Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(7):1472-1487. doi:10.7150/ijbs.33817 This issue Cite
Research Paper
Hypericin maintians PDX1 expression via the Erk pathway and protects islet β-cells against glucotoxicity and lipotoxicity
Chen Liang1,2*, Fang Hao1*, Xinlei Yao1, Ye Qiu1, Lei Liu2, Shuyue Wang2, Chunlei Yu1, Zhenbo Song2, Yongli Bao1, Jingwen Yi2, Yanxin Huang1, Yin Wu1, Lihua Zheng1, Ying Sun2, Guannan Wang1, Xiaoguang Yang1,3, Shaonian Yang1,4, Luguo Sun1 ![]() , Yuxin Li2
, Yuxin Li2 ![]()
1. National Engineering Laboratory for Druggable Gene and Protein Screening, Northeast Normal University, Changchun 130024, China
2. Research Center of Agriculture and Medicine gene Engineering of Ministry of Education, Northeast Normal University, Changchun 130024, China
3. School of Physics, Northeast Normal University, Changchun 130024, China
4. The Rolf Luft Research Center for Diabetes and Endocrinology, Karolinska Institutet, SE-171 76 Stockholm, Sweden.
* Co-first author
Received 2019-2-3; Accepted 2019-4-30; Published 2019-6-2
Abstract

A decrease in islet β-cell mass is closely associated with the development and progression of diabetes. Therefore, protection against β-cell loss is an essential measure to prevent and treat diabetes. In this study, we investigated the protective effects of non-photoactivated hypericin, a natural compound, on β-cells both in vitro and in vivo. In vitro, hypericin greatly improved INS-1 cell viability under high-glucose and high-fatty-acid conditions by inhibiting glucotoxicity- and lipotoxicity-induced apoptosis and nitric oxide (NO) production. Then, we further demonstrated that hypericin elicited its protective effects against glucotoxicity and lipotoxicity in INS-1 cells by attenuating the reduction in pancreatic duodenal homeobox-1 (PDX1) expression and Erk activity. In vivo, prophylactic or therapeutic use of hypericin inhibited islet β-cell apoptosis and enhanced the anti-oxidative ability of pancreatic tissue in high-fat/high-sucrose (HFHS)-fed mice, thus alleviating β-cell loss and maintaining or improving β-cell mass and islet size. More importantly, hypericin treatment decreased fasting blood glucose, improved glucose intolerance and insulin intolerance, and alleviated hyperinsulinaemia in HFHS-fed mice. Therefore, hypericin showed preventive and therapeutic effects against HFHS-induced onset of type II diabetes in mice. Hypericin possesses great potential for development as an anti-diabetes drug in the future.
Keywords: Hypericin, β-cell protection, apoptosis, diabetes
Introduction
Pancreatic β-cells are the only type of mammalian cells that synthesize and secrete insulin, a hormone to reduce blood glucose concentration. β-cells can release their stored insulin quickly in response to high blood glucose levels. β-cell function and mass are diminished in patients with diabetes mellitus (DM), leading to insufficient insulin secretion and hyperglycaemia [1]. Type I diabetes results from autoimmune destruction of β-cells [2]. Type II diabetes, the most common form of diabetes, is initiated by insulin resistance, which causes a high demand for insulin production by β-cells. This high demand on β-cells frequently causes the cells to malfunction, de-differentiate and die [3]. A progressive loss of β-cell mass occurs in type II diabetes. A decrease in β-cell mass of up to 60% has been reported in type II diabetes [4]. A reduction in the number of β-cells is closely associated with diabetes development and progression. Therefore, strategies aiming to protect β-cells from impairment and apoptosis may delay the onset of diabetes and may restrict the continuous deterioration of metabolic control after clinical onset.
Several factors are known to contribute to the progressive loss of β-cell mass in type II diabetes, such as high blood glucose (glucotoxicity), increased levels of plasma fatty acids (lipotoxicity), and chronic inflammation [5-6]. These factors may impair β-cell function and lead to β-cell apoptosis through multiple mechanisms, among which oxidative stress is considered the major cause of cell death [5]. Islets isolated from the pancreas of subjects with type II diabetes show increased markers of oxidative stress [7]. Oxidative stress activates stress-induced pathways that damage β-cells [8]. Pancreatic duodenal homeobox-1 (PDX1) is a key transcription factor for pancreas development and mature β-cell function [9]. It also plays a pro-survival role in adult β-cells and its partial deficiency increases β-cell apoptosis, leading to decreased β-cell mass and diabetes in rodent and human [10-12]. Some studies have shown that Glucotoxicity and lipotoxicity result in β-cell apoptosis and dysfunction via oxidative-stress-mediated decrease in the expression of PDX1 [13]. MEK/Erk has been implicated in cell survival. Accumulating researches have shown that MEK/Erk activation protects against pancreatic β-cell apoptosis [14-18] and its protective effect on β-cells is associated with inhibiting ROS [14-15]. However, Erk activation is always attenuated when islet β-cells are exposed to high glucose and high fatty acids [17,19], implying that high glucose and high fatty acids may inhibit Erk activation, which subsequently induces oxidative stress and leads to β-cell apoptosis.
Currently, treatment of type I diabetes requires continuous administration of exogenous insulin, whereas type II diabetes can be controlled by oral or injected non-insulin drugs. These drugs may act on β-cells mainly to promote insulin secretion or may act on other peripheral tissues. However, these non-insulin drugs are usually effective for glycaemic control only in the early stages of the disease. Due to the progressive nature of type II diabetes, most patients will eventually require insulin [20]. Moreover, these therapies are used for treatment after the onset of diabetes and not for prevention. We speculate that effective early prevention might be of greater significance than treatment, especially for people with a high propensity to develop type II diabetes. More importantly, neither exogenous insulin nor non-insulin drugs match the precision of endogenous β-cells; consequently, their use is accompanied by risks of hypoglycaemia and ketosis [3]. Therefore, protecting β-cells and maintaining β-cell mass is an ideal option for both preventive and therapeutic treatment. To our knowledge, few drugs for this purpose have been developed to date.
Hypericin (4,5,7,4′,5′,7′-hexahydroxy-2,2′-dimethylnaphthodianthrone) is a naturally occurring chromophore synthesized by some species of the genus Hypericum, particularly Hypericum perforatum L. Hypericin is a potent natural photosensitizing agent. In recent decades, the pharmacological activities of photoactivated hypericin have been extensively studied. Photoactivated hypericin possesses great potential in anticancer photodynamic therapy (PDT) and photodynamic diagnosis (PDD), and its antibacterial, antifungal and antiviral effects have also been reported [21-22]. Comparatively, studies on the light-independent activity of hypericin are insufficient. In the dark, it possesses minimal or no toxicity and can be used to treat fatigue, weakness, and mild depression [23]. Here, for the first time, we reveal the protective effects of non-photoactivated hypericin in islet β-cells against the toxicity of high glucose and fatty acid levels both in vitro and in vivo. Additionally, we demonstrated the preventive and therapeutic effect of hypericin against high-fat/high-sucrose (HFHS)-induced type II diabetes in mice.
