Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(6):1299-1309. doi:10.7150/ijbs.32677 This issue Cite
Research Paper
Novel Role of Heterogeneous Nuclear Ribonucleoprotein E1 in Regulation of Apoptosis and Autophagy by a Triazole Derivative in Vascular Endothelial Cells
Ning Meng1 ![]() , Yan Gong1, Xin Mu1, Yan Hong Wang1, Le Su2, Cheng Shi Jiang1
, Yan Gong1, Xin Mu1, Yan Hong Wang1, Le Su2, Cheng Shi Jiang1 ![]() , Hua Zhang1
, Hua Zhang1 ![]()
1. School of Biological Science and Technology, University of Jinan, Jinan 250022, China.
2. State Key Laboratory of Biobased Material and Green Papermaking, School of Bioengineering, Qilu University of Technology, Shandong Academy of Sciences, Jinan, 250353, China
Received 2018-12-30; Accepted 2019-4-4; Published 2019-5-12
Abstract

Vascular endothelial cell (VEC) apoptosis and autophagy play an important role in the maintenance of vascular homeostasis. However, the association of molecular mechanisms between vascular endothelial cell apoptosis and autophagy has not been clarified. Here, we identified a novel triazole derivative, JL014, which could inhibit human umbilical vein vascular endothelial cell (HUVEC) apoptosis induced by deprivation of serum and fibroblast growth factor 2 and maintain HUVEC survival by promoting autophagy. Importantly, JL014 increased the mRNA and protein level of heterogeneous nuclear ribonucleoprotein E1 (hnRNP E1) in HUVECs. In addition, knockdown of hnRNP E1 by RNA interference inhibited the effects of JL014 on VEC apoptosis and autophagy. Furthermore, we investigated the effect of JL014 on the expression of HMBOX1, a key VEC apoptosis inhibitor and autophagy inducer by inhibiting mTOR signaling and the level of cleaved caspase-3. Our results demonstrated that JL014 enhanced mRNA transcription and increased protein synthesis of HMBOX1. JL014 also inhibited mTOR signaling and the cleaved caspase-3 level. Mechanistic studies revealed that hnRNP E1 could bind to the promoter and 5'UTR of HMBOX1 and active HMBOX1 expression. Therefore, our results firmly establish hnRNP E1 as a new regulator of VEC apoptosis and autophagy through mediating HMBOX1 expression, and opened the door to a novel therapeutic drug for related vascular diseases.
Keywords: heterogeneous nuclear ribonucleoprotein E1, autophagy, apoptosis, vascular endothelial cell
Introduction
Vascular endothelial cells (VECs), lying in the inner of blood vessels, are the most abundant component of blood vessel and participate in the exchange of substances between blood and surrounding tissue. In addition to acting as a barrier to substance exchange, VECs produce and secrete a variety of bioactive substances and regulate vascular physiological functions such as hemostasis, angiogenesis, hormone trafficking, inflammation, among others [1]. Due to direct contact to blood, chemical and physical stress from circulation system can easily impair the function and structural integrity of nendothelium, resulting in a cascade of events of cardiovascular diseases, which in turn worsen the injury of VEC and cause the apoptosis of endothelial cells [2-5]. Research evidences have proven that the VEC apoptosis is an initiating factor in numerous cardiovascular diseases, such as arteriosclerosis, arteritis, thrombus and ischemic stroke [6]. Therefore, inhibiting endothelial cell apoptosis is an efficient strategy to prevent cardiovascular diseases.
Autophagy, literally meaning 'self-eating', is a cellular process of degradation of the internal material, such as proteins or organelles, in lysosomes [7, 8]. Autophagy is of vital importance for the maintenance of cellular homeostasis and organelle function in most cells, including cardiomyocytes, endothelial cells and arterial smooth muscle cells [9]. Increasing research evidences revealed that autophagy was dysfunctional under numerous pathological conditions and moderate or adequate autophagy could protect VECs from injury [10-12]. On the contrary, autophagy can induce cell death while it exceeds a certain level [13]. Nowadays, researchers have demonstrated that there were crosstalk between autophagy and apoptosis and the two phenomena involved common molecular mediators [14]. However, the double-edge sword roles of autophagy make it difficult to clarify the molecular mechanisms between VEC apoptosis and autophagy.
Recent years, using small chemical molecules to modulate biological phenomenon and reveal insights into diverse biological questions has made significant influence on various areas of development biology and cell biology [15]. Here, using deprivation of serum and fibroblast growth factor 2 condition, a classic VEC apoptosis model [16-18], we identified a 1,2,3-triazole derivative (5-(1-(3,4-difluorobenzyl)-1H- 1,2,3-triazol-4-yl)-2-methyl-N-(2-morpholinoethyl)benzamide) named JL014 which could inhibit VEC apoptosis and maintain VECs survival by promoting autophagy. Interestingly, compound JL014 increased heterogeneous nuclear ribonucleoprotein E1 (hnRNP-E1) mRNA and protein levels. HnRNP E1, a poly(C)-binding protein, has been identified to be ubiquitously expressed in various tissues and to regulate different steps of gene expression, including transcription, mRNA stability and translation [19]. Over the last two decades researches have demonstrated the multifunction of hnRNP E1 plays important roles in maintaining cellular homeostasis during different physiological processes. HnRNP E1 has been proven to promote cell apoptosis in several cancer cell lines [20-22]. Recently, a research group reported that hnRNP E1 repressed autophagy through downregulation of LC3B to promote tumor cell apoptosis [23]. In endothelial cells, hnRNP E1 serves as a key protective mechanism through increasing human endothelial nitric oxide synthase (eNOS) mRNA stability and we found hnRNP E1 is a new regulator of endothelium-protein disulphide isomerase translation in oxLDL-activated VECs [24, 25]. Although hnRNPE1 possess these important roles in VECs, whether hnRNP E1 mediates the association of VEC apoptosis and autophagy is still unknown. In this study, we explored the role and mechanism of hnRNP E1 in regulating VEC apoptosis and autophagy using the small chemical molecule JL014.
Materials and Methods
Synthesis of JL014
Compound JL014 was synthesized by the CuI-catalyzed Azide-Alkyne cycloddition reaction of 4-(bromomethyl)-1,2-difluorobenzene with 5-ethynyl- 2-methyl-N-(2-morpholinoethyl) benzamide, which was obtained from methyl 5-bromo-2-methylbenzoate as starting material according to our previous report [26]. Compound JL014 was dissolved in DMSO (0.1 M, Sigma-Aldrich, D2650) as a stock solution, and its chemical structure was shown in supplemental Fig.1A.
