↓, IL-10↑ | Nrf2/HO-1 ↑ |
| HRM | Yu et. al 2015[38] | HUVECs-12 | HUVECs (LPS) | HRM 0.6 mmol/L | Leukocyte coagulation ↓ | N/A |
| HRM | Chen et. al 2103[64] | RAW264.7 | Macrophages (LPS) | HRM 0.6 mmol/L | TNF-α↓, IL-1β↓ HMGB1↓, IL-10↑ | HO-1 ↑ |
HRS/W/M: hydrogen rich saline/water/media, LPS: Lipopolysaccharide, CLP: cecal ligation and puncture, i.p.: Intraperitoneal injection, i.v.: Intravenous injection, po.: oral administration, HUVECs: human umbilical vein endothelial cells, HMGB1: high mobility group box 1, PMNs: polymorphonuclear neutrophils.
The anti-inflammatory effects of molecular hydrogen in sepsis are mainly represented by the followings: (1) reducing the release of LPS-induced pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, and HMGB1[8, 16, 35, 36], and increasing the level of the anti-inflammatory cytokine IL-10[36]; (2) reducing the release of chemokines, such as macrophage inflammatory protein 1 (MIP1) and MIP2[8]; (3) reducing the aggregation and infiltration of neutrophils and macrophages[8, 13, 20, 22]; (4) alleviating vascular endothelial injury induced by inflammatory cells adhesion through reducing LPS-induced adhesion molecules production[38].
These anti-inflammatory effects of molecular hydrogen may be related to the following pathways (Fig. 5):
Anti-inflammation mechanisms of molecular hydrogen during sepsis. (a) Molecular hydrogen suppresses the NF-κB pathway to reduce pro-inflammatory cytokines by inhibiting IκB-a phosphorylation or suppressing ERK activation mediated by upstream and ASK1. (b) Molecular hydrogen reduces neutrophil aggregation by inhibiting activation of ROS-ASK1-MAPK. (c) Molecular hydrogen attenuates the inflammatory response by activating the Nrf2-mediated HO-1 signaling pathway. ASK1: apoptotic signal-regulated kinase 1; ROS: reactive oxygen species; ERK extracellular regulated kinase; MAPK: mitogen activated protein kinase.
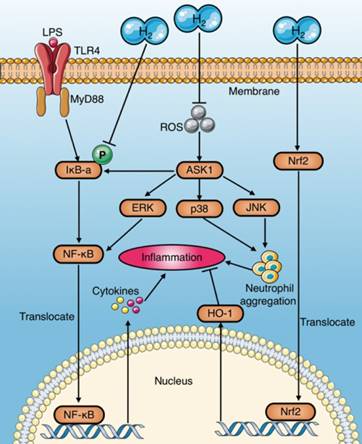
(1) NF-κB: HRS has been demonstrated to play a protective role by inhibiting activation of the NF-κB pathway during sepsis[8, 27], which may be achieved by inhibiting IκBα phosphorylation or suppressing ROS and its downstream related signaling pathways [8].
(2) MAPK (p38, ERK, and JNK): Tao et al. reported that HRS reduces LPS-induced neutrophil aggregation by inhibiting activation of the p38 MAPK and JNK pathways [14]. Similarly, Liang et al.[15] and Xu et al.[22] demonstrated the inhibitory effect of HRS on the p38 MAPK pathway. Activation of ERK promotes separation of IκBα from NF-κB complexes in an acute liver injury model of septic rats, subsequently activating NF-κB and promoting the release of inflammatory factors[65]. However, HRS reduces LPS-induced activation of the ERK pathway [65]. Itoh et al. found that HRS inhibits activation of apoptotic signal-regulated kinase 1 (ASK1) and its downstream signaling pathways p38 MAPK, JNK, and IκBα[58]. (3) HO-1: Chen et al. reported that HRS attenuates the inflammatory response during sepsis by activating the Nrf2-mediated HO-1 signaling pathway[36]. Similarly, the HO-1 pathway mediates the anti-inflammatory effects of molecular hydrogen in LPS-challenged RAW264.7 macrophages[64].
Anti-apoptosis
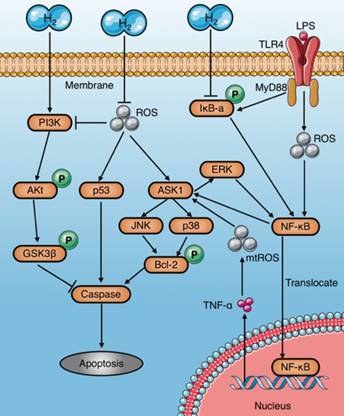
Apoptosis induced by sepsis aggravates tissue damage and organ dysfunction[8, 26]. In particular, immunosuppression caused by apoptosis of immune cells, such as macrophages, is a major cause of deterioration in sepsis[66]. Several studies have demonstrated that molecular hydrogen alleviates septic injury in rats by reducing apoptosis (Table 3). The last common pathway of apoptosis is activation of a series of proteases called caspases. Studies have shown that molecular hydrogen suppresses activation of caspases-3, 8, and 9 and inhibits apoptosis through multiple signaling pathways[67]. The inhibitory mechanisms of molecular hydrogen on caspase activation are very complex and may be involved multiple upstream pathways (Fig. 6): (1) Molecular hydrogen Inhibits the activation of ASK1-MAPKs (p38/JNK/ERK)-Bcl-2 signaling pathways network [65, 68, 69] by ROS neutralization[13, 21, 22] or NF-κB suppression[8, 22, 68]. However, study also found HRS reduces LPS-induced hepatocyte apoptosis by inhibiting ERK-mediated activation of NF-κB[65]; (2) Molecular hydrogen Inhibits the ROS-p53-caspase-3 signaling pathway[70]. It has been proved that ROS inhibit p53 phosphorylation, and lead to cleavage of cytochrome c and caspase-3 in mitochondria, which finally promote apoptosis[70]; (3) Molecular hydrogen activates the PI3K/Aκt/GSK3β signaling pathway[71, 72], which is essential for cell survival[73]. GSK3β phosphorylation occurs after activation of PI3K/Aκt, and subsequently regulates the endogenous apoptotic pathway and its downstream molecules involving MCL-1 (Bcl-2 family member) and BAD (Bcl-Xl/Bcl-2 related death promoting factors)[74, 75].