Materials and Methods
Reagents and Antibodies
Hypericin (HPLC≥97%) was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Hypericin stock solution was prepared in DMSO and stored in a brown bottle. Specific antibodies against B-cell lymphoma-2 (Bcl-2, Cat#sc-7382) and Bcl-2 associated X protein (Bax, Cat#sc-20067) were provided by Santa Cruz Biotechnology (Santa Cruz, CA, USA). Specific antibodies against caspase-3 (C3, Cat#9662S), cleaved caspase-3 (CC3, Cat#9661) and Erk (Cat#5013S) were obtained from Cell Signaling Technology (Beverly, MA, USA). Anti-phospho (p)-Erk antibodies (Cat#ab47339) were obtained from Abcam (Cambridge, UK). Anti-GAPDH antibodies (Cat#60004-1-Ig), PDX1 (Cat#10951-1-AP) and goat anti-rabbit (Cat#SA00001-2) or anti-mouse IgG-horseradish peroxidase (IgG-HRP, Cat#SA00001-1) secondary antibodies were purchased from Proteintech (Proteintech, Wuhan, China). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT, M2128), palmitic acid (PA, P5585), and U0126 monoethanolate (U120) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 4',6-Diamidino-2-phenylindole (DAPI, C1002), NO (S0021) and enhanced chemiluminescence (ECL, P0018) reagent were obtained from Beyotime (Shanghai, China). An apoptosis assay kit (556547) was obtained from BD ( San Jose, CA, USA). Total antioxidant capacity (T-AOC, A015-1), glutathione peroxidase (GSH-PX, A005), superoxide dismutase (SOD, A001-3) and malondialdehyde (MDA, A003-1) were detected with kits from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Cell Culture
INS-1 cells (a rat insulinoma cell line) were obtained from the Chinese Academy of Sciences Shanghai Institute for Biological Sciences Cell Resource Center and cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (R8758, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10 mM HEPES (H8090, Solarbio, Beijing, China), 1 mM sodium pyruvate, 50 μM β-mercaptoethanol and 10% foetal bovine serum (FBS, 10099-141, Gibco, Thermo Fisher, Shanghai, China) at 37℃ with 5% CO2. For cells with hypericin treatment, we prevented photoactivation of hypericin by covering the cell culture dish with tin foil and switching off the lights when manipulating cells in the culture hood.
Cell Viability
The protective effects of hypericin on the viability of INS-1 cells under glucotoxicity and lipotoxicity were assessed by an MTT assay. Briefly, INS-1 cells were seeded in 96-well plates (5 × 104 cells/well) and then incubated with different concentrations of hypericin (20 nM, 200 nM, 2000 nM) in culture media containing high (33 mM) glucose for 72 h [24] or 200 μM PA for 24 h [25-26]. Thereafter, 20 µl of MTT (2.5 mg/mL) was added to each well for 4-6 h of incubation. Subsequently, the medium was discarded, and 100 µl of DMSO was added to dissolve the formazan crystals. The optical densities were measured at 492 nm using a spectrophotometric plate reader (Bio-Rad, Hercules, CA, USA). The experiment was repeated at least three times.
DAPI staining
DAPI staining was performed as reported previously [27]. Briefly, INS-1 cells were seeded in 6-well plates with coverslip and treated with or without 200nM hypericin for 24h. Medium was then removed and cells were washed three times with PBS followed by fixation with 4 % paraformaldehyde for 15 min at 37℃. Then, cells were stained with DAPI (1:10000) for 10 min in the dark room after PBS washing. Cell morphology was observed using an Olympus BX50 fluorescence microscope (Olympus Tokyo, Japan).
RT-qPCR
INS-1 cells were seeded in six-well plates and treated with hypericin in the absence or presence of 33 mM glucose or 200 μM PA. Total RNA was extracted with TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions and reverse transcribed into cDNA using a real-time PCR kit (TransGen Biotech, Beijing, China). RT-qPCR was performed using the SYBR Green I PCR master mix kit (TAKARA, Beijing, China). The amplification protocol was carried out as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 10 s, 60°C for 1 min, and 72°C for 10 min. The PCR primers for specific mouse genes are as follows: PDX1 (F: 5'-CCGAATGGAACCGAGACTGG-3'; R: 5'-GGGTCCTCTTATTCT CCTCC-3'), inducible nitric oxide synthase (iNOS) (F: 5'-GGGTCGTAATGTCCAGGAAGT-3'; R: 5'-TCTTGGAGCGAGTTGTGGAT-3'), and GAPDH (F: 5'-AAATGGTGAAGGTCGGTGTG-3'; R: 5'-TGAAGGGGTCGTTGATGG-3').
NO Assay
The cell culture supernatant (50 µL) was collected and mixed with Griess reagent (Beyotime, Shanghai, China). The optical density was read at 540 nm on a spectrophotometric plate reader. The NO concentration was estimated against a standard curve, which was calibrated using sodium nitrite standards.
Western blot analysis
Total protein was extracted by cell lysis. The extracts were electro-blotted onto a polyvinylidene fluoride (PVDF) membrane following separation by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The PVDF membrane was incubated with a blocking solution of 5% skim milk for 2 h at room temperature, followed by incubation with a specific antibody overnight at 4℃ and then with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Finally, the blots were visualized with ECL reagent.
Animals and feeding management
One hundred twenty pathogen-free male C57BL/6J mice (18-22 g, 4-5 weeks old) were purchased from the Experimental Animal Center, Medical College of Norman Bethune, Jilin University. On arrival, mice were acclimatized for at least one week prior to experimentation. The mice were weighed, coded and randomly divided into five groups of 12 mice: one group that was fed a normal diet (normal, 4.5 kcal% fat diet, SPF growth feed, Beijing Keao Xieli Feed Co.,Ltd.), a control group that received an HFHS diet (HFHS, 45 kcal% fat diet, 20% sucrose, Medicience Ltd, Yangzhou, China) and HFHS groups treated with hypericin at 0.5 mg/kg, 1 mg/kg and 2 mg/kg. There was no significant difference in body weight or food intake among the groups before the experiments were initiated. The mice in the normal group were fed a normal diet throughout the entire experiment, while the other groups of mice, including the HFHS control and hypericin-treated mice, were fed an HFHS diet for the duration of the experiment. For the prevention group, hypericin was administered every other day by intraperitoneal (i.p.) injection after one month of HFHS feeding until the fasting blood glucose of the HFHS control mice was over 7 mM/L, which occurred after approximately four months of HFHS feeding [28-31]. For the treatment group, hypericin was administered every other day by i.p. injection after four months of HFHS feeding and continued for 30 days. Simultaneously, normal and HFHS control mice were administered equal amounts of 0.9% NaCl (wt/vol) by i.p. injection every other day. After 2 months of HFHS feeding, fasting blood glucose was tested twice a week. Throughout the entire experiment, all mice were raised under a normal laboratory light cycle and were not deliberately sheltered from light. The experimental protocols were approved by the Institutional Animal Care and Use Committee of Northeast Normal University.