Cell culture and treatment
Human umbilical vascular endothelial cells (HUVECs) were obtained from human umbilical veins as described [27]and were cultured on gelatin-coated plastic dishes in M199 medium (Gibco Laboratories, Grand Island, NY) with 10% (v/v) fetal bovine serum (Gibco Laboratories, Grand Island, NY) and 10 IU/mL fibroblast growth factor 2. The cells were maintained at 37 °C under humidified conditions and 5% CO2. HUVECs at no more than passage 10 were used in the experiments. Confluent HUVECs were treated with 0.01% (v/v) DMSO (control group), 5 μM or 10 μM JL014 and cultured in basal M199 medium without FGF-2 for the indicated time. Morphologic changes of HUVECs were observed by inverted phase-contrast microscopy (ZEISS, Primovert, Germany).
Hoechst 33258 staining
Nuclear fragmentation of cells was examined by Hoechst 33258 staining. Briefly, after treatment, HUVECs were incubated with Hoechst 33258 (2 μg/ml, Sigma-Aldrich, USA) away from light for 15 min at 37 °C. Stained cells were washed twice with PBS and then viewed under a Leica DMI8 (Germany) fluorescence microscope.
Cell viability assay
Cell viability was detected using Sulforhodamine B assay (SRB; Sigma-Aldrich, USA). Briefly, HUVECs were plated in 96-well plates. After the specific treatments, they were precipitated for 1 h at 4°C with 100 μl 10% trichloroacetic acid (Shenggong Biotech, Shanghai, China) and gently rinsed with deionized water for 5 times. Then the cells were stained with 50 μl sulforhodamine B and rinsed 5 times with 1% glacial acetic acid solution. Finally, 100 μl of 10 mM Trisbase (pH 10.5) was added to dissolve the conjugation. The absorbance was detected at 540 nm wavelength using a microplate spectrophotometer (TECAN, USA). Cell viability (%) = (OD of treated group/OD of control group) × 100.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay
The TUNEL assay was performed as described. Briefly, cells were fixed with 4% paraformaldehyde (Dingguo, Beijing, China) at 37°C for 15min. After washing with PBS three times, cells were permeabilized in 0.1% Triton X-100 on the ice board for 2 min. Then the cells were incubated with TUNEL reaction mixture for 1 h at 37°C in the moist box. TUNEL-positive cells were assessed by laser scanning confocal microscopy (Leica, DMI8, Germany) after washing in PBS three times.
Western blotting
Protein was extracted from cells using radio immunoprecipitation assay (RIPA) lysis buffer (Dingguo, Beijing, China) containing proteinase inhibitors (Sigma-Aldrich, USA). The lysate was centrifuged at 12,000 rpm for 15 min at 4°C. The supernatant was then collected and the protein concentration was determined using BCA Protein Assay kits (Beyotime, China). Equal amounts of protein lysates (15 μg per lane) were resolved on 15% SDS-PAGE gels and transferred onto PVDF membrane (Millipore, Madison, WI, USA). The membrane was blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h to reduce non-specific binding. The membrane was incubated overnight at 4°C with primary antibodies. After washing with TBST three times, membranes were incubated with HRP-conjugated secondary antibody (Dingguo, Beijing) for 1 h at room temperature. Immune complexes were detected by enhanced chemiluminescence (Millipore Corporation, Billerica, USA). Integrated densities of bands were quantified by Image J software. Antibodies for hnRNP-E1 (#8534), LC3B (#2775), β-actin (#4970), phospho-p70 S6 Kinase (#9205), and Phospho-4EBP1 (#2855) were brought from Cell Signaling Technology (USA). 4EBP1 (60246-1-lg), p70S6K (14485-1-AP), HMBOX1 (16123-1-AP), caspase-3 (19677-1-AP) and Peroxidase-conjugated AffiniPure Goat Anti-Rabbit/ Mouse IgG (H+L) were all from Jackson ImmunoResearch (USA).
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde, blocked with normal donkey serum (1:30) for 30 min and incubated with LC3B (#2775,1:100) antibody from Cell Signaling Technology (USA) for 18 h at 4°C. Then, cells were incubated with Alexa Fluor 488/546-conjugated Affini Pure Goat Anti-Rabbit IgG (H+L) (1:200) for 1 h at 37°C in the dark. After washing with PBS, the immunofluorescence was examined by a Leica DMI8 inverted fluorescent microscope.
RNA interference (RNAi)
Cells at 50% to 60% confluence were transfected with hnRNP E1 (Santa Cruz Biotechnology, sc-38268), HMBOX1 which was designed and synthesized by Invitrogen, or scrambled siRNA (Santa Cruz Biotechnology, sc-37007) at 20-80 nM for 24 h by use of lipofectamine 2000 according to the manufacturer's protocol (Invitrogen, USA). The efficiency of RNA interference was determined by western blotting and 60 nM siRNA against HMBOX1 and 20 nM siRNA against hnRNP E1 were used in the following knockdown experiments.
Quantitative real-time PCR (qPCR)
Total RNA was extracted using RNAiso Plus (TaKaRa, Japan) in an RNAase-free environment. The reverse transcription of cDNA was synthesized with PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, Japan) from 1μg RNA. The product was performed for qPCR in PCR 8-TUBE STRIP in a volume of 20 μl including UltraSYBR Mixture (cwbio, China). Primers were used as follows: Fwd hnRNP-E1: 5'-GGCGGGTGTAAGATCAAAGA-3'; Rev hnRNP-E1: 5'-GAGCGGAGAAATGGTGTGTT -3'; Fwd HMBOX1: 5'-AGCATGGGTCAGAGGTCATACAG-3'; Rev HMBOX1: 5'-GGAAAGTACCAGATGTGGCAG-3'; Fwd GAPDH: 5'-GAAGTGTGACGTGGACATCC-3'; Rev GAPDH: 5'-CCGATCCACACGGAGTACTT-3'.