Studies of molecular hydrogen against sepsis through an anti-apoptosis effect
| Drug | Authors | Animal/cell | Disease model | Administration | Effects | Signaling pathways |
|---|---|---|---|---|---|---|
| HRS | Sun et. al 2011[21] | C57BL/6 mice | Septic liver injury (GalN/LPS) | HRS 8ml/kg/3h × 3 i.p. | TUNEL (+)↓, caspase-3 ↓, cPARP ↓ | ROS-JNK-Caspase3 ↓ |
| 2% H2 | Qiu et. al 2011[13] | C57BL/6 mice | Septic lung injury (LPS) | 2% H2 inhalation | TUNEL (+) ↓, Bax ↓, caspase-3↓, Bcl-xl ↑ | ROS-JNK-Caspase3 ↓ |
| HRS 2% H2 | Xie et. al 2012[8] | C57BL/6 mice | Septic lung injury (LPS) | 2% H2 inhalation/HRS 10 ml/kg i.p. | TUNEL (+) ↓, caspase-3 ↓ | NF-κB ↓ |
| HRS | Zhou et. al 2012[113] | Wistar rats | Sepsis (CLP) | HRS 2.5 or 10 ml/kg i.p. | TUNEL (+) ↓, caspase-3 ↓ | N/A |
| HRS | Li et. al 2013[23] | Sprague-Dawley rats | Sepsis (CLP) | HRS 5 ml/kg i.p. | TUNEL (+) ↓ | N/A |
| HRS | Xu et. al 2013[22] | Sprague-Dawley rats | Septic liver injury (LPS) | HRS 8 ml/kg/h i.v.× 6h | TUNEL (+) ↓, Smac ↓, caspase-3 ↓ | NF-κB ↓, Smac ↓, MAPK (JNK/P38) ↓ |
| 2% H2 | Liu et. al 2014[26] | ICR mice | Septic brain injury (CLP) | Inhale 2% H2 1h (post-operation 1h and 6h) | TUNEL (+) ↓ | Nrf2/HO-1 ↑ |
| HRS HRM | Chen et. al 2015[36] | ICR mice, UVECs-12 | Sepsis (CLP) HUVECs (LPS) | HRS 5 ml/kg i.p., HRM 0.6 mmol/L | Annexin-V/PI ↓, caspase-3 ↓ | Nrf2/HO-1 ↑ |
| HRS | Zhang et. al 2015[11] | Sprague-Dawley rats | Septic lung injury (LPS) | HRS 8 ml/kg/h i.v.× 6h | TUNEL (+) ↓, caspase-3 ↓ | P38 MAPK ↓ |
| HW | Iketani et. al 2017[6] | C57BL/6 mice | Sepsis (LPS) | HW oral (72h before and after LPS) | TUNEL (+) ↓ | HO-1 ↑, ET-1 ↑ |
HRS: hydrogen rich saline, HRM: hydrogen rich medium, HW: hydrogen water, LPS: Lipopolysaccharide, CLP: cecal ligation and puncture, GalN: D-galactosamine, i.p.: Intraperitoneal injection, i.v.: Intravenous injection, TUNEL: terminal deoxynucleotidyl transferase mediated dUTP nick end labeling, cPARP: cytoplasm poly ADP-ribose polymerase, ROS: reactive oxygen species, Smac: second mitochondria-derived activator of caspases.
Anti-apoptosis mechanisms of molecular hydrogen in sepsis. Molecular hydrogen suppresses caspases through several pathways: (a) inhibiting ASK1-MAPKs(p38/JNK/ERK)-Bcl-2 activation by neutralizing ROS or suppressing NF-κB; (b) inhibiting p53 activation induced by ROS; (c) activating the PI3K-AKt-GSK3β signaling pathway. PI3K: phosphatidylinositide 3-kinase; AKt: protein kinase B; GSK3β: glycogen synthase kinase 3 beta; ROS: reactive oxygen species.
Anti-shock
Endotoxin shock is one of the most important causes of death in septic patients[76]. A large quantity of NO is an important cause of septic shock[77, 78]. Carnio et al. reported that LPS-induced hypotension decreases significantly in iNOS knockout mice[78].
As a natural antioxidant, molecular hydrogen selectively scavenges ROS[2]. Although it does not react directly with NO [2], molecular hydrogen plays an anti-shock role by indirectly eliminating NO through various mechanisms[23, 29, 58, 59, 79-81] (Table 4) Saramago et al. determined that LPS leads to severe hypotension, which coincides with a sharp increase in NO production during sepsis[79]. In contrast, molecular hydrogen alleviates LPS-induced hypotension by significantly reducing NO production[79]. Up to now, the mechanism of NO removal by molecular hydrogen can be summarized in two ways (Fig. 7A): (1) Reduced expression of iNOS and eNOS, which are key NO production enzymes. LPS-induced phosphorylation of p38 MAPK, JNK, and IκBα, leading to activation of transcription factors involving AP1, ELK and NF-KB, which bind to the promoter region of iNOS and increase its expression [58]. Molecular hydrogen counteracts the effect of LPS on iNOS and reduces NO production[58]. Ikeda et al. demonstrated that HRS reduces iNOS expression in intestinal epithelial cells of septic rats[29]. In addition, Zheng et al. reported that HRS reduces eNOS expression in vascular endothelial cells of spontaneously hypertensive rats and alleviates vascular dysfunction [59]. (2) Positive feedback consumption of NO by eliminating NO-derived peroxynitrite: Under various stress conditions, NADPH oxidase is rapidly activated and becomes the main source of ROS[63]. Superoxide (·O2-) produced by catalysis of NADPH oxidase reacts with NO to form peroxynitrite (·ONOO-). Molecular hydrogen selectively scavenges ONOO-, and may consume NO in a positive feedback manner[81].
Studies of molecular hydrogen against sepsis through an anti-shock effect
| Drug | Authors | Animal/cell | Disease model | Administration | Effects | Signaling pathways |
|---|---|---|---|---|---|---|
| HRS | Li et. al 2013[23] | Sprague-Dawley rats | Sepsis (CLP) | HRS 5 ml/kg i.p. (0,6,18h after CLP) | NO↓, | N/A |
| HRS | Ikeda et. al 2018[29] | C57BL/6 mice | Intestinal barrier dysfunction (CLP) | HRS 8 ml/kg/d × 7d gavage | Permeability ↓, | N/A |
| HRM | Itoh et. al 2011[58] | RAW264 | Macrophages (LPS) | HRM 0.3 mmol/L | NO ↓ | iNOS ↓ |
| 2% H2 | Saramago et. al 2019[79] | Wistar rats | Sepsis (LPS) | 2% H2 inhalation× 6h | MAP↑, HR ↓,NO ↓ | PGE2 ↓ |
| HRS | Tao et. al 2015[7] | Sprague-Dawley rats | Heart dysfunction (LPS) | HRS 10 ml/kg i.p. (1h,4h after LPS) | ATP ↑ | PGC-1α↑, PPARα↑, ERRα↑, JNK↓ |
| HRM | Xie et. al 2015[30] | HUVECs U937 | Endothelial dysfunction (LPS) | HRM 0.6 mmol/L | VCAM-1↓, E-selectin ↓, TEER ↑, E-cadherin ↑ | Rho/ROCK ↓ |
| HRM | Wang et. al 2013[35] | HUVEC-12 U937 | Endothelial dysfunction (LPS) | HRM 0.6 mmol/L | VCAM-1↓, E-selectin ↓, VE-cadherin ↑ | N/A |
| HRS HRM | Chen et. al 2015[36] | ICR mice, HUVECs-12 | Sepsis (CLP), HUVECs (LPS) | HRS 5 ml/kg i.p., HRM 0.6 mmol/L | ICAM-1 ↓,VCAM-1 ↓ | Nrf2/HO-1 ↑ |
| HRM | Yu et. al 2015[38] | HUVECs-12 | HUVECs (LPS) | HRM 0.6 mmol/L | VCAM-1↓, ICAM-1↓, E-selectin ↓, TEER ↑, VE-cadherin ↑ | N/A |
HRS/M: hydrogen rich saline/media, LPS: Lipopolysaccharide, CLP: cecal ligation and puncture, i.p.: Intraperitoneal injection, HUVECs: human umbilical vein endothelial cells, VCAM-1: vascular cell adhesion molecule -1, ICAM-1: intercellular adhesion molecule-1, TEER: trans-endothelial electrical resistance, PGE2: Prostaglandin E2, PGC-1α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha, PPARα: Peroxisome proliferator-activated receptor alpha, ERRα: Estrogen-related receptor alpha, Rho: Ras homolog gene, ROCK: Rho-associated coiled-coil protein kinase.