IPGTT and IPITT
An intraperitoneal glucose tolerance test (IPGTT) and an intraperitoneal insulin tolerance test (IPITT) were carried out before the termination of the experiments. After an 8 h fast, mice received an i.p. injection of 2 g/kg glucose or 0.5 IU/kg insulin dissolved in 0.9% NaCl, and the glucose concentration was measured in blood samples collected from the tip of the tail before (0 min) and 15, 30, 45, 60 and 90 min after treatment. Glucose levels were detected using glucose strips and an Abbott FreeStyle glucometer (TheraSense, USA).
Tissue sample collection
Venous blood samples were drawn and placed into centrifuge tubes for serum preparation after the mice were anaesthetized (4% chloral hydrate, 0.1 ml/10 g of body weight). The pancreas was rapidly removed in an aseptic manner and weighed after the excess fat was removed. A portion of the pancreatic tissue was stored at -80°C for further analysis. The rest of the tissue was fixed in Bouin's fixation fluid for 12 h and washed with 70% ethanol. After the tissue was dehydrated in ascending ethanol solutions and rendered transparent in xylene, it was embedded in paraffin.
ELISA
The insulin level was measured in each drawn blood sample using a commercial ELISA kit (ml001983, Shanghai Enzyme-linked Biotechnology Co., Ltd, Shanghai, China) according to the manufacturer's instructions.
Biochemical assay
The pancreatic tissue was washed with ice-cold PBS to remove the blood and wiped dry with filter paper. PBS (pH 7.4) was added to the tissue at a ratio of 9:1 by weight. The mixture was homogenized with a cold glass homogenizer in an ice bath. Subsequently, the pancreatic tissue homogenate was centrifuged at 3,000 rpm for 10 min at 4°C. The supernatant was collected to measure T-AOC, MDA content and GSH-PX and SOD activity using the appropriate assay kits according to the manufacturer's instructions.
Haematoxylin-eosin staining and immunohistochemistry
Five serial paraffin sections of pancreatic tissue were obtained from mice and processed for routine haematoxylin-eosin (HE) staining or immunohistochemical (IHC) staining according to the manufacturer's protocols. IHC staining kits were obtained from Beijing Biosynthesis Biotechnology Co. Ltd. (Beijing, China). The tissue sections were stained with DAB, counterstained with haematoxylin and observed under a microscope. Brown particles indicated positive staining for target proteins. Three tissue sections were randomly chosen for observation, with 5-10 different fields of view captured for each section. All tissue sections were imaged with an Olympus DP72 microscope (Olympus Optical Co. Ltd, Japan). Pancreatic β-cell mass was calculated by dividing the area of C-peptide-positive cells by the total area of pancreatic tissue and multiplying by the weight of the pancreas. Islet area was calculated by dividing the total area covered by islets by the total area of the pancreatic tissue.
Statistical analysis
The Experimental data were analysed using one-way ANOVA with the software SPSS 16.0 software (SPSS Inc, Chicago, US), and all data shown are presented as the mean±SD of three or more independent experiments. p<0.001, p<0.01, and p<0.05 were considered statistically significant.
Results
Hypericin inhibits apoptosis and NO production in INS-1 cells under high-glucose toxicity
Maintaining islet β-cell function and mass is essential to curb the development and progression of diabetes. To elucidate whether the natural compound hypericin (Figure 1A) has any therapeutic potential in this respect, we first established a glucotoxicity model using INS-1 cells to investigate the protective effect of hypericin on islet β-cells. By using an MTT assay, we determined that 33 mM glucose significantly decreased INS-1 cell viability in a time-dependent manner compared with normal control cells cultured in medium with 11 mM glucose at the 48 h, 72 h and 96 h timepoints (Figure S1A). These data indicated that the glucotoxicity model was successfully established. On this basis, we further demonstrated that hypericin effectively improved the viability of INS-1 cells under glucotoxic conditions in a dose-dependent manner (Figure 1B), suggesting that hypericin effectively alleviated the cell damage caused by high-glucose toxicity. High glucose impairs cell viability in islet β-cells by inducing apoptosis. Indeed, in our glucotoxicity model, incubation with high glucose for 72 h induced apoptosis in INS-1 cells, as detected by flow cytometry using Annexin V/PI staining (Figure S1B). Therefore, we investigated whether hypericin alleviated the glucotoxic effects on INS-1 cells by inhibiting the induction of apoptosis. Since hypericin is autofluorescent, we could not apply flow cytometry to analyse the effect of hypericin on apoptosis. Therefore, we detected the production of apoptotic bodies by DAPI staining and the levels of apoptosis-related proteins by Western blotting to evaluate apoptosis.
Hypericin inhibits apoptosis and NO production in INS-1 cells under high-glucose toxicity. (A) Chemical structure of hypericin. (B) Measurement of cell viability by an MTT assay. INS-1 cells were treated with 33 mM glucose in the absence or presence of hypericin (20, 200, and 2000 nM) for 72 h and then subjected to the MTT assay. INS-1 cells treated with 11 mM glucose were used as the control. Data are presented as the mean ± S.D. (n = 3). (C) Detection of apoptotic bodies by DAPI staining. INS-1 cells were treated with 33 mM glucose, 200 nM hypericin or a combination of the two for 72 h. The cells were then stained with DAPI and observed under a fluorescence microscope. Arrows indicate the apoptotic bodies. Scale bar 50 μm. The results are representative of three independent experiments. (D) Detection of apoptosis-related proteins by Western blot. INS-1 cells were treated as in (C). Then, the protein levels of cleaved-caspase-3, Bax and Bcl-2 were determined by Western blot; GAPDH was used as a loading control. Density ratios of CC3 to GAPDH or Bax to Bcl-2 as measured with ImageJ are shown in the two right-hand panels. The experiment was repeated three times. (E) Detection of NO production. INS-1 cells were treated as in (C). Production of NO was detected by the Griess reaction in culture medium. Data are presented as the mean ± S.D. (n = 3). (F) Detection of iNOS mRNA levels by RT-qPCR. INS-1 cells were treated as in (E), after which total RNA was extracted and iNOS mRNA was amplified by conventional SYBR Green real-time PCR analysis. Data are presented as the mean ± S.D. (n = 3). NO, nitric oxide; iNOS, inducible nitric oxide synthase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; DAPI, 4',6-diamidino-2-phenylindole; Bax, Bcl-2-Associated X protein; Bcl-2, B-cell lymphoma-2. Experiments were repeated three times. * p<0.05; ** p<0.01; *** p<0.001 versus the high-glucose group.