RNA-chromatin immunoprecipitations (RNA-ChIP) assay
RNA-ChIP involved using of the RNA ChIP-IT Kit (Active Motif, Carlsbad, CA, USA, 53024). HUVECs were fixed with 1% formaldehyde and gathered in PBS containing complete protease inhibitor and PMSF and 1U/μL RNase inhibitor. Cells were pelleted by centrifugation for 15 min at 1000 g and resuspended with lysis buffer, then chromatin was sheared by ultrasonication. After spinning at 18 000 g for 15 min, supernatant was treated with DNaseI, then used for precipitation of the hnRNP E1-RNA complexes with hnRNP E1 or control IgG antibodies. Co-precipitated RNA was purified with TRIZOL reagent, treated with DNase I. RNA was reverse-transcribed to cDNA and quantified by qPCR with seven primers separately (Supplementary Table S1 A-G).
Chromatin immunoprecipitation (ChIP)
ChIP involved using of the ChIP kits (Millipore, 17-10086). Briefly, cells were fixed with 1% formaldehyde for 10 min and washed with cold PBS containing 5μL/ml cocktail II. Lysed cells were centrifugalized with 800 g at 4°C and sonication was used to shear chromatin to an approximate length of 500 bp. An amount of 50 μl of chromatin was immunoprecipitated with 6 μg hnRNP E1 antibody and 1 μg normal rabbit IgG for negative control at 4°C with rotation for 18 h, then, 20 μl fully resuspended protein A/G magnetic beads were added. The Protein A/G bead-antibody/chromatin complexes were washed and eluted. Then the cross-links of protein/DNA complexes were reversed to free DNA. Finally, 50 μl DNA solution was obtained for further analysis. Six primers (a-e) for PCR were provided in Supplementary Table S1.
Statistical analysis
Data are presented as mean ± SEM. Statistical analyses involved SPSS 11.5 (SPSS Inc., Chicago, IL). Images were processed by use of Graphpad Prism 5 (GraphPad Software, La Jolla, CA, USA) and Adobe Photoshop (Adobe, San Jose, USA). At least three independent replications were performed. Data were compared by one-way ANOVA. p <0.05. was considered statistically significant.
Results
JL014 inhibited HUVEC apoptosis and maintained HUVEC survival by promoting autophagy
Deprivation of serum and fibroblast growth 2 was used to induce VECs apoptosis. The SRB assay results showed that 5-20 μM JL014 significantly increased cell viability (Supplemental Fig.1B). Then, hoechst33258 staining was performed to detect nuclear DNA condensation and fragmentation, characteristics of apoptosis. We found that in the control groups, about 60% cells with nuclear condensation and fragmentation, while after JL014 treatment, the percentage of apoptosis cells significantly decreased to around 20% (Fig.1A and B). Results from TUNEL assay showed that JL014 significantly reduced the number of TUNEL-positive cells (Fig.1A). The protein level of cleaved-PARP, one of the main characteristics of cell apoptosis, was decreased after treatment with compound JL014, confirming that JL014 could inhibit HUVECs apoptosis (Fig.1D and E).
JL014 inhibited HUVEC apoptosis and maintained HUVEC survival by promoting autophagy. HUVECs were treated with 5 μM or 10 μM JL014 in basal M199 medium with serum and FGF-2 deprivation for 12 h. (A) Microscopy images of the morphological changes of HUVECs (10×). Nuclear fragmentation of cells by Hoechst 33258 and TUNEL staining (10×). Immunostaining of LC3 puncta (20×); (B) Bar chart showing quantification of HUVEC apoptosis percentage according to Hoechst 33258 staining; (C) Bar chart showing quantification of LC3 puncta per cell (D) Western blot analysis of LC3 and PARP protein levels; (E) The protein level of LC3-II and cleaved-PARP were calculated based on densitometry analysis. (F) Western blot analysis of LC3 protein level with 10 μM JL014, 100 nM bafilomycin A1 (Baf A1), or JL014 + Baf A1 for 12 h; (G) The protein level of LC3-II was calculated based on densitometry analysis. Data are means ± SEM. *P < 0.05 and **P < 0.01. n=3.
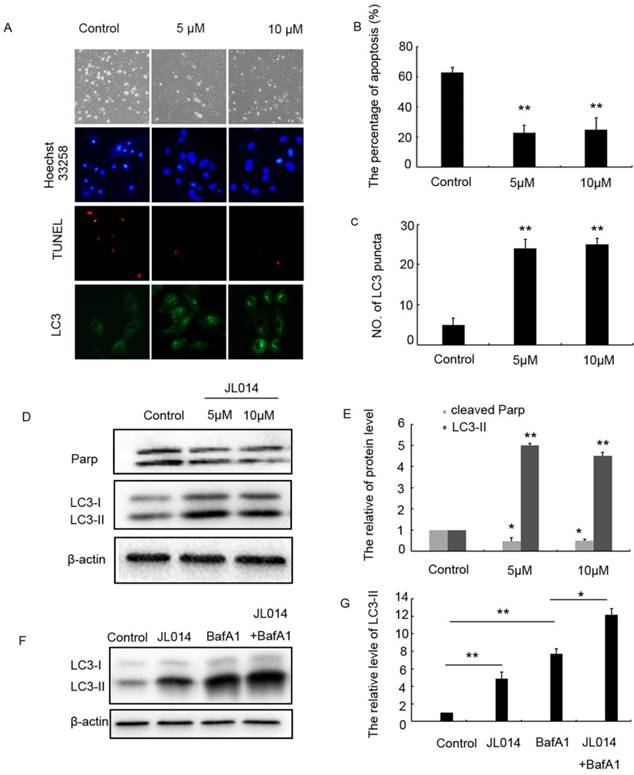
In light of the relationship between apoptosis and autophagy, we investigated the effect of JL014 on LC3 processing and LC3-II accumulation that specific hallmarks of autophagy. Immunofluorescence and western blot assay results demonstrated in the cells with different concentration of JL014 treatment, the numbers of LC3 puncta in cytoplasm were greatly increased and the LC3-II level was elevated significantly (Figs.1 C, 1D and 1 E). Moreover, in the presence of Bafilomycin A1 (Baf A1) which could prevent fusion of autophagosomes with lysosomes and was used to detect autophagic flux, JL014 could still increase the protein level of LC3-II (Figs.1F and 1G). These results suggested that JL014 induced VEC autophagy. Autophagy could act as cytoprotective response or death phenomenon, we further examined whether JL014-induced autophagy maintained VEC survival. Using autophagy pharmacological inhibitor 3-MA and ATG5 siRNA to inhibit autophagy procession, we found that in 3-MA treated cells or ATG5 siRNA treatment groups, the increased cell viability by JL014 was inhibited (Supplemental Figs.1C and 1D).