Anti-shock mechanisms of molecular hydrogen during sepsis. (A) Molecular hydrogen inhibits production of the vasodilator nitric oxide and promotes its consumption. (B) Molecular hydrogen increases ATP production mediated by OXPHOS activation in cardiomyocytes. (C) Molecular hydrogen alleviates vascular endothelial barrier damage mediated by the RhoA/ROCK/mDia signaling pathway and adhesion molecules. AP1: activator protein 1; ELK1: ETS domain-containing protein; OXPHOS: oxidative phosphorylation; Cplx: mitochondrial redox carrier (complex); GLUT: Glucose transporters; ghrelin: growth hormone releasing peptide; FGF21: fibroblast growth factor 21.
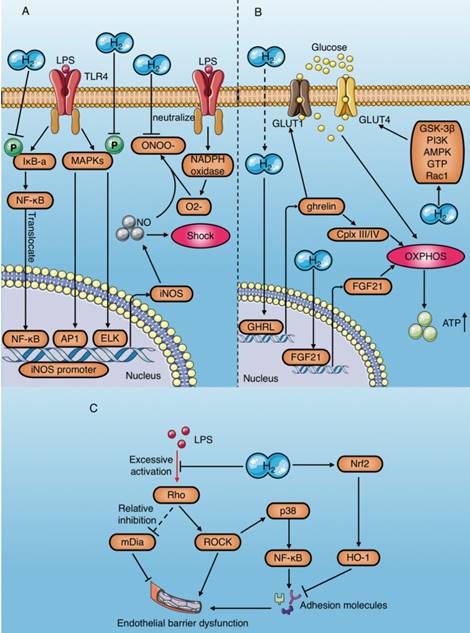
In addition to NO-induced hypotension, the decline in cardiac pump function caused by insufficient ATP production in cardiomyocytes is an important reason for endotoxic shock[28]. Mitochondria are the main organelles of ATP production. Several studies have proved that molecular hydrogen improves mitochondrial function through preserving the mitochondrial membrane potential (ΔΨ)[82] and alleviating mitochondrial swelling[59, 61] (Table 4). Tao et al. demonstrated that HRS reverses LPS-induced phosphocreatine (PCr)/ATP decline and improves the myocardial energy supply[7]. In addition, multiple studies reported HRS increases ATP production in cardiomyocytes by restoring electron transport chain enzyme activity[59, 61, 83]. The increase of ATP production by molecular hydrogen is achieved by activating mitochondrial oxidative phosphorylation (OXPHOS)[84], and the mechanisms include upregulating the expression of a growth hormone releasing peptide (ghrelin) and fibroblast growth factor 21 (FGF21) and increasing glucose metabolism [85]. Moreover, the regulation of molecular hydrogen on glucose metabolism can be divided into the insulin-dependent pathway (glucose transporter 4 [GLUT4]) and the insulin-independent pathway (GLUT1) (Fig. 7B). In addition, crosstalk occurs in the regulatory mechanism of molecular hydrogen on ghrelin and glucose metabolism (Fig. 7B). Ghrelin has recently been acknowledged as a major modulator of mitochondrial bioenergetics, as it increases mitochondrial energy production by increasing protein complexes III and IV[86]. GLUT4 is an insulin- regulated glucose transporter that plays a carrier role during cellular uptake of glucose. FGF21 is a hepatic hormone that enhances utilization of fatty acids and glucose. Therefore, positive regulation of GLUT4 and FGF21 by molecular hydrogen may play a role in enhancing energy metabolism.
Moreover, as previously mentioned, hyperpermeability of the vascular endothelium may aggravate endotoxin shock[34] (Table 4). However, molecular hydrogen can alleviate LPS-induced vascular endothelial injury by regulating the RhoA/mDia/ ROCK signaling pathway and down-regulating the expression of adhesion molecules (Fig. 7C).
In conclusion, molecular hydrogen alleviates LPS-induced hypotension or shock by eliminating NO, increasing ATP production in cardiomyocytes, and decreasing vascular endothelium permeability.
Regulation of autophagy
Autophagy is one of the most important functions of eukaryotic cells, which encapsulates substrates within a double-membrane-bound vesicle, termed an autophagosome that fuses with lysosomes for degradation and recycling of the sequestered substrates[87]. Cells clear damaged organelles, abnormal proteins, and extracellular pathogens in the cytoplasm by autophagy, which is essential for maintaining cell homeostasis [87]. Autophagy is involved in the regulation of immunity and inflammation [87-91], and plays an important role in sepsis [92-95].
Studies have demonstrated that molecular hydrogen plays a protective role by modulating autophagy in multiple diseases and conditions, including sepsis [11, 12, 20], ischemia- reperfusion injury[96-98], organ transplantation [70, 99-102], and pathological neuralgia [103-105] (Table 5). However, the regulation of autophagy by molecular hydrogen remains controversial, and the regulatory mechanism is very complex (Fig. 8).
Studies and mechanisms of molecular hydrogen modulating autophagy
| Drug | Authors | Animal/cell | Disease model | Administration | Effects | Signaling pathways |
|---|---|---|---|---|---|---|
| HRS | Zhang et. al 2015[11] | Sprague-Dawley rats | Septic lung injury (LPS) | HRS 8 ml/kg/h i.v. × 6h | Autophagosome ↓, LC3II/I ↓ | P38 MAPK ↓ |
| 2% H2 | Dong et. al 2017[20] | ICR mice | Sepsis (CLP) | 2% H2 inhalation | LC3II/I ↑ | N/A |
| HRS | Bai et. al 2016[96] | BALB/c mice | HIBD | HRS 5 ml/kg/d i.p. × 3d | LC3II/I ↑, Beclin1 ↑ | mTOR ↓, stat3 ↓, ERK1/2 ↓ |
| HRS | Pan et. al 2015[97] | Sprague-Dawley rats | Myocardial injury after IR | HRS 10 ml/kg i.p. | LC3II/I ↓, Beclin1 ↓ | AMPK ↓, mTOR ↑ |
| HRS | Chen et. al 2017[98] | C57BL/6 mice | CKD after IR | HRS 1 ml/kg i.p. | LC3II/I ↑, Beclin1 ↑ | Klotho |
| HRS | Du et. al 2016[70] | Sprague-Dawley rats | AKI after OLT | HRS 6 ml/kg i.v. | LC3II/I ↑, Autophagosome ↑, Beclin1 ↑, P62 ↓ | p53 ↑ |
| HRS | Shi et. al 2016[99] | Sprague-Dawley rats | IRI after OLT | HRS 6 ml/kg i.v. | LC3II ↓, Beclin1 ↓ | N/A |
| HRS | Shi et. al 2016[100] | Sprague-Dawley rats | OLT | HRS 6 ml/kg i.v. | LC3II ↓, Beclin1 ↓ | p53 ↑ |
| HRS | Wu et. al 2015[101] | Sprague-Dawley rats | AKI after OLT | HRS 6 ml/kg i.v. | LC3II ↑, Beclin1 ↑ | p53 ↑ |
| HRS | Chen et. al 2017[102] | Sprague-Dawley rats | HNA after OLT | HRS 6 ml/kg i.v. | LC3II ↑, Beclin1 ↑ | p53 ↑ |
| HRS | Wang et. al 2016[103] | Sprague-Dawley rats | Neuropathic pain (CCI) | HRS 5 ml/kg/d i.p. × 7d | LC3II/I ↑, Beclin1 ↑, P62 ↓ | N/A |
| HRS | Ma et. al 2017[104] | Sprague-Dawley rats | PHN | HRS 10 ml/kg/12h i.p. × 14d | LC3II ↑, Beclin1 ↑, P62 ↓ | N/A |
| HRS | Wang et. al 2018[105] | Sprague-Dawley rats | Neuropathic pain (CCI) | HRS 10 ml/kg/d i.p.× 15d | Beclin1 ↑, P62 ↓, Autophagosome ↑ | HIF-1α ↑ |
HRS: hydrogen rich saline, LPS: Lipopolysaccharide, CLP: cecal ligation and puncture, HIBD: hypoxic-ischemic brain damage, IR: ischemia-reperfusion, IRI: ischemia-reperfusion injury, CKD: chronic kidney disease, AKI: acute kidney injury, OLT: orthotropic liver transplantation, HNA: hippocampal neuron apoptosis, CCI: chronic constriction injury, PHN: post-herpetic neuralgia, i.p.: Intraperitoneal injection, i.v.: Intravenous injection.