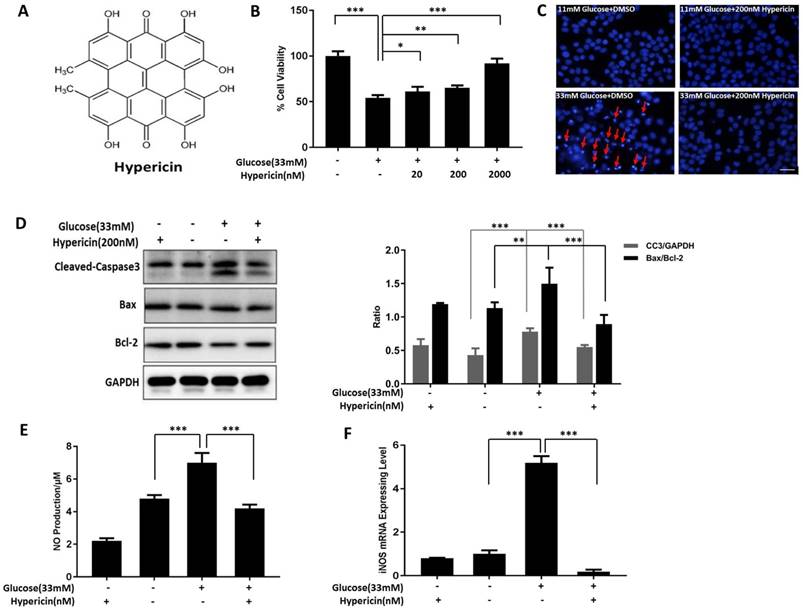
As shown in Figure 1C, high glucose induced the formation of large numbers of apoptotic bodies in INS-1 cells, as expected, while no apoptotic bodies were observed in control cells cultured under normal conditions. However, treatment with 200 nM hypericin dramatically reduced the number of apoptotic bodies in glucotoxic INS-1 cells. Along similar lines, the high-glucose-induced elevation of CC3 levels and high-glucose-induced reduction in the ratio of Bax to Bcl-2 were also partially or completely reversed by hypericin in INS-1 cells (Figure 1D). From these results combined with the results shown in Figure 1C, we concluded that hypericin suppressed the occurrence of glucotoxicity-induced apoptosis in INS-1 cells. NO, generated by iNOS, is an oxidant and an important inflammatory mediator. Excess NO production is involved in oxidative-stress-related cell injury and apoptosis [32]. Here, we showed that iNOS expression and corresponding NO production in INS-1 cells were both significantly increased under high-glucose conditions in comparison with control conditions (p<0.001,n=3). However, this increase was completely abolished by hyperin treatment (Figure 1E-F, glucose+hypericin VS glucose alone, P<0.001, n=3).
Hypericin inhibits apoptosis and NO production in INS-1 cells under high-lipid toxicity
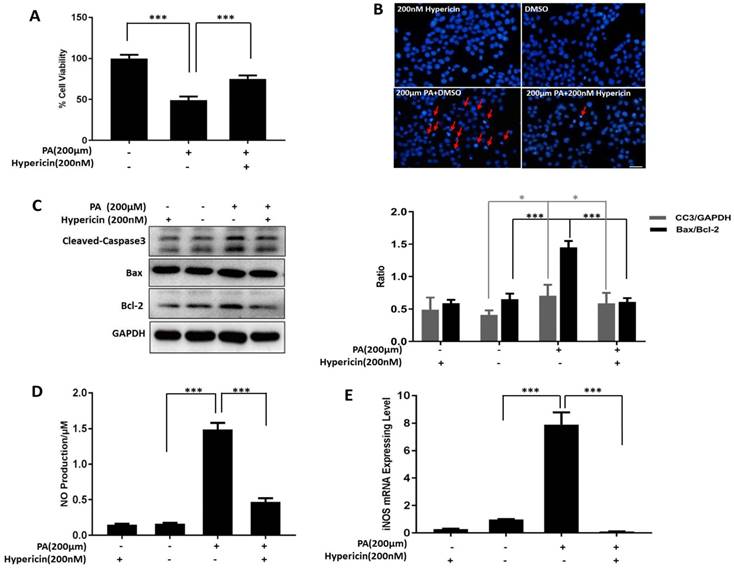
We also established a lipotoxicity cell model to test the protective effect of hypericin on β-cells. We examined the effects of different concentrations of PA on INS-1 cell viability and found that both 200 µM and 250 µM PA significantly decreased cell viability at the 24 h time point compared to the control treatment (Figure S2). In the subsequent experiments, 200 µM PA was used for the lipotoxicity model. Consistent with the results of the glucotoxicity protection assay, 200 nM hypericin improved the viability of PA-treated INS-1 cells (Figure 2A, p<0.001). Similar to glucotoxicity, lipotoxicity can cause apoptosis in β-cells. Similarly, hypericin also protected INS-1 cells from lipotoxicity-induced apoptosis, dramatically decreasing the number of apoptotic bodies marked by DAPI staining (Figure 2B), reducing the levels of the apoptotic protein CC3 and lowering the ratio of Bax to Bcl-2 (Figure 2C) in lipotoxicity-exposed INS-1 cells. These results suggest that hypericin effectively alleviated the cell damage caused by high-lipid toxicity. Similarly, we also examined NO production and iNOS expression under high-lipid conditions and showed that the NO and iNOS expression levels in PA-treated INS-1 cells were significantly increased compared to those in control cells (p<0.001). Again, hypericin completely restored NO production and iNOS expression to normal levels in control cells (Figure 2D-E, p<0.001).
Hypericin inhibits apoptosis and NO production in INS-1 cells under high-lipid toxicity. (A) INS-1 cells were treated with 200 μM PA, 200 nM hypericin or a combination of the two for 24 h. Then, cell viability (B), apoptotic bodies (C), apoptosis-related proteins (D), NO production (E) and iNOS mRNA (F) were detected or analysed as in Fig. 1. *p<0.05, ***p < 0.001 versus the PA-treated group. PA, palmitic acid.
Hypericin protects INS-1 cells from glucotoxicity and lipotoxicity by maintaining PDX1 expression and Erk activation
The pancreatic β-cell-specific transcription factor PDX1 is involved in the development and differentiation of the pancreas, promotes the proliferation of β-cells and inhibits their apoptosis. Therefore, we tested whether the protective effects of hypericin on β-cells were correlated with PDX1 expression. The results showed that both the mRNA and protein levels of PDX1 were significantly decreased in INS-1 cells under high-glucose toxicity; however, hypericin alleviated the glucotoxicity-induced downregulation of PDX1 expression (Figure 3A-B, p<0.001). Then, we further investigated the molecular mechanisms underlying this effect.
Hypericin protects INS-1 cells from glucotoxicity and lipotoxicity via Erk signaling and maintianing PDX1 expression. (A) Effects of hypericin on the PDX1 mRNA level in INS-1 cells under glucotoxicity. INS-1 cells were treated with high (33 mM) glucose, 200 nM hypericin or a combination of the two for 72 h. Total RNA was extracted, and PDX1 mRNA was amplified by conventional SYBR Green real-time PCR analysis. Data are presented as the mean ± S.D. (n = 3). (B) Effects of hypericin on the protein levels of PDX1, Erk1/2 and p-Erk in INS-1 cells under glucotoxicity. INS-1 cells were treated as in (A). Cell lysates were prepared and subjected to Western blots using the indicated antibodies. GAPDH was used as a loading control. The density ratios of PDX1 to GAPDH or p-Erk to Erk were measured by ImageJ as shown in the right panel. The experiment was repeated three times. *** p<0.001 versus the 33 mM glucose-treated group. (C) Blockade of hypericin-mediated effects by U0126 in INS-1 cells under glucotoxicity. INS-1 cells were treated with different combinations of high (33 mM) glucose, 200 nM hypericin and U0126 as indicated for 72 h. Then, target proteins were detected by Western blot using the indicated antibodies. GAPDH was used as a loading control. The density ratios of PDX1 to GAPDH, p-Erk to Erk or CC3 to GAPDH were measured by ImageJ as shown in the right-hand panel. The experiment was repeated three times. **p<0.01, ***p<0.001 versus the 33 mM glucose-treated group; ##p<0.01, ###p<0.001 versus the 33 mM glucose+200 nM hypericin-treated group. (D) Effects of hypericin on the Erk pathway in INS-1 cells under lipotoxicity. INS-1 cells were treated with 200 μM PA, 200 nM hypericin or a combination of the two for 24 h. Cell lysates were prepared and subjected to Western blots using anti-Erk and anti-p-Erk antibodies. GAPDH was used as a loading control. The density ratios of p-Erk to Erk were measured by ImageJ as shown in the right panel. The experiment was repeated three times. **p<0.01, *** p<0.001 versus the 200 μM PA-treated group. (E) Blockade of hypericin-mediated effects by U0126 in INS-1 cells under lipotoxicity. INS-1 cells were treated with different combinations of 200 μM PA, 200 nM hypericin and U0126 as indicated for 24 h. Then, target proteins were detected and analysed as in (c). *** p<0.001 versus the 200 μM PA-treated group; ## p<0.01 versus the 200 μM PA+200 nM hypericin-treated group.