hnRNP E1 mediated the inhibition of apoptosis and promotion of autophagy induced by JL014
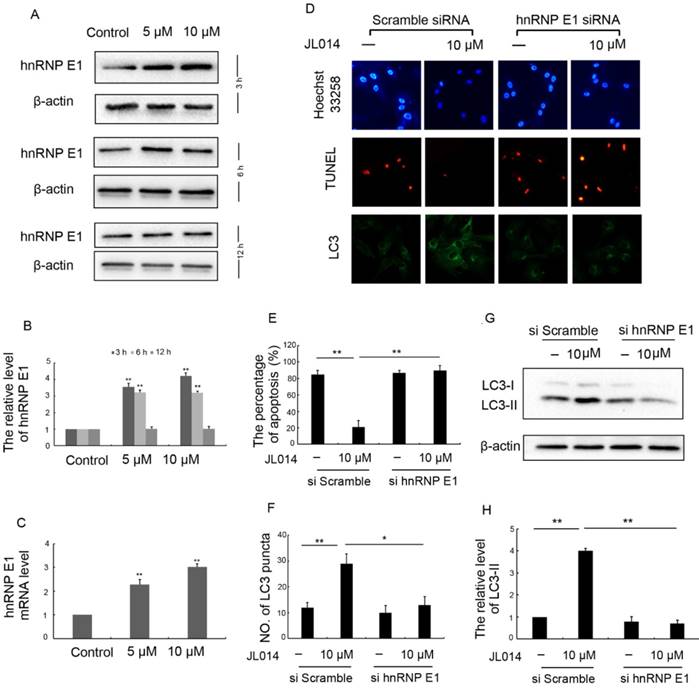
We first examined the effect of JL014 on hnRNP E1 expression at the mRNA and protein levels. Results revealed that JL014 increased the hnRNP E1 mRNA level and promoted the protein expression of hnRNP E1 (Figs.2A, 2B and 2C). To further determine whether the induction of autophagy and inhibition of apoptosis by JL014 is mediated by hnRNP E1, we knocked down the expression of hnRNP E1 using RNA interference. Compared with scramble siRNA groups, JL014 could not reduce the apoptosis cell numbers in hnRNP E1 siRNA group (Figs. 2D and 2E). In addition, knockdown of hnRNP E1 significantly inhibited the endogenous LC3 aggregation and reduced the LC3-II protein level (Figs. 2D, 2F, 2G and 2H).
hnRNP E1 mediated the inhibition of apoptosis and promotion of autophagy induced by JL014. (A) HUVECs were treated with 5 μM or 10 μM JL014 in basal M199 medium with serum and FGF-2 deprivation for 3h, 6h and 12 h and western blot analysis of hnRNP E1 protein levels; (B) The protein level of hnRNP E1 were calculated based on densitometry analysis; (C) qPCR analysis of hnRNP E1 mRNA levels after JL014 treated HUVECs 6 h; (D-H) HUVECs were treated with 20 nM hnRNP E1 siRNA or scramble siRNA for 36 h and stimulated with 10 μM JL014 for up 12 h and (D) Nuclear fragmentation of cells by Hoechst 33258 and TUNEL staining (10×). Immunostaining of LC3 puncta (20×); (E) Bar chart showing quantification of HUVEC apoptosis percentage according to Hoechst 33258 staining; (F) Bar chart showing quantification of LC3 puncta per cell; (G) Western blot analysis of LC3 protein level; (H) The protein level of LC3-II were calculated based on densitometry analysis. Data are means ± SEM. *P < 0.05 and **P < 0.01. n=3.
JL014 inhibited HUVEC apoptosis and promoted autophagy through enhancing HMBOX1 expression
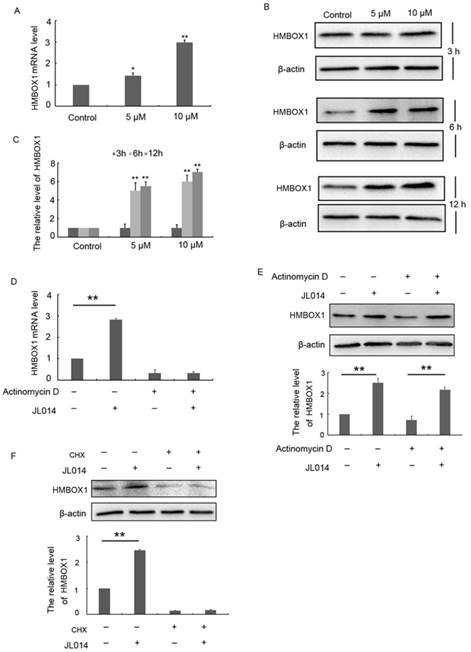
Recently, HMBOX1 was identified as a key VEC apoptosis inhibitor and autophagy inducer [18]. Here, we examined whether JL014 inhibited VEC apoptosis and promoted autophagy through HMOBX1. Results from qPCR and Western blotting analysis demonstrated that the mRNA and protein levels of HMBOX1 were increased by compound JL014 (Figs.3A, 3B and 3C). The up-regulated HMBOX1 mRNA and protein level evoked by JL014 could be either due to enhanced mRNA stability, transcription or increased protein synthesis. We examined the effect of JL014 on HMBOX1 mRNA and protein level in the presence or absence of actinomycin D, a widely used intercalating transcription inhibitor. JL014 failed to increase HMBOX1 mRNA level in the presence of actinomycin D, suggesting that JL014 did not affect HMBOX1 mRNA stability but increased HMBOX1 transcription. Moreover, JL014 could elevate HMBOX1 protein level in the presence of actinomycin D (Figs. 3D and 3 E). However, cycloheximide (CHX, 10 μg/ml), an inhibitor of protein biosynthesis in eukaryotic organisms, completely inhibited JL014-increased HMBOX1 protein level (Fig. 3F). These results suggested JL014 might enhance transcription and protein synthesis of HMBOX1.