Autophagy regulatory mechanisms of molecular hydrogen. (A) Molecular hydrogen has a dual effect on the regulation of autophagy during sepsis. On the one hand, molecular hydrogen suppresses excessive autophagy by inhibiting NF-κB and p38 MAPK, enhancing HO-1, and reducing mTOR suppression induced by reactive oxygen species. On the other hand, molecular hydrogen may enhance autophagy through an unknown mechanism. (B) In studies of ischemia-anoxia reperfusion injury, molecular hydrogen enhances autophagy by inhibiting activation of ERK, mTOR, and Stat3, or by restraining Klotho gene expression. However, molecular hydrogen inhibits AMPK activation by increasing ATP production, which subsequently reduces mTOR suppression and inhibits autophagy. (C) Molecular hydrogen promotes autophagy in orthotopic liver transplantation by activating p53 in the nucleus and increasing target gene expression involving DRAM and ULK. In contrast, molecular hydrogen suppresses autophagy by activating p53 in the cytoplasm. (D) In pathological neuralgia, molecular hydrogen activates HIF-1α and subsequently increases BNIP3 expression or inhibits mTOR, both of which promote autophagy. HI: hypoxia-ischemia; OLT: orthotopic liver transplantation; HIF-1α: hypoxia inducing factor-1; BNIP3: Bcl-2 19 kilodalton interacting protein 3; mTOR: mammalian target of rapamycin; Stat3: signal transducer and activator of transcription 3; AMPK: AMP-activated protein kinase; DRAM: damage regulated autophagy modulator; ULK: Unc-51 like autophagy activating kinase.
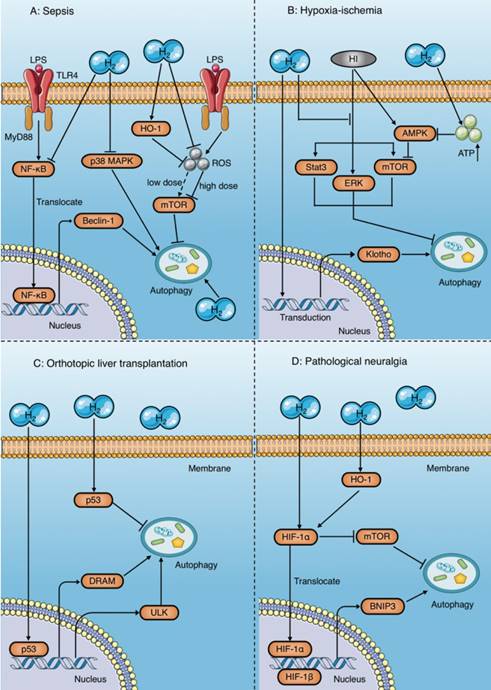
Autophagy is a double-edged sword. Moderate autophagy helps cells survive harsh environments by reducing apoptosis and necrosis, while excessive autophagy leads to autophagic death[106]. Zhang et al. reported that HRS attenuates lung injury by inhibiting autophagy in alveolar epithelial cells of septic rats, which might be related to inhibition of the p38 MAPK signaling pathway[11] (Fig. 8A). Similarly, other studies have found that molecular hydrogen inhibits excessive autophagy to attenuate the ALI induced by sepsis by activating HO-1 and the mammalian target of rapamycin (mTOR) pathway, and inhibiting the NF-κB pathway[12, 107] (Fig. 8A). In contrast, Dong et al. demonstrated that inhaling molecular hydrogen enhances the autophagy level of lung tissue in septic mice, which improves mitochondrial function, and protects the lungs [20] (Fig. 8A).
The same controversy has also occurred in studies of animal organ ischemia-anoxia reperfusion injury models[96-98] and orthotopic liver transplantation (OLT) models[70, 99-102]. Bai et al. reported that molecular hydrogen enhances autophagy in neurons and attenuates brain damage by suppressing mTOR and signal transducer and activator of transcription 3 (Stat3) phosphorylation, and preventing the activation of ERK1/2 induced by hypoxia-ischemia [96] (Fig. 8B). A study by Chen et al. showed that HRS reduces renal fibrosis after ischemia-reperfusion injury by increasing autophagy, which may be related to the retention of Klotho gene expression [98] (Fig. 8B). However, in a rat model of myocardial ischemia-reperfusion injury, HRS was demonstrated to reduce myocardial injury by inhibiting cardiomyocyte autophagy[97], the mechanism of which was reduced suppression of the mTOR signaling pathway mediated by HRS-induced inhibition of AMP-activated protein kinase activation[97] (Fig. 8B).
Results in a rat model of OLT show that HRS exerts a protective effect by inhibiting autophagy in orthotopic liver tissue[99, 100], but enhances autophagy in other distant tissues, such as the brain and kidney[70, 101, 102], both of which are associated with p53 activation (Fig. 8C). Tumor suppressor gene p53 is widely involved in regulation of the cell cycle, apoptosis, and autophagy induction. However, the regulatory mechanism of p53 in autophagy is very complex and not fully understood. Researchers believe that the effect of p53 on autophagy may depend on its location within the cell[108]. p53 upregulates autophagy levels by acting as a transcriptional activator in the nucleus, while inhibiting autophagy in the cytoplasm[108]. This viewpoint has been validated in several studies. Shi et al.[100] reported that HRS inhibits autophagy by increasing p53 phosphorylation in the hepatic cytoplasm after OLT, and reduces hepatocyte damage after transplantation (Fig. 8C). In contrast, the results of Du et al.[70], Wu et al.[101], and Chen et al.[102] suggest that HRS promotes autophagy to protect the kidney and brain after OLT by activating the p53 signaling pathway (Fig. 8C).
In addition, a number of studies have shown that HRS promotes autophagy in rats with neuropathic pain and plays a protective role[103-105]; the underlying mechanism of which is activation of hypoxia inducing factor-1 (HIF-1α) by HRS[105] (Fig. 8D). HIF-1α induces autophagy by increasing the expression of its target gene BNIP3[105] and inhibiting mTOR[109]. Wang et al. also showed that HRS promotes autophagy in CCI rats by further increasing activation of HIF-1α and expression of BNIP3[105]. In addition, studies have shown that HRS increases HIF-1α expression by promoting HO-1 activation[110, 111], which may also be involved in the regulation of autophagy by HRS (Fig. 8D).
Notably, there is crosstalk between the regulatory mechanisms of molecular hydrogen on autophagy and apoptosis, which involve MAPK[11, 65, 69, 96] and p53[70, 100-102]. Therefore, we can infer that HRS may change the fate of cells and the prognosis of diseases by regulating the balance of apoptosis and autophagy, which requires further research.