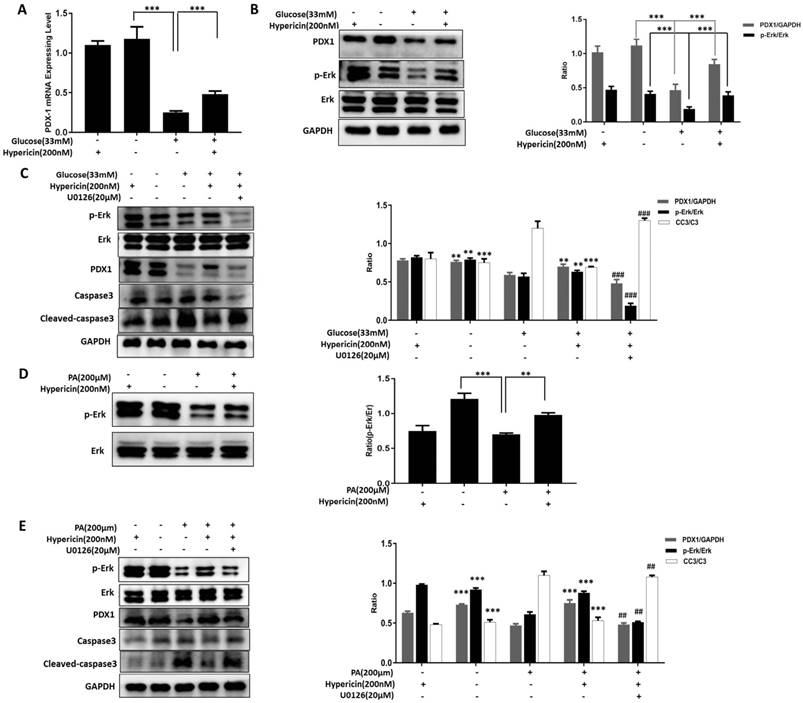
As shown in Figure 3B, compared with the control cells, glucotoxicity-exposed INS-1 cells had a significantly decreased p-Erk level, and hypericin partially restored this value. This indicates that hypericin attenuates the high glucose-induced down-regulation of Erk activity. The Erk signalling pathway is involved in maintaining cell survival, promoting cell proliferation and resisting apoptosis. We hypothesized that restoring the Erk activity is responsible for the protective effects of hypericin on β-cells. To test this hypothesis, we co-treated cells with the Erk inhibitor U0126. We found that U0126 caused a sharp reduction in p-Erk levels, and the hypericin-mediated increase in PDX1 expression and reduction in CC3 levels in glucotoxicity-exposed INS-1 cells were almost completely abolished (Figure 3C). These results suggested that high glucose interferes with Erk signalling to cause β-cell impairment and death, whereas hypericin prevents glucotoxic damage by enhancing Erk signalling and elevating PDX1 expression. Next, we asked whether the same mechanism occurs in lipotoxic INS-1 cells protected by hypericin. To this end, we performed similar experiments, and we found that the p-Erk level was also decreased in PA-treated INS-1 cells, while hypericin restored the p-Erk level to normal (Figure 3D). Likewise, U0126 treatment completely eliminated the effects of hypericin in lipotoxic INS-1 cells, including increased PDX1 expression and decreased CC3 levels (Figure 3E). Therefore, we concluded that hypericin exerts its protective effects on β-cells by attenuating glucotoxicity- and lipotoxicity-induced reduction in PDX1 expression and Erk activity.
Prophylactic use of hypericin ameliorates diabetic phenotypes in HFHS-fed mice
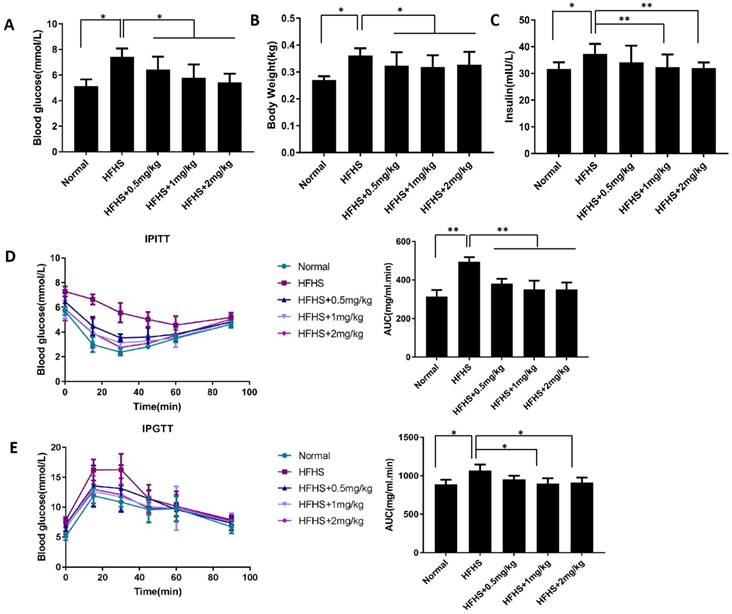
Since hypericin showed dramatic and well-defined protective effects on islet β-cells in vitro, we next sought to explore whether it had the same effects in vivo. To this end, we established an HFHS-induced diabetic mouse model to test whether hypericin could exhibit preventive effects against the onset of diabetes in mice, since it might curb lipotoxicity and glucotoxicity and maintain islet β-cell mass. As shown in Figure 4A, when the fasting blood glucose levels of the HFHS control mice were approximately 8.0 mmol/L, prophylactic use of hypericin dramatically inhibited the increase in fasting blood glucose levels in HFHS-fed mice in a dose-dependent manner. Likewise, continuous HFHS feeding led to body weight gain in mice, as shown in Figure 4B. However, prophylactic use of hypericin partially prevented weight gain in a dose-independent manner. Since treatment with hypericin improves the fasting blood glucose levels of HFHS-fed mice (Figure 4A), we further investigated the enhancing effects of hypericin on glucose tolerance and insulin tolerance. As shown in the IPITT and IPGTT, HFHS control mice developed glucose intolerance and exhibited disturbed insulin sensitivity compared to mice fed a normal diet, as detected by the augmented area under the glucose excursion curve (AUC) in both IPITT and IPGTT conditions (Figure 4D-E). This result suggests that HFHS-fed mice have defects in both insulin secretion and insulin sensitivity, which are consistent with type II diabetic phenotypes. Moreover, in the early stages of type II diabetes, insulin resistance, a defect in insulin sensitivity, causes hyperinsulinaemia. As expected, HFHS control mice in our study displayed elevated levels of serum insulin compared to the mice fed a normal diet (Figure 4C). However, these diabetic phenotypes were ameliorated in a dose-dependent manner by prophylactic use of hypericin, which reduced the levels significantly, nearly to their baseline values, when used at its maximum dose. These data indicate that hypericin can prevent the onset of HFHS in diabetic mice.