JL014 enhanced transcription and protein synthesis of HMBOX1. (A) HUVECs were treated with 5 μM or 10 μM JL014 in basal M199 medium with serum and FGF-2 deprivation for 6 h and qPCR analysis of HMBOX1 mRNA levels; (B) HUVECs were treated with 5 μM or 10 μM JL014 in basal M199 medium with serum and FGF-2 deprivation for 3 h, 6 h and 12 h and western blot analysis of HMBOX1 protein levels; HUVECs were treated with 2 μg/ml actinomycin D for 3 h and then were treated with 10 μM JL014 for 12 h and (C) qPCR analysis of HMBOX1 mRNA level and (D) western blot analysis of HMBOX1 protein level; (E) HUVECs were treated with 10 μg/ml cycloheximide (CHX), 10 μM JL014 or both for 12 h. Western blot analysis of HMBOX1 protein level. Data are means ± SEM. *P < 0.05 and **P < 0.01. n=3.
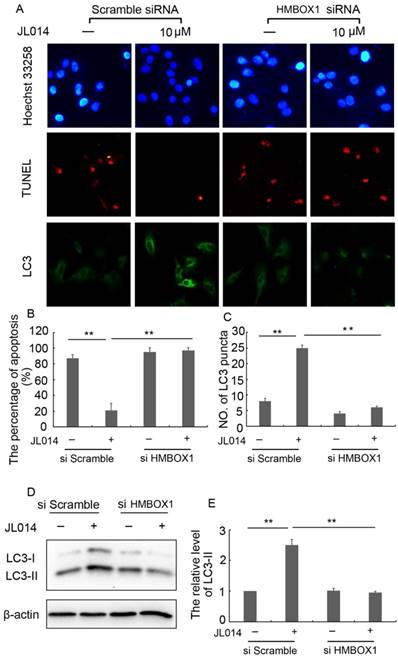
Moreover, in scramble siRNA treatment groups, JL014 significantly inhibited the VEC nuclear condensation, reduced the number of TUNEL-positive cells and induced LC3 processing and LC3-II accumulation. However, these effects of compound JL014 were inhibited by knockdown of HMBOX1 (Fig.4.), suggesting HMBOX1 was involved in JL014-induced autophagy and apoptosis inhibition.
HMBOX1 was involved in JL014-induced autophagy and apoptosis inhibition. HUVECs were treated with 60 nM HMBOX1 siRNA or scramble siRNA for 36 h and stimulated with 10 μM JL014 for up to 12 h. (A) Nuclear fragmentation of cells by Hoechst 33258 and TUNEL staining (10×). Immunostaining of LC3 puncta (20×); (B) Bar chart showing quantification of HUVEC apoptosis percentage according to Hoechst 33258 staining; (C) Bar chart showing quantification of LC3 puncta per cell (D) Western blot analysis of LC3 protein levels; (E) The protein level of LC3-II were calculated based on densitometry analysis. Data are means ± SEM. *P < 0.05. n=3.
hnRNP E1 regulated the HMBOX1 mRNA transcription and protein translation
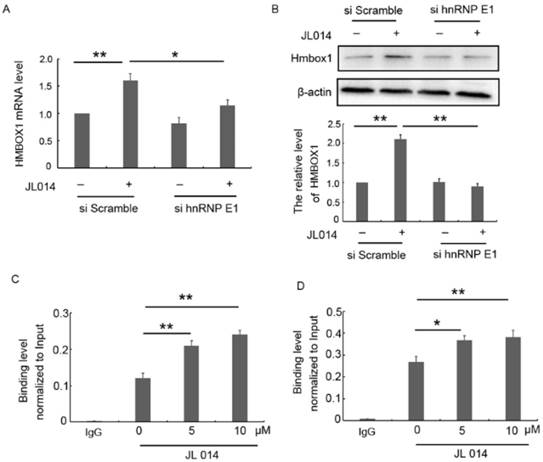
hnRNP E1, a poly(C)-binding protein, has been shown to function as a transcriptional activator and a translational coactivator through directly binding to promoter region or mRNA. Here, we found that siRNA-mediated silencing of hnRNP E1 significantly inhibited the elevated HMBOX1 mRNA and protein level induced by JL014 (Figs.5A and 5B.), suggesting that hnRNP E1 regulated HMBOX1 expression. Subsequently, we predicted six binding sites 2-kb upstream of the HMBOX1 promoter region and seven regions across HMBOX1 5'UTR and 3'UTR. ChIP and RNA-ChIp were used to detect hnRNP E1 occupancy at each of these regions with primers specific for the predicted regions. ChIp results showed that hnRNP E1 bound to the region (e) predicted binding site (Fig. 5C), with no binding ability with the other 5 sites (Supplementary Figs. 2A and 2B). The results from RNA-chip showed that hnRNP E1 directly bound to HMBOX1 5' UTR (region A), in which cytosine is rich (Fig. 5D and Supplementary Figs. 2C and 2D). Moreover, after JL014 treatment, with the increased hnRNP E1 protein level, the binding level of HMBOX1 promoter and 5' UTR to hnRNP E1 were also significantly increased. These results indicated that hnRNP E1 might regulate HMBOX1 transcription and protein synthesis through binding to the promoter and 5' UTR of HMBOX1.
hnRNP E1 regulated the HMBOX1 mRNA transcription and protein translation. (A-B) HUVECs were treated with 20 nM hnRNP E1 siRNA or scramble siRNA for 36 h and stimulated with 10 μM JL014 for up 12 h and (A) qPCR analysis of HMBOX1 mRNA levels; (B) Western blot analysis of HMBOX1 protein levels; (C-D) HUVECs were treated with 5 μM or 10 μM JL014 in basal M199 medium with serum and FGF-2 deprivation for 3 h and submitted to (C) ChIP-qPCR and (D) RNA-ChIP-qPCR analysis. Data are means ± SEM. *P < 0.05 and **P < 0.01. n=3.
JL 014 inhibited mTOR signaling and the cleaved caspase-3 level through hnRNP E1
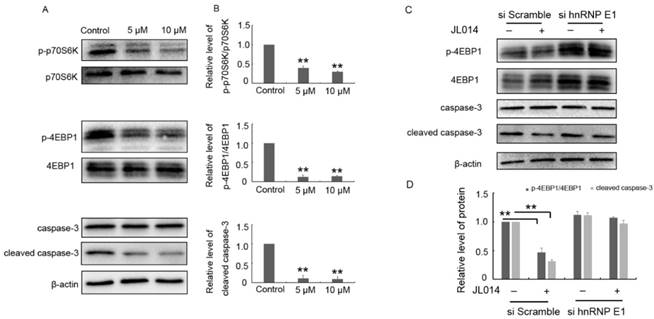
Previously studies have clarified HMBOX1 regulated VEC apoptosis and autophagy through inhibiting mTOR and cleaved caspase-3 level [18]. Here, the results from western blotting analysis demonstrated that JL014 inhibited the phosphorylation of mTOR downstream targets p70S6K and 4E-binding protein 1 and decreased the cleaved caspase-3 level (Fig.6 A and B.). Moreover, after siRNA hnRNP E1, JL014 could not inhibited the phosphorylation of 4E-binding protein 1 and decreased the cleaved caspase-3 level, suggesting that JL014 affected mTOR and apoptosis signaling pathway through the hnRNP E1 (Fig.6 C and D.).