Effective concentration and dose of molecular hydrogen against sepsis
HRS/HRW was generally prepared as the method originally described by Ohsawa et. al[112]. Briefly, hydrogen gas was dissolved in NS or water under 0.4 MPa pressure to be oversaturated (>0.6 mmol/L) and then stored in a sealed container without dead volume at 4°C under atmospheric pressure followed by γ-radiation sterilization before use. According to the method described by Ohsawa et. al[2], HRM was prepared through dissolving hydrogen gas into medium under 0.4 MPa pressure to achieve supersaturated status (>0.6 mmol/L) and stored in a hydrogen-rich closed culture flask. Moreover, the method of inhaling 2% hydrogen gas referred to the research of Xie et. al[8]. The various concentrations and doses of molecular hydrogen, which have been proved effective in organs and cells protection in sepsis, were summarized in Table 6.
Effective concentration and dose of molecular hydrogen against sepsis
| Organ/cell | Administration | Effective concentration | References |
|---|---|---|---|
| Lung | H2 inhalation | 2% | [8, 13, 15, 20] |
| HRS 8 ml/kg i.v. | > 0.6 mmol/L | [11] | |
| HRS 10 ml/kg i.p. | > 0.6 mmol/L | [14] | |
| HRW po. | 0.25-1.6 mmol/L | [16] | |
| Liver | HRW po. | 0.6 mmol/L | [6] |
| HRS 8 ml/kg i.p. | > 0.6 mmol/L | [21] | |
| HRS 8 ml/kg i.v. | > 0.6 mmol/L | [22] | |
| Kidney | H2 inhalation | 2% | [24, 25] |
| HRS 5 ml/kg i.p. | > 0.6 mmol/L | [23] | |
| Brain | H2 inhalation | 2% | [26] |
| Pancreas | HRW 6 ml/kg po. | 0.62-0.82 mmol/L | [27] |
| Heart | HRS 10 ml/kg i.p. | > 0.6 mmol/L | [7] |
| Eye | HRS 10 ml/kg i.p. | > 0.6 mmol/L | [10] |
| Endothelial cell | HRM incubation | 0.6 mmol/L | [30, 35, 36, 38] |
| HRS 5 ml/kg i.p. | > 0.6 mmol/L | [36] | |
| Epithelial cell | HRS 8 ml/kg gavage | 0.7 mmol/L | [29] |
| HRS 8 ml/kg i.v. | > 0.6 mmol/L | [11] | |
| HRM incubation | 0.6 mmol/L | [9] |
Conclusion and perspectives
In summary, molecular hydrogen exhibits multiple advantages in the treatment of sepsis due to its unique physicochemical properties. Molecular hydrogen scavenges free radicals selectively, modulates signaling transduction, and enters the nucleus to regulate transcription. Recent studies have shown that molecular hydrogen has a significant protective effect on multiple organs and physiological barriers in septic animal models. In addition to the well-known anti-oxidative stress effects, the mechanisms of molecular hydrogen against sepsis include anti-inflammation, anti-apoptosis, anti-shock and regulation of autophagy, each of which involves multiple signaling pathways and crosstalk. However, the potential molecular mechanisms are still not completely clear, and some results remain controversial, which need further research. Moreover, the current research results are mainly based on animal experiments. Whether these findings are equally applicable to humans is not yet known, which also requires further clinical studies to validate. Nevertheless, the advantages of molecular hydrogen have provided important means and optimistic prospects for treating sepsis.
Abbreviations
HRS: hydrogen-rich saline; HRW: hydrogen-rich water; HRM: hydrogen-rich medium; LPS: lipopolysaccharide; ALI: acute lung injury; BPD: bronchopulmonary dysplasia; FGFR4: fibroblast growth factor receptor 4; VEGFR2: vascular endothelial growth factor receptor 2; HO-1: heme oxygenase 1; EMT: epithelial-mesenchymal transition; ROS: reactive oxygen species; TGF-β: transforming growth factor-β; EVLW: extravascular lung water; AQP1,5: Aquaporin 1,5; MAPK: mitogen-activated protein kinase; JNK: c-Jun N-terminal kinase; ERK: extracellular regulated protein kinases; NF-κB: nuclear factor kappa-light- chain-enhancer of activated B cells; Smac: second mitochondria-derived activator of caspase; ET-1: endothelin-1; Nrf2: nuclear factor erythroid 2-related factor 2; LVD: left ventricular diameter; FS: fractional shortening; FAO: fatty acid oxidation; EIU: endotoxin- induced uveitis; AqH: aqueous humor; VCAM-1: vascular cell adhesion molecule -1; ICAM-1: intercellular adhesion molecule-1; TEER: trans-endothelial electrical resistance; TER: trans-epithelial resistance; AJ: adherens junction; TJ: tight junction; FITC- dextran: fluorescein isothiocyanate-dextran; MLC: myosin light chain; TNF-α: tumor necrosis factor alpha; IL-1β: interleukin 1 beta; IL-10: interleukin 10; HMGB1: high mobility group box 1; HUVECs: human umbilical vein endothelial cells; CLP: cecal ligation and puncture; Cdc42: Cell division control protein 42 homolog; Rac1: Ras-related C3 botulinum toxin substrate 1; RhoA: Ras homolog gene family member A; mDia1: mammalian diaphanous-related formin 1; ROCK: Rho-associated coiled-coil protein kinase; RNS: reactive nitrogen species; ·O2-: superoxide anion; ·OH: hydroxyl radical; H2O2: hydrogen peroxide; ·NO: RNS include nitric oxide; ·NO2: nitrogen dioxide; ·ONOO-: peroxynitrite anion; SOD: superoxide dismutase; CAT: catalase; MOD: myeloperoxidase; 8-iso-PGF2α: 8-iso-prostaglandinF2α; MDA: malondialdehyde; iNOS: inducible nitric oxide synthase; eNOS: endothelial nitric oxide synthase; NADPH oxidase: nicotinamide adenine dinucleotide phosphate oxidase; BPD: bronchopulmonary dysplasia; EMT: epithelial-mesenchymal transition; PF: pulmonary fibrosis; TGF-β: transforming growth factor-β; MPO: myeloperoxidase; PMNs: polymorphonuclear neutrophils; BALF: bronchoalveolar lavage fluid; AP1: activator protein 1; ELK1: ETS domain- containing protein; OXPHOS: oxidative phosphorylation; Cplx: mitochondrial redox carrier (complex); GLUT: Glucose transporters; ghrelin: growth hormone releasing peptide; FGF21: fibroblast growth factor 21; MIP: macrophage inflammatory protein; IκBα: nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha; ASK1: apoptotic signal-regulated kinase 1; GSK3β: Glycogen synthase kinase 3 beta; HI: hypoxia-ischemia; OLT: orthotopic liver transplantation; HIF-1α: hypoxia inducing factor-1; BNIP3: Bcl-2 nineteen kilodalton interacting protein 3; mTOR: mammalian target of rapamycin; Stat3: signal transducer and activator of transcription 3; AMPK: AMP-activated protein kinase; DRAM: damage regulated autophagy modulator; ULK: Unc-51 like autophagy activating kinase.