Prophylactic use of hypericin ameliorates diabetic phenotypes in HFHS-fed mice. (A) Fasting blood glucose levels of mice. After 1 month on an HFHS diet, mice were injected i.p. with hypericin or 0.9% NaCl (HFHS control) every other day for nearly three months. After 8 h of fasting, blood samples collected from the tip of the tail were used to measure blood glucose levels. Mice that were fed a normal diet and injected with 0.9% NaCl were used as the normal control. Data are presented as the mean ± S.D. (n = 8). (B) Body weight of mice. Before sacrifice of the mice in (A), their body weights were recorded. Data are presented as the mean ± S.D. (n = 8). (C) Blood insulin levels of mice. Insulin levels were determined by ELISA in venous blood from the mice in (A). Data are presented as the mean ± S.D. (n = 6). (D) IPITT. After 8 h of fasting, mice in (A) received an i.p. administration of 0.5 IU/kg insulin in 0.9% NaCl, and their blood glucose was measured as in (a) before (0 min) and 15, 30, 45, 60 and 90 min after the treatment. The AUC was calculated and is shown in the right-hand panel. Data are presented as the mean ± S.D. (n = 6). (E) IPGTT. The experiment was conducted as in (C), but 2 g/kg glucose instead of insulin was injected intraperitoneally; *p<0.05, **p<0.01 versus the HFHS group. HFHS, high-fat/high-sucrose; IPGTT, intraperitoneal glucose tolerance test; IPITT, intraperitoneal insulin tolerance test; AUC, area under the glucose excursion curve.
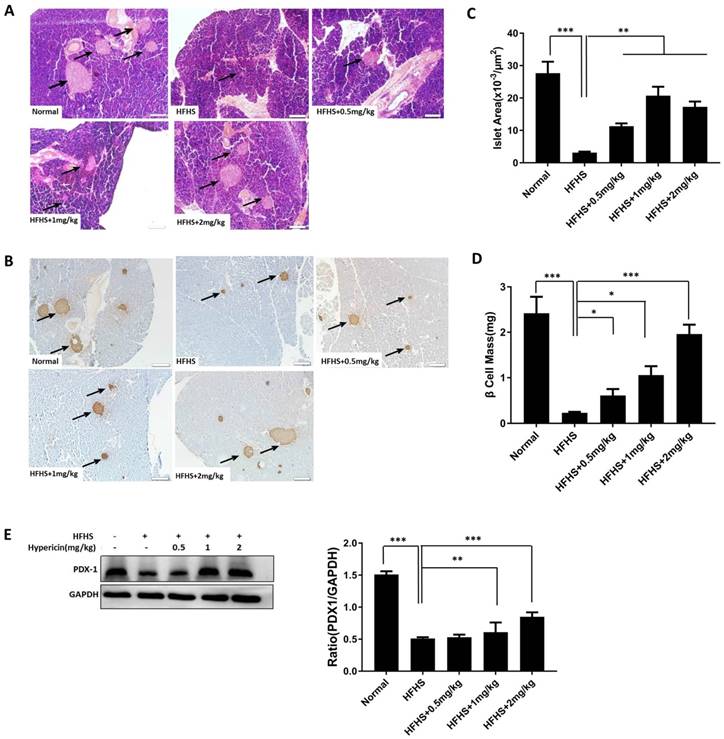
Prophylactic use of hypericin decreases β-cell loss, maintains islet mass and improves insulin expression in HFHS-fed mice
Next, we sought to investigate the effects of hypericin treatment on the pancreas of HFHS-fed mice. First, through histologic analysis, we explored the protective effects of hypericin on islets and β-cells, which might mediate the preventive effects of hypericin on diabetes. Preliminary HE staining of pancreatic slices revealed severely shrunken islets and decreased islet counts in the HFHS control mice compared to the mice that received a normal diet, indicating that HFHS conditions led to islet damage involving glucotoxicity and lipotoxicity (Figure 5A). This is consistent with previous reports [29-30]. In contrast, prophylactic hypericin treatment improved both the size and the number of islets in HFHS-fed mice in a dose-dependent manner (Figure 5A). We then conducted IHC staining with antibodies against C-peptide (Figure 5B). Again, we observed the same alterations in pancreatic islets as we observed with HE staining in each group of mice, only more clearly. In particular, the HFHS mice treated with the highest concentration of hypericin maintained almost the same islet size as the mice that received a normal diet. Moreover, islet area and β-cell mass were measured by morphometric analyses in C-peptide-positive islets. Indeed, significant increases in both islet area and β-cell mass were observed in the pancreas of hypericin-treated HFHS-fed mice compared to HFHS control mice (Figure 5C-D). These data suggest that prophylactic hypericin treatment indeed protects β-cells and islets under HFHS conditions. Additionally, the PDX1 expression level in pancreatic tissue was analysed by Western blot. As shown in Figure 5E, PDX1 expression was significantly decreased in the pancreases of HFHS control mice. However, hypericin significantly elevated pancreatic PDX1 levels in a dose-dependent manner, which was consistent with the increase in β-cell mass and islet size observed in hypericin-treated HFHS mice and the in vitro data.
Prophylactic use of hypericin decreases β-cell loss and maintains islet mass in HFHS-fed mice. (A) Histological sections of mouse pancreatic tissue. After sacrifice, the mouse pancreases were removed and weighed. Portions of the mouse pancreases from (A) were fixed and subjected to HE staining. The scale bar represents 100 μm. Arrows indicate pancreatic islets. (B) IHC analysis of the mouse pancreas using anti-C-peptide antibodies. Portions of the mouse pancreases from (A) were fixed and subjected to IHC analysis. The scale bar represents 100 μm. Arrows indicate positively stained cells. (C) Measurement of islet area in the mouse pancreas. Pancreatic sections subjected to IHC staining with an anti-C-peptide antibody in (B) were used to measure the islet area of the pancreas. Data are presented as the mean ± S.D. (n = 8). (D) Calculation of β-cell mass of the pancreas. Pancreatic sections that were IHC stained with an anti-C-peptide antibody in (B) were used to calculate the β-cell mass of the pancreas. Data are presented as the mean ± S.D. (n = 8). (E) PDX1 protein levels in the mouse pancreas. Portions of the mouse pancreases from (A) were homogenized, and total cellular lysates were prepared and subjected to Western blots using anti-PDX1 antibodies. GAPDH was used as a loading control. The density ratios of PDX1 to GAPDH were measured by ImageJ, and the fold change relative to the normal group is shown in the right-hand panel. Data are presented as the mean ± S.D. (n = 6). * p< 0.05, **p<0.01, ***p<0.001 versus the HFHS group.