JL014 inhibited mTOR signaling and the cleaved caspase-3 level. (A-B) HUVECs were treated with 5 μM or 10 μM JL014 in basal M199 medium with serum and FGF-2 deprivation for 6 h and (A) Western blot analysis of p70S6K, phosphorylated p70S6K (p-p70S6K, Thr389), 4EBP1, phosphorylated4EBP1 (p-4EBP1, Thr37/46), caspase 3 and cleaved caspase 3 protein levels; (B) Densitometry results of ratio of p-p70S6K to total p70S6K, p-4EBP1 to total 4EBP1 and cleaved caspase 3. (C-D) HUVECs were treated with 20 nM hnRNP E1 siRNA or scramble siRNA for 36 h and stimulated with 10 μM JL014 for up 6 h and (C)Western blot analysis of 4EBP1, p-4EBP1(Thr37/46), caspase 3 and cleaved caspase 3 protein levels; (D) Densitometry results of ratio of p-4EBP1 to total 4EBP1 and cleaved caspase 3. Data are means ± SEM. **P < 0.01. n=3.
Discussion
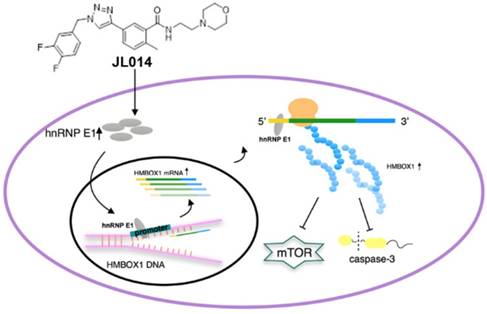
In this study, we identified compound JL014, 1,2,3-triazole derivative, as a potent VEC apoptosis inhibitor and autophagy inducer. Using JL 014 as a tool, we demonstrated that hnRNP E1 was a key factor in association of VEC apoptosis and autophagy. The major mechanism underlying this process might be that hnRNP E1 could bind to the promoter and 5'UTR of HMBOX1, and thus stimulated HMBOX1 expression and subsequently inhibited mTOR signaling and cleaved caspase-3 level.
Vascular endothelial cells which line the interior surface of blood vessels are involved in various aspects of vascular functions, including vasoconstriction, barrier function, angiogenesis, etc. Thus, endothelial cell injury and dysfunction play important roles in the development and pathogenesis of cardiovascular disease. VEC apoptosis, a hallmark of endothelium injury, can destroy the structure and function of the endothelium and thus accelerate the progression of cardiovascular disease including atherosclerosis. Recent years, autophagy, an evolutionarily conserved catabolic pathway, has attracted increasing attention. Under basal conditions, autophagy maintains cellular homeostasis by degradation and the recycling of damaged organelles and misfolded proteins. Under stress conditions, moderate autophagy could act as a protective mechanism preventing cell death[10, 28]. Current evidences suggest apoptosis and autophagy are two closely processes that maintain vascular homeostasis. Recently, some small chemical molecule were found to alleviate the development of some vascular disease through inhibiting VEC apoptosis and promoting VEC autophagy [29-32]. Therefore, exploring novel VEC apoptosis inhibitor and autophagy inducer has great significance. In this study, we discovered a novel 1,2,3-triazole derivative (JL014) which could inhibit HUVEC apoptosis induced by deprivation of serum and fibroblast growth factor 2 and maintain HUVEC survival by enhancing autophagy. JL014 was firstly synthesized in our research group and it exhibited neuroprotective activity against Aβ25-35-induced neurotoxicity in SH-SY5Y cells. Moreover, JL014 did not exhibit significant cytotoxic effects on both HaCaT and NIH-3T3 cell lines [26]. These findings demonstrated that JL014 might be a cell protective agent and could be used as a useful tool to explore the association of VEC apoptosis and autophagy mechanisms.
HnRNP E1, Poly(rC)-binding protein 1 (PCBP1), was abundantly expressed in various tissues and played different physiological roles. A growing body of evidence has shown that hnRNP E1 is a tumor suppressor through regulating many cancer-related genes expression [33]. Especially, hnRNP E1 repressed autophagy and promoted apoptosis in ovary tumors [23]. In normal endothelial cells, hnRNP E1 increased the eNOS, which was involved in the maintenance of homeostasis in the blood vessel wall, mRNA stability and protein expression [25]. In oxLDL activated VECs, phosphorylated hnRNP E1 triggered PDI overproduction which might contribute to thrombus formation[24]. These research studies revealed that hnRNP E1 played important roles in mediating VEC physiological functions. Here, we found that during the process of JL014-induced inhibition of VEC apoptosis and promotion of VEC autophagy, hnRNP E1 mRNA and protein level significantly increased and knockdown of hnRNP E1 inhibited the effects of JL014 on VEC apoptosis and autophagy, suggesting hnRNP E1 might be an important factor in the association of VEC apoptosis and autophagy. Interestingly, our results demonstrated that hnRNP E1 might has opposite roles in regulating VEC autophagy and apoptosis compared with tumor cells. This might be due to the different mechanisms by which hnRNP E1 regulates autophagy and apoptosis under different conditions or in different cell types.