Acknowledgements
This study was supported by the Shenyang Municipal Science and Technology Commission (Grant Number 17-230-9-45); the Liaoning Education Department (Grant Number LK201634); and the National Natural Science Foundation of China (Grant Number 81701951).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Singer M, Deutschman CS, Seymour CW. et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315:801-10
2. Ohsawa I, Ishikawa M, Takahashi K. et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007;13:688-94
3. Ichihara M, Sobue S, Ito M. et al. Beneficial biological effects and the underlying mechanisms of molecular hydrogen - comprehensive review of 321 original articles. Med Gas Res. 2015;5:12
4. Shen MH, Zang HY, Yu CY. et al. A review of experimental studies of hydrogen as a new therapeutic agent in emergency and critical care medicine. Medical Gas Research. 2014;4:17
5. Qian L, Shen J, Chuai Y. et al. Hydrogen as a New Class of Radioprotective Agent. International Journal of Biological Sciences. 2013;9:887-94
6. Iketani M, Ohshiro J, Urushibara T. et al. Preadministration of Hydrogen-Rich Water Protects Against Lipopolysaccharide-Induced Sepsis and Attenuates Liver Injury. Shock. 2017;48:85-93
7. Tao B, Liu L, Wang N. et al. Hydrogen-Rich Saline Attenuates Lipopolysaccharide-Induced Heart Dysfunction by Restoring Fatty Acid Oxidation in Rats by Mitigating C-Jun N-Terminal Kinase Activation. Shock. 2015;44:593-600
8. Xie K, Yu Y, Huang Y. et al. Molecular hydrogen ameliorates lipopolysaccharide-induced acute lung injury in mice through reducing inflammation and apoptosis. Shock. 2012;37:548-55
9. Yang T, Wang L, Sun R. et al. Hydrogen-Rich Medium Ameliorates Lipopolysaccharide-Induced Barrier Dysfunction Via Rhoa-Mdia1 Signaling in Caco-2 Cells. Shock. 2016;45:228-37
10. Yan WM, Zhang L, Chen T. et al. Effects of hydrogen-rich saline on endotoxin-induced uveitis. Med Gas Res. 2017;7:9-18
11. Zhang Y, Liu Y, Zhang J. Saturated hydrogen saline attenuates endotoxin-induced lung dysfunction. J Surg Res. 2015;198:41-9
12. Liu Y, Zhang J. Saturated hydrogen saline ameliorates lipopolysaccharide-induced acute lung injury by reducing excessive autophagy. Exp Ther Med. 2017;13:2609-15
13. Qiu X, Li H, Tang H. et al. Hydrogen inhalation ameliorates lipopolysaccharide-induced acute lung injury in mice. Int Immunopharmacol. 2011;11:2130-7
14. Tao B, Liu L, Wang N. et al. Effects of hydrogen-rich saline on aquaporin 1, 5 in septic rat lungs. J Surg Res. 2016;202:291-8
15. Liang CX, Liu XW, Liu L. et al. Effect of hydrogen inhalation on p38 MAPK activation in rats with lipopolysaccharideinduced acute lung injury. J South Med Univ. 2012;32:1211-7
16. Muramatsu Y, Ito M, Oshima T. et al. Hydrogen-rich water ameliorates bronchopulmonary dysplasia (BPD) in newborn rats. Pediatr Pulmonol. 2016;51:928-35
17. Li R, Herriges JC, Chen L. et al. FGF receptors control alveolar elastogenesis. Development. 2017;144:4563-72
18. Delaney C, Wright RH, Tang JR. et al. Lack of EC-SOD worsens alveolar and vascular development in a neonatal mouse model of bleomycin-induced bronchopulmonary dysplasia and pulmonary hypertension. Pediatr Res. 2015;78:634-40
19. Ishikawa F, Kaneko E, Sugimoto T. et al. A mitochondrial thioredoxin-sensitive mechanism regulates TGF-beta-mediated gene expression associated with epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2014;443:821-7
20. Dong W-W, Zhang Y-Q, Zhu X-Y. et al. Protective Effects of Hydrogen-Rich Saline Against Lipopolysaccharide-Induced Alveolar Epithelial-to-Mesenchymal Transition and Pulmonary Fibrosis. Medical Science Monitor. 2017;23:2357-64
21. Sun H, Chen L, Zhou W. et al. The protective role of hydrogen-rich saline in experimental liver injury in mice. J Hepatol. 2011;54:471-80
22. Xu XF, Zhang J. Saturated Hydrogen Saline Attenuates Endotoxin-Induced Acute Liver Dysfunction in Rats. Physiol Res. 2013;62:395-403
23. Li G, M, Ji M. et al. Effects of hydrogen-rich saline treatment on polymicrobial sepsis. J Surg Res. 2013;181:279-86
24. Xie K, Yu Y, Pei Y. et al. Protective effects of hydrogen gas on murine polymicrobial sepsis via reducing oxidative stress and HMGB1 release. Shock. 2010;34:90-7
25. Liu W, Dong XS, Sun YQ. et al. A novel fluid resuscitation protocol: provide more protection on acute kidney injury during septic shock in rats. Int J Clin Exp Med. 2014;7:919-26
26. Liu L, Xie K, Chen H. et al. Inhalation of hydrogen gas attenuates brain injury in mice with cecal ligation and puncture via inhibiting neuroinflammation, oxidative stress and neuronal apoptosis. Brain Res. 2014;1589:78-92
27. Zhang J, Wu Q, Song S. et al. Effect of hydrogen-rich water on acute peritonitis of rat models. Int Immunopharmacol. 2014;21:94-101
28. Zaky A, Deem S, Bendjelid K. et al. Characterization of cardiac dysfunction in sepsis: an ongoing challenge. Shock. 2014;41:12-24
29. Ikeda M, Shimizu K, Ogura H. et al. Hydrogen-Rich Saline Regulates Intestinal Barrier Dysfunction, Dysbiosis and Bacterial Translocation in a Murine Model of Sepsis. Shock. 2018;50:640-7
30. Xie K, Wang W, Chen H. et al. Hydrogen-Rich Medium Attenuated Lipopolysaccharide-Induced Monocyte-Endothelial Cell Adhesion and Vascular Endothelial Permeability via Rho-Associated Coiled-Coil Protein Kinase. Shock. 2015;44:58-64
31. Chua J, Soon AS, Becker D. Connexins in endothelial barrier function - novel therapeutic targets countering vascular hyperpermeability. Thrombosis and Haemostasis. 2017;116:852-67
32. Niño CA, Sala S, Polo S. When ubiquitin meets E-cadherin: Plasticity of the epithelial cellular barrier. Seminars in Cell & Developmental Biology. 2018
33. Zheng S, Lin Z, Liu Z. et al. Lipopolysaccharide Mediates the Destruction of Intercellular Tight Junction among Renal Tubular Epithelial Cells via RhoT1/SMAD-4/JAM-3 Pathway. Int J Med Sci. 2018;15:595-602
34. Gulati A. Vascular Endothelium and Hypovolemic Shock. Curr Vasc Pharmacol. 2016;14:187-95
35. Wang WN. et al. Regulative effects of hydrogen-rich medium on monocytic adhesion and vascular endothelial permeability. Natl Med J China. 2013;93:3467-9
36. Chen H, Xie K, Han H. et al. Molecular hydrogen protects mice against polymicrobial sepsis by ameliorating endothelial dysfunction via an Nrf2/HO-1 signaling pathway. Int Immunopharmacol. 2015;28:643-54
37. Liu LD, Wu XY, Tao BD. et al. Protective effect and mechanism of hydrogen treatment on lung epithelial barrier dysfunction in rats with sepsis. Genet Mol Res. 2016:15