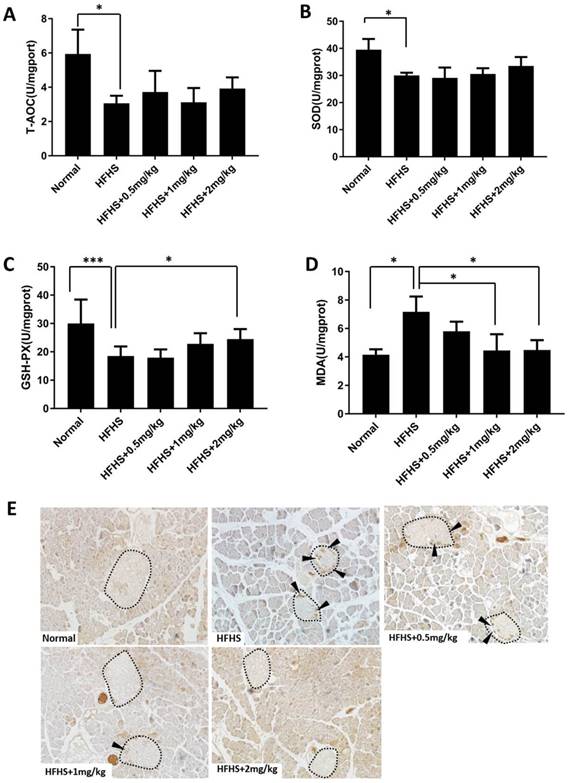
Prophylactic use of hypericin enhances the anti-oxidative ability of the pancreas and blocks islet β-cell apoptosis in HFHS-fed mice
To further elucidate the mechanisms underlying the protective effects of hypericin on β-cells under HFHS conditions in vivo, we investigated the effects of hypericin on anti-oxidative function, which fights against damage mediated by oxidative stress. We assessed T-AOC, SOD and GSH-PX activity, which represent anti-oxidative function, as well as MDA content, which represents the level of oxidative products, in mouse pancreatic homogenate. As shown in Figure 6A-C, T-AOC and the activity levels of SOD and GSH-PX were significantly lower in the pancreases of HFHS control mice than in those of mice that received a normal diet (p<0.05). Consequently, the pancreatic MDA content in HFHS control mice was much higher than that in normal mice (Figure 6D). These data indicated that the anti-oxidative function of the HFHS pancreas was impaired. However, in HFHS-fed mice that received prophylactic hypericin treatment, the T-AOC levels and the activity levels of SOD and GSH-PX were increased to varying degrees in the pancreas depending on the concentration of hypericin, but only the increases in GSH-PX activity with 1 mg/kg and 2 mg/kg hypericin treatment were statistically significant (Figure 6A-6C). The MDA content was significantly decreased in a dose-dependent manner in the hypericin-treated mice compared to the HFHS control mice (Figure 6D). These results suggest that prophylactic use of hypericin may enhance the anti-oxidative function of the pancreas to some extent under HFHS conditions. Furthermore, we detected apoptosis of β-cells using IHC staining for CC3 in the pancreas. As shown in Figure 6E, positive CC3 signal was visible in the form of round yellow or brown particles in the pancreatic islets of HFHS control mice, whereas no CC3 staining was observed in mice fed a normal diet. In HFHS-fed mice treated with 1 mg/kg and 2 mg/kg prophylactic hypericin, we observed much less CC3 staining in the pancreatic islets than in those of HFHS-fed control mice. This indicates that prophylactic hypericin treatment decreased HFHS-induced apoptosis of β-cells, in agreement with the in vitro data.
Prophylactic use of hypericin enhances the anti-oxidative ability of the pancreas and blocks islet β-cell apoptosis in HFHS-fed mice. (A-D) Assessment of anti-oxidative function in the mouse pancreas. Portions of the mouse pancreases from Fig. 5A were homogenized, and the homogenate supernatant was collected to measure T-AOC (A), SOD (B) and GSH-PX activity (C), and MDA content (D). Data are presented as the mean ± S.D. (n=6). *p<0.05, ***p<0.001 versus the HFHS group. (E) IHC staining of the mouse pancreas with the anti-CC3 antibody. Portions of the mouse pancreases from Fig. 5A were fixed and subjected to IHC analysis. The scale bar represents 50 μm. Islets are circled with dashed lines. Cells positive for CC3 are indicated by arrowheads.
Hypericin exhibits therapeutic effects on mice with HFHS-induced diabetes
Since hypericin showed strong preventive effects against the onset of diabetes in HFHS-fed mice, we further explored the therapeutic effects of hypericin on diabetes. Using HFHS-induced diabetic mice, we demonstrated that hypericin treatment markedly decreased the fasting blood glucose levels (Figure 7A) and body weight (Figure 7B) of HFHS-induced diabetic mice. Additionally, hypericin showed a tendency to reduce blood insulin levels in diabetic mice, although the difference was not statistically significant (Figure 7C). As expected, hypericin treatment significantly improved the state of glucose intolerance and insulin insensitivity of diabetic mice, as shown in the IPITT and IPGTT (Figure 7D-E). Furthermore, we showed that therapeutic hypericin treatment augmented both the size and the number of islets in the diabetic mouse pancreas in a dose-dependent manner as observed through HE and C-peptide IHC staining of pancreatic slices (Figure 8A-B), which was in agreement with the significantly increased islet area and β-cell mass in hypericin-treated diabetic mice compared to HFHS control mice (Figure 8C-D). Finally, as shown in Figure 8E, therapeutic hypericin treatment dramatically elevated pancreatic PDX1 levels in diabetic mice, which was consistent with the results observed in the prophylactic model. These data indicate that hypericin displayed strong therapeutic effects on HFHS-induced diabetes; these effects might be related to the amelioration of β-cell loss.
Therapeutic use of hypericin improves the diabetic phenotype of HFHS-fed mice. (A-E) After 4 months on an HFHS, mice were injected intraperitoneally with hypericin or 0.9% NaCl (HFHS control) every other day for nearly one month. The fasting blood glucose levels (A), body weight (B), blood insulin levels C), IPITT results (D) and IPGTT results (E) of the mice were then detected or analysed as in Fig. 4. *p<0.05, **p<0.01 versus the HFHS group.

Therapeutic use of hypericin decreases β-cell loss and maintains islet mass in mice with HFHS-induced diabetes. (A) Histological sections of mouse pancreatic tissue. The mice in Fig. 7 were sacrificed, and the pancreases were removed and weighed. Portions of the mouse pancreases were fixed and subjected to HE staining as in Fig. 5A. Scale bar represents 100 μm. Arrows indicate pancreatic islets. (B-D) Fixed pancreas tissue from A was subjected to IHC analysis using anti-C-peptide antibodies (B) as shown in Fig. 5B, after which IHC-stained pancreatic sections were used to calculate the islet area (C) and β-cell mass (D) of the pancreas. The scale bar represents 100 μm. Arrows indicate positively stained cells. Data are presented as the mean ± S.D. (n = 8). (E) PDX1 protein levels in the mouse pancreas. Portions of the mouse pancreases from (A) were homogenized. PDX1 levels in the homogenate were detected by Western blot, and the density ratios of PDX1 to GAPDH were measured as in Fig. 5E. Fold change relative to the normal group is shown in the right-hand panel. Data are presented as the mean ± S.D. (n = 6). *p<0.05, **p<0.01 ***p<0.001, versus the HFHS group.