Researchers have confirmed that hnRNP E1 participates in stability, transcriptional and translational regulation of target gene through binding to DNA or RNA and thus control the cell physiological process[19]. Here, we found JL014 enhanced HMBOX1 transcription and protein syntheses and HMBOX1 was involved in JL014-induced VEC autophagy and apoptosis inhibition. HMBOX1, first known as a transcription repressor, is abundantly expressed in the cytoplasm of VECs. Moreover, HMBOX1 has been shown to maintain VEC survival by inducing autophagy and inhibiting apoptosis [18]. Interestingly, results from our studies demonstrated that knockdown of hnRNPE1 inhibited the mRNA and protein increased by JL014, suggesting hnRNP E1 might be an upstream mediator of HMBOX1 expression. It is reported that hnRNP E1 functions as a transcriptional activator of the mouse mo-pioid receptor gene by binding to a polypyrimidine stretch within the proximal promoter. In addition, hnRNP E1 has been proven to function as both translational coactivator and co-repressor. For example, hnRNP E1 could bind to the 5'-UTR of Bag-1 mRNA and stimulate it translation [34]. On the other hand, hnRNP E1 causes translational repression of genes through binding to the 5'-UTR or 3'-UTR of mRNA [24, 35, 36]. Our studies showed that hnRNPE1 bound to the HMBOX1 promoter region and 5'UTR region, in which cytosine is rich, and JL014 increased the binding levels. Based on these results, we speculated hnRNP E1 might function through binding to HMBOX1 DNA or mRNA to induce HMBOX1 expression.
Additionally, the recently studies have clarified the mechanisms of HMBOX1 regulating autophagy and apoptosis in VECs. HMBOX1 elevated the level of intracellular free zinc by interacting with MT2A, the up-regulated zinc level inhibited mTOR signaling and promoted VEC autophagy. Moreover, the altered zinc level affects the level of cleaved caspase-3 and thus inhibits VEC apoptosis[18]. Here, we found that JL014 inhibited the phosphorylation of mTOR downstream targets p70S6K and 4E-BP1 and decreased the cleaved caspase-3 level through hnRNP E1. These results further confirmed hnRNP E1 associated VEC apoptosis and autophagy through mediating HMBOX1 expression and thus inhibited downstream mTOR signal and the level of cleaved caspase-3.
In summary, our study revealed that a novel 1,2,3-triazole derivative named JL014 could inhibit VEC apoptosis and maintained VEC survival by promoting autophagy. JL014 increased the mRNA and protein level of hnRNP E1. With the hnRNP E1 protein increased, the binding level of HMBOX1 promoter and 5' UTR to hnRNP E1 were also significantly increased. These bindings might mediate the HMBOX1 transcription and protein synthesis. Furthermore, the elevated HMBOX1 inhibited mTOR signaling and the cleaved caspase 3 level (Fig.7.). By identifying hnRNP E1 as a regulator of HMBOX1 expression, we demonstrated how hnRNP E1 regulated apoptosis and autophagy in HUVECs and compound JL014 is a useful tool to study the association molecular mechanisms of apoptosis and autophagy in VECs. There are some issues should be addressed in the future studies. One is that as JL014 is a VEC apoptosis inhibitors and apoptosis is also a normal and important physiological process, whether JL014 affects the apoptosis of other cell types in vivo. In addition, whether hnRNP E1 is a key VEC apoptosis and autophagy regulator in some pathological conditions such as atherosclerosis and whether it is a useful way to inhibit the progression of related cardiovascular diseases through modulating hnRNP E1 by using of JL014.
Conceptual schematic of JL014 and hnRNP E1 action mechanism in regulating VEC apoptosis and autophagy. JL014 increased the mRNA and protein level of heterogeneous nuclear ribonucleoprotein E1 (hnRNP E1) in HUVECs. hnRNP E1 could bind to the promoter and 5'UTR of HMBOX1 and active HMBOX1 expression. The elevated HMBOX1 inhibited mTOR signaling and promoted VEC autophagy. On the other hand, the elevated HMBOX1 inhibited the cleaved caspase 3 level and inhibited VEC apoptosis.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This research work was financially supported by the National Natural Science Foundation of China (No. 31671214), the Young Taishan Scholars Program (No. tsqn20161037), Shandong Talents Team Cultivation Plan of University Preponderant Discipline (No. 10027) and The Natural Science Foundation of Shandong Province (No. ZR2016CM01). The authors declare no competing financial interests.
Abbreviations
HUVECs, human umbilical vein endothelial cells; RNAi, RNA interference; siRNA, small interfering RNA; VECs, Vascular endothelial cells; hnRNP E1, heterogeneous nuclear ribonucleoprotein E1; eNOS, human endothelial nitric oxide synthase; TBST, Tris-buffered saline containing 0.1% Tween-20; RNA- ChIP, RNA-chromatin immunoprecipitations; ChIP, Chromatin immunoprecipitation; Baf A1, Bafilomycin A1; CHX, cycloheximide.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Khaddaj Mallat R, Mathew John C, Kendrick DJ, Braun AP. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit Rev Clin Lab Sci. 2017;54:458-70
2. Luchetti F, Crinelli R, Cesarini E, Canonico B, Guidi L, Zerbinati C. et al. Endothelial cells, endoplasmic reticulum stress and oxysterols. Redox Biol. 2017;13:581-7
3. Ghosh A, Gao L, Thakur A, Siu PM, Lai CWK. Role of free fatty acids in endothelial dysfunction. J Biomed Sci. 2017;24:50
4. Dong Y, Fernandes C, Liu Y, Wu Y, Wu H, Brophy ML. et al. Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis. Diab Vasc Dis Res. 2017;14:14-23
5. Bordy R, Totoson P, Prati C, Marie C, Wendling D, Demougeot C. Microvascular endothelial dysfunction in rheumatoid arthritis. Nat Rev Rheumatol. 2018;14:404-20
6. Xu K, Liu P, Zhao Y. Upregulation of microRNA-876 Induces Endothelial Cell Apoptosis by Suppressing Bcl-Xl in Development of Atherosclerosis. Cell Physiol Biochem. 2017;42:1540-9
7. Kim J, Lim YM, Lee MS. The Role of Autophagy in Systemic Metabolism and Human-Type Diabetes. Mol Cells. 2018;41:11-7