38. Yu Y, Wang WN, Han HZ. et al. Protective effects of hydrogen-rich medium on lipopolysaccharide-induced monocytic adhesion and vascular endothelial permeability through regulation of vascular endothelial cadherin. Genet Mol Res. 2015;14:6202-12
39. Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103-12
40. Schlegel N, Meir M, Spindler V. et al. Differential role of Rho GTPases in intestinal epithelial barrier regulation in vitro. Journal of Cellular Physiology. 2011;226:1196-203
41. Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol. 2002;4:408-15
42. Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell. 2008;14:25-36
43. Chaturvedi LS, Marsh HM, Basson MD. Role of RhoA and its effectors ROCK and mDia1 in the modulation of deformation-induced FAK, ERK, p38, and MLC motogenic signals in human Caco-2 intestinal epithelial cells. Am J Physiol Cell Physiol. 2011;301:C1224-38
44. Lu Q, Lu L, Chen W. et al. RhoA/mDia-1/profilin-1 signaling targets microvascular endothelial dysfunction in diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2015;253:669-80
45. Carramusa L, Ballestrem C, Zilberman Y. et al. Mammalian diaphanous-related formin Dia1 controls the organization of E-cadherin-mediated cell-cell junctions. J Cell Sci. 2007;120:3870-82
46. Thumkeo D, Shinohara R, Watanabe K. et al. Deficiency of mDia, an actin nucleator, disrupts integrity of neuroepithelium and causes periventricular dysplasia. PLoS One. 2011;6:e25465
47. Huang Y, Tan Q, Chen R. et al. Sevoflurane prevents lipopolysaccharide-induced barrier dysfunction in human lung microvascular endothelial cells: Rho-mediated alterations of VE-cadherin. Biochem Biophys Res Commun. 2015;468:119-24
48. He F, Peng J, Deng XL. et al. RhoA and NF-kappaB are involved in lipopolysaccharide-induced brain microvascular cell line hyperpermeability. Neuroscience. 2011;188:35-47
49. Joshi AD, Dimitropoulou C, Thangjam G. et al. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am J Respir Cell Mol Biol. 2014;50:170-9
50. Soliman M, Cho EH, Park JG. et al. Rotavirus-Induced Early Activation of the RhoA/ROCK/MLC Signaling Pathway Mediates the Disruption of Tight Junctions in Polarized MDCK Cells. Sci Rep. 2018;8:13931
51. Guo F, Zhou Z, Dou Y. et al. GEF-H1/RhoA signalling pathway mediates lipopolysaccharide-induced intercellular adhesion molecular-1 expression in endothelial cells via activation of p38 and NF-kappaB. Cytokine. 2012;57:417-28
52. Mei Y, Feng G, Rahimi N. et al. Loss of mDia1 causes neutropenia via attenuated CD11b endocytosis and increased neutrophil adhesion to the endothelium. Blood Adv. 2017;1:1650-6
53. Hoetzenecker W, Echtenacher B, Guenova E. et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med. 2011;18:128-34
54. Ohta S. Molecular hydrogen as a novel antioxidant: overview of the advantages of hydrogen for medical applications. Methods Enzymol. 2015;555:289-317
55. Shinbo T, Kokubo K, Sato Y. et al. Breathing nitric oxide plus hydrogen gas reduces ischemia-reperfusion injury and nitrotyrosine production in murine heart. Am J Physiol Heart Circ Physiol. 2013;305:H542-50
56. Cai WW, Zhang MH, Yu YS. et al. Treatment with hydrogen molecule alleviates TNFalpha-induced cell injury in osteoblast. Mol Cell Biochem. 2013;373:1-9
57. Xie K, Liu L, Yu Y. et al. Hydrogen gas presents a promising therapeutic strategy for sepsis. Biomed Res Int. 2014;2014:807635
58. Itoh T, Hamada N, Terazawa R. et al. Molecular hydrogen inhibits lipopolysaccharide/interferon gamma-induced nitric oxide production through modulation of signal transduction in macrophages. Biochem Biophys Res Commun. 2011;411:143-9
59. Zheng H, Yu Y-S. Chronic hydrogen-rich saline treatment attenuates vascular dysfunction in spontaneous hypertensive rats. Biochemical Pharmacology. 2012;83:1269-77
60. Itoh T, Fujita Y, Ito M. et al. Molecular hydrogen suppresses FcεRI-mediated signal transduction and prevents degranulation of mast cells. Biochemical and Biophysical Research Communications. 2009;389:651-6
61. Yu Y-S, Zheng H. Chronic hydrogen-rich saline treatment reduces oxidative stress and attenuates left ventricular hypertrophy in spontaneous hypertensive rats. Molecular and Cellular Biochemistry. 2012;365:233-42
62. Liu A, Fang H, Wei W. et al. G-CSF pretreatment aggravates LPS-associated microcirculatory dysfunction and acute liver injury after partial hepatectomy in rats. Histochem Cell Biol. 2014;142:667-76
63. Geiszt M. NADPH oxidases: new kids on the block. Cardiovasc Res. 2006;71:289-99
64. Chen HG, Xie KL, Han HZ. et al. Heme oxygenase-1 mediates the anti-inflammatory effect of molecular hydrogen in LPS-stimulated RAW 264.7 macrophages. Int J Surg. 2013;11:1060-6
65. Shi Q, Chen C, Deng WH. et al. Hydrogen-Rich Saline Attenuates Acute Hepatic Injury in Acute Necrotizing Pancreatitis by Inhibiting Inflammation and Apoptosis, Involving JNK and p38 Mitogen-Activated Protein Kinase-dependent Reactive Oxygen Species. Pancreas. 2016;45:1424-31
66. Wang TS, Deng JC. Molecular and cellular aspects of sepsis-induced immunosuppression. J Mol Med (Berl). 2008;86:495-506
67. Li J. et al. Hydrogen-Rich Saline Promotes the Recovery of Renal Function after Ischemia/Reperfusion Injury in Rats via Anti-apoptosis and Anti-inflammation. Front Pharmacol. 2016;7:106
68. Liu YQ, Liu YF, Ma XM. et al. Hydrogen-rich saline attenuates skin ischemia/reperfusion induced apoptosis via regulating Bax/Bcl-2 ratio and ASK-1/JNK pathway. J Plast Reconstr Aesthet Surg. 2015;68:e147-56
69. Guo SX, Fang Q, You CG. et al. Effects of hydrogen-rich saline on early acute kidney injury in severely burned rats by suppressing oxidative stress induced apoptosis and inflammation. J Transl Med. 2015;13:183
70. Du H, Sheng M, Wu L. et al. Hydrogen-Rich Saline Attenuates Acute Kidney Injury After Liver Transplantation via Activating p53-Mediated Autophagy. Transplantation. 2016;100:563-70
71. Hong Y, Shao A, Wang J. et al. Neuroprotective effect of hydrogen-rich saline against neurologic damage and apoptosis in early brain injury following subarachnoid hemorrhage: possible role of the Akt/GSK3beta signaling pathway. PLoS One. 2014;9:e96212
72. Chen KY. et al. Hydrogen-Rich Saline Attenuates Brain Injury Induced by Cardiopulmonary Bypass and Inhibits Microvascular Endothelial Cell Apoptosis Via the PI3K/Akt/GSK3β Signaling Pathway in Rats. Cell Physiol Biochem. 2017;43:1634-47
73. Yu H, Zhang H, Zhao W. et al. Gypenoside Protects against Myocardial Ischemia-Reperfusion Injury by Inhibiting Cardiomyocytes Apoptosis via Inhibition of CHOP Pathway and Activation of PI3K/Akt Pathway In Vivo and In Vitro. Cell Physiol Biochem. 2016;39:123-36