8. Tan Y, Gong Y, Dong M, Pei Z, Ren J. Role of autophagy in inherited metabolic and endocrine myopathies. Biochim Biophys Acta Mol Basis Dis. 2018;1865:48-55
9. Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and Mitophagy in Cardiovascular Disease. Circ Res. 2017;120:1812-24
10. Vion AC, Kheloufi M, Hammoutene A, Poisson J, Lasselin J, Devue C. et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc Natl Acad Sci U S A. 2017;114:E8675-E84
11. Liu WJ, Huang WF, Ye L, Chen RH, Yang C, Wu HL. et al. The activity and role of autophagy in the pathogenesis of diabetic nephropathy. Eur Rev Med Pharmacol Sci. 2018;22:3182-9
12. Gao Y, Zhu H, Yang F, Wang Q, Feng Y, Zhang C. Glucocorticoid-activated IRE1alpha/XBP-1s signaling: an autophagy-associated protective pathway against endotheliocyte damage. Am J Physiol Cell Physiol. 2018;315:C300-C9
13. Yang G, Wang N, Seto SW, Chang D, Liang H. Hydroxysafflor yellow a protects brain microvascular endothelial cells against oxygen glucose deprivation/reoxygenation injury: Involvement of inhibiting autophagy via class I PI3K/Akt/mTOR signaling pathway. Brain Res Bull. 2018;140:243-57
14. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81-94
15. Spring DR. Chemical genetics to chemical genomics: small molecules offer big insights. Chem Soc Rev. 2005;34:472-82
16. Araki S, Shimada Y, Kaji K, Hayashi H. Apoptosis of vascular endothelial cells by fibroblast growth factor deprivation. Biochem Biophys Res Commun. 1990;168:1194-200
17. Relou IA, Damen CA, van der Schaft DW, Groenewegen G, Griffioen AW. Effect of culture conditions on endothelial cell growth and responsiveness. Tissue Cell. 1998;30:525-30
18. Ma H, Su L, Yue H, Yin X, Zhao J, Zhang S. et al. HMBOX1 interacts with MT2A to regulate autophagy and apoptosis in vascular endothelial cells. Sci Rep. 2015;5:15121
19. Chaudhury A, Chander P, Howe PH. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1's multifunctional regulatory roles. RNA. 2010;16:1449-62
20. Shi H, Li H, Yuan R, Guan W, Zhang X, Zhang S. et al. PCBP1 depletion promotes tumorigenesis through attenuation of p27(Kip1) mRNA stability and translation. J Exp Clin Cancer Res. 2018;37:187
21. Jiang P, Li Z, Tian F, Li X, Yang J. Fyn/heterogeneous nuclear ribonucleoprotein E1 signaling regulates pancreatic cancer metastasis by affecting the alternative splicing of integrin beta1. Int J Oncol. 2017;51:169-83
22. Ishii T, Hayakawa H, Igawa T, Sekiguchi T, Sekiguchi M. Specific binding of PCBP1 to heavily oxidized RNA to induce cell death. Proc Natl Acad Sci U S A. 2018;115:6715-20
23. Zhang W, Shi H, Zhang M, Liu B, Mao S, Li L. et al. Poly C binding protein 1 represses autophagy through downregulation of LC3B to promote tumor cell apoptosis in starvation. Int J Biochem Cell Biol. 2016;73:127-36
24. Meng N, Peng N, Huang S, Wang SQ, Zhao J, Su L. et al. Heterogeneous nuclear ribonucleoprotein E1 regulates protein disulphide isomerase translation in oxidized low-density lipoprotein-activated endothelial cells. Acta Physiol (Oxf). 2015;213:664-75
25. Ho JJ, Robb GB, Tai SC, Turgeon PJ, Mawji IA, Man HS. et al. Active stabilization of human endothelial nitric oxide synthase mRNA by hnRNP E1 protects against antisense RNA and microRNAs. Mol Cell Biol. 2013;33:2029-46
26. Li JC, Zhang J, Rodrigues MC, Ding DJ, Longo JP, Azevedo RB. et al. Synthesis and evaluation of novel 1,2,3-triazole-based acetylcholinesterase inhibitors with neuroprotective activity. Bioorg Med Chem Lett. 2016;26:3881-5
27. Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745-56
28. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845-6
29. Zheng XT, Wu ZH, Wei Y, Dai JJ, Yu GF, Yuan F. et al. Induction of autophagy by salidroside through the AMPK-mTOR pathway protects vascular endothelial cells from oxidative stress-induced apoptosis. Mol Cell Biochem. 2017;425:125-38
30. Zhang S, Guo C, Chen Z, Zhang P, Li J, Li Y. Vitexin alleviates ox-LDL-mediated endothelial injury by inducing autophagy via AMPK signaling activation. Mol Immunol. 2017;85:214-21
31. Han J, Pan XY, Xu Y, Xiao Y, An Y, Tie L. et al. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy. 2012;8:812-25
32. Li H, Huang S, Wang S, Zhao J, Su L, Zhao B. et al. Targeting annexin A7 by a small molecule suppressed the activity of phosphatidylcholine-specific phospholipase C in vascular endothelial cells and inhibited atherosclerosis in apolipoprotein E(-)/(-)mice. Cell Death Dis. 2013;4:e806
33. Guo J, Jia R. Splicing factor poly(rC)-binding protein 1 is a novel and distinctive tumor suppressor. J Cell Physiol. 2018;234:33-41
34. Pickering BM, Mitchell SA, Evans JR, Willis AE. Polypyrimidine tract binding protein and poly r(C) binding protein 1 interact with the BAG-1 IRES and stimulate its activity in vitro and in vivo. Nucleic Acids Res. 2003;31:639-46
35. Chaudhury A, Hussey GS, Ray PS, Jin G, Fox PL, Howe PH. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat Cell Biol. 2010;12:286-93
36. Kosturko LD, Maggipinto MJ, Korza G, Lee JW, Carson JH, Barbarese E. Heterogeneous nuclear ribonucleoprotein (hnRNP) E1 binds to hnRNP A2 and inhibits translation of A2 response element mRNAs. Mol Biol Cell. 2006;17:3521-33
Author contact
![]() Corresponding authors: Ning Meng, School of Biological Science and Technology, University of Jinan, Jinan 250022, China, Tel/fax, 86-531-89736199, mengning20com; Cheng Shi Jiang, School of Biological Science and Technology, University of Jinan, Jinan 250022, China, Tel/fax, 86-531-89736199, jiangchengshi-20com and Hua Zhang, School of Biological Science and Technology, University of Jinan, Jinan 250022, China, Tel/fax, 86-531-89736199, bio_zhanghedu.cn
Corresponding authors: Ning Meng, School of Biological Science and Technology, University of Jinan, Jinan 250022, China, Tel/fax, 86-531-89736199, mengning20com; Cheng Shi Jiang, School of Biological Science and Technology, University of Jinan, Jinan 250022, China, Tel/fax, 86-531-89736199, jiangchengshi-20com and Hua Zhang, School of Biological Science and Technology, University of Jinan, Jinan 250022, China, Tel/fax, 86-531-89736199, bio_zhanghedu.cn