74. Endo H, Nito C, Kamada H. et al. Akt/GSK3beta survival signaling is involved in acute brain injury after subarachnoid hemorrhage in rats. Stroke. 2006;37:2140-6
75. Teng LS. et al. Liquiritin modulates ERK- and AKT/GSK-3β-dependent pathways to protect against glutamate-induced cell damage in differentiated PC12 cells. Mol Med Rep. 2014;10:818-24
76. Cawcutt KA, Peters SG. Severe Sepsis and Septic Shock: Clinical Overview and Update on Management. Mayo Clinic Proceedings. 2014;89:1572-8
77. Miranda M, Balarini M, Caixeta D. et al. Microcirculatory dysfunction in sepsis: pathophysiology, clinical monitoring, and potential therapies. Am J Physiol Heart Circ Physiol. 2016;311:H24-35
78. Carnio EC, Stabile AM, Batalhao ME. et al. Vasopressin release during endotoxaemic shock in mice lacking inducible nitric oxide synthase. Pflugers Arch. 2005;450:390-4
79. Saramago EA, Borges GS, Singolani-Jr CG. et al. Molecular hydrogen potentiates hypothermia and prevents hypotension and fever in LPS-induced systemic inflammation. Brain, Behavior, and Immunity. 2019;75:119-28
80. Liu H, Liang X, Wang D. et al. Combination Therapy With Nitric Oxide and Molecular Hydrogen in a Murine Model of Acute Lung Injury. Shock. 2015;43:504-11
81. Sakai T, Sato B, Hara K. et al. Consumption of water containing over 3.5 mg of dissolved hydrogen could improve vascular endothelial function. Vasc Health Risk Manag. 2014;10:591-7
82. Ren JD, Wu XB, Jiang R. et al. Molecular hydrogen inhibits lipopolysaccharide-triggered NLRP3 inflammasome activation in macrophages by targeting the mitochondrial reactive oxygen species. Biochim Biophys Acta. 2016;1863:50-5
83. Noda K, Tanaka Y, Shigemura N. et al. Hydrogen-supplemented drinking water protects cardiac allografts from inflammation-associated deterioration. Transpl Int. 2012;25:1213-22
84. Murakami Y, Ito M, Ohsawa I. Molecular hydrogen protects against oxidative stress-induced SH-SY5Y neuroblastoma cell death through the process of mitohormesis. PLoS One. 2017;12:e0176992
85. Ostojic SM. Does H2 Alter Mitochondrial Bioenergetics via GHS-R1alpha Activation? Theranostics. 2017;7:1330-2
86. Martins AD, Sa R, Monteiro MP. et al. Ghrelin acts as energy status sensor of male reproduction by modulating Sertoli cells glycolytic metabolism and mitochondrial bioenergetics. Mol Cell Endocrinol. 2016;434:199-209
87. Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16:661-75
88. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323-35
89. Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol. 2016;138:28-36
90. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722-37
91. Deretic V, Kimura T, Timmins G. et al. Immunologic manifestations of autophagy. J Clin Invest. 2015;125:75-84
92. Saitoh T, Fujita N, Jang MH. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264-8
93. Lin CW, Lo S, Perng DS. et al. Complete activation of autophagic process attenuates liver injury and improves survival in septic mice. Shock. 2014;41:241-9
94. Mei S, Livingston M, Hao J. et al. Autophagy is activated to protect against endotoxic acute kidney injury. Sci Rep. 2016;6:22171
95. Unuma K, Aki T, Funakoshi T. et al. Extrusion of mitochondrial contents from lipopolysaccharide-stimulated cells: Involvement of autophagy. Autophagy. 2015;11:1520-36
96. Bai X, Liu S, Yuan L. et al. Hydrogen-rich saline mediates neuroprotection through the regulation of endoplasmic reticulum stress and autophagy under hypoxia-ischemia neonatal brain injury in mice. Brain Res. 2016;1646:410-7
97. Pan ZH. et al. Effect of hydrogen-rich saline on cardiomyocyte autophagy during myocardial ischemia-reperfusion in aged rats. Natl Med J China. 2015;95:2022-6
98. Chen J, Zhang H, Hu J. et al. Hydrogen-Rich Saline Alleviates Kidney Fibrosis Following AKI and Retains Klotho Expression. Front Pharmacol. 2017;8:499
99. Shi DJ. et al. Effects of hydrogen-rich saline on autophagy in rats underwent orthotopic liver transplantation. Pract J Organ Transplant. 2016;4:18-22
100. Shi DJ. et al. Role of autophagy in reduction of apoptosis in liver cells by hydrogen-rich saline in rats undergoing liver transplantation. Chin J Anesthesiol. 2016;36:546-50
101. Wu L, Du H. et al. Effect of hydrogen-rich saline on acute kidney injury after orthotopic liver transplantation and the role of autophagy. Chin J Anesthesiol. 2015;35:1202-6
102. Chen MX. et al. Role of autophagy in hydrogen-induced inhibition of apoptosis in hippocampal neurons in a rat model of orthotopic liver transplantation. Chin J Anesthesiol. 2017;37:184-7
103. Wang HX. et al. Effect of hydrogen-rich saline on autophagy in spinal neurons of rats with neuropathic pain. Chin J Anesthesiol. 2016;36:693-6
104. Ma HT. et al. Hydrogen-rich saline attenuates hyperalgesia and reduces cytokines in rats with post-herpetic neuralgia via activating autophagy. Chin J Cell Mol Immunol. 2017;33:155-63
105. Wang H, Huo X, Chen H. et al. Hydrogen-Rich Saline Activated Autophagy via HIF-1alpha Pathways in Neuropathic Pain Model. Biomed Res Int. 2018;2018:4670834
106. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22:367-76
107. Gao Y, Wang N, Liu L. et al. Relationship between mammalian target of rapamycin and autophagy in lipopolysaccharide-induced lung injury. J Surg Res. 2016;201:356-63
108. Maiuri MC, Galluzzi L, Morselli E. et al. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181-5
109. Moon JH, Lee JH, Lee YJ. et al. Hinokitiol protects primary neuron cells against prion peptide-induced toxicity via autophagy flux regulated by hypoxia inducing factor-1. Oncotarget. 2016;7:29944-57
110. Chen Y, Chen H, Xie K. et al. H2 Treatment Attenuated Pain Behavior and Cytokine Release Through the HO-1/CO Pathway in a Rat Model of Neuropathic Pain. Inflammation. 2015;38:1835-46
111. Malec V, Gottschald OR, Li S. et al. HIF-1 alpha signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic Biol Med. 2010;48:1626-35
112. Ohsawa I, Nishimaki K, Yamagata K. et al. Consumption of hydrogen water prevents atherosclerosis in apolipoprotein E knockout mice. Biochem Biophys Res Commun. 2008;377:1195-8
113. Zhou J, Chen Y, Huang GQ. et al. Hydrogen-rich saline reverses oxidative stress, cognitive impairment, and mortality in rats submitted to sepsis by cecal ligation and puncture. J Surg Res. 2012;178:390-400
Author contact
![]() Corresponding author: Jin Zhang, Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang 110004, China. E-mail: zhangj_sjcom Tel.: +86 18940259646
Corresponding author: Jin Zhang, Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang 110004, China. E-mail: zhangj_sjcom Tel.: +86 18940259646