Discussion
The pathogenesis and development of type I and type II diabetes are both closely related to β-cell failures, including decreased β-cell mass and β-cell dysfunction [33]. Protecting β-cells from loss and dysfunction is an essential way to prevent or treat diabetes [34]. Here, for the first time, we elucidated the protective role of hypericin against glucotoxicity and lipotoxicity in β-cells, both in vitro and in vivo. In that role, hypericin maintained and improved pancreatic β-cell mass and the size and number of islets in HFHS-induced diabetic mice, exhibiting both preventive and therapeutic effects on type II diabetes. Therefore, hypericin or derivatives discovered by using hypericin as a lead compound possess the potential to be developed as anti-diabetic drugs in the future.
Glucotoxicity and lipotoxicity are diabetes-related phenomena that generate oxidative stress, which is implicated in the pathogenesis of β-cell dysfunction and apoptosis and is therefore associated with diabetes [35-36]. For example, the overproduction of NO, an oxidant, can lead to oxidative stress and β-cell dysfunction [37]. Reports indicate that glucotoxicity and lipotoxicity result in β-cell apoptosis and dysfunction via an oxidative-stress-mediated decrease in the expression of PDX1 [38]. PDX1, as a housekeeping gene for pancreatic islet β-cells, is a key regulator of insulin gene expression [39] and is essential for the function and survival of mature β-cells as well [40-41]. Downregulation of PDX1 expression in β-cells may underlie the pathogenesis of β-cell failure and type II diabetes. In this study, we demonstrated that hypericin could attenuate oxidative stress triggered by high glucose or high fat and enhance the expression of PDX1 in β-cells exposed to glucotoxicity and lipotoxicity. Through that effect, hypericin reduced β-cell apoptosis in in vitro glucotoxicity and lipotoxicity models as well as in the pancreatic islets of HFHS-fed mice and effectively protected β-cells against death and loss. Thus, HFHS-fed mice with hypericin treatment had increased β-cell mass, islet size and pancreatic indices compared to HFHS control mice. Furthermore, prophylactic use of hypericin prevented the onset of diabetes to some extent in HFHS-fed mice, and hypericin also exerted strong therapeutic effects on HFHS-induced diabetes. Combining these data, we conclude that hypericin may elicit its protective effects on islet β-cells in part through its biological functions of attenuating oxidative stress and thereby preventing oxidative-stress-induced reduction in PDX1 expression, and we also conclude that hypericin is a promising drug candidate for the prevention and treatment of diabetes. In addition, our research suggests possible applications of hypericin against other oxidative-stress-related diseases, such as obesity, neurodegenerative disorders, ageing, and cardiovascular diseases, a possibility that deserves further exploration in the future.
Furthermore, we demonstrated that the Erk signalling pathway was involved in hypericin-mediated restoration of PDX1 expression and protection of β-cells from glucotoxicity- and lipotoxicity-induced apoptosis. The Erk signalling pathway plays an important role in the pathogenesis of diabetes, especially in diabetic complications and glucose metabolism [42]. For example, studies have shown that β1 integrity regulates glucose metabolism and changes PDX1 expression in vivo through Erk signalling [43]. Here, we found that the Erk phosphorylation level was significantly decreased under high-glucose and high-lipid conditions. However, hypericin treatment reversed the decrease in p-Erk levels. Utilization of the inhibitor U0126 to block activation of the Erk pathway completely abolished the increase in PDX1 expression and decrease in CC3 levels induced by hypericin in INS-1 cells by both glucotoxicity and lipotoxicity. These results provide direct evidence that hypericin may maintain the expression of PDX1 and protect β-cells by attenuating the high glucose- or high lipid-induced reduction in Erk activity. However, how hypericin rescues Erk activation in β-cells under glucotoxic and lipotoxic conditions and how Erk activation regulates PDX1 expression in β-cells remain elusive. So far, the molecular targets of hypericin have never been reported. Identification and validation of hypericin targets would help better understand working mechanisms of hypericin and deserves further investigation. Interestingly, photoactivated hypericin has been demonstrated to inhibit Erk signaling, induce ROS and activate the caspase pathway, resulting in cell apoptosis [44-45]. We speculate that photoactivation may change the structure of hypericin, altering its interaction with target proteins, or photoactivated hypericin may modify its interacting proteins in ways such as oxidation [46], which may lead to distinct biological effects. However, the underlying molecular mechanisms accounting for the opposite effects of photoactivated and non-photoactivated hypericin merit further investigation.
In vitro studies have shown that hypericin, even at high concentrations, lacks toxicity in the dark [47]. Non-photoactivated hypericin is non-mutagenic and does not alter the frequency of chromosome aberrations either [48]. Therefore, non-photoactivated hypericin is nontoxic and relatively safe. However, a major concern when hypericin is applied to the human body is its photoactivated cytotoxic effects on skin exposed to sunlight, which cause various types of photosensitivity reactions, such as paresthesias, dermatitis, and pruritic nodules [49]. These photosensitivity reactions are dose-dependent and are resolved after the cessation of hypericin treatment. Another concern regarding hypericin is its effect on nerve transmission, since hypericin shows an antidepressant effect and may affect the uptake and release of neurotransmission. Thus far, hypericin has been reported to show micromolar affinities for neuropeptide Y (NPY)-Y1, NPY-Y2, sigma and N-methyl-D-aspartate (NMDA) receptors in vitro [50-51], and hypericin has also been demonstrated to inhibit the release of glutamate from purified nerve terminals [23]. However, except for skin photosensitivity reactions, no other frequently occurring serious adverse events, including nervous system side effects, have been reported thus far in association with hypericin use. In our study, mouse experiments were conducted under a normal light cycle, and we did not observe any skin photosensitivity reactions at the concentrations of hypericin we used. Moreover, there were no pathological changes in the main organs of hypericin-treated mice (data not shown). To diminish the toxic side effects of hypericin for clinical acceptance, we next plan to chemically modify hypericin in an effort to obtain hypericin analogues that lose the ability to be photoactivated and/or cross the blood-brain barrier but retain the pharmacological activities of non-photoactivated hypericin. We believe that such hypericin analogues would merit extensive exploration of their clinical application values.
In conclusion, hypericin, as an agent to promote PDX1 expression, showed protective effects on islet β-cells and prevented the onset of type II diabetes in mice. The lead compound hypericin and its analogues possess great potential to be developed as preventive or therapeutic anti-diabetic drugs in the future. Our research expands the range of future clinical applications for hypericin.
Abbreviations
AUC: Area Under Curve; Bax: Bcl-2-Associated X protein; Bcl-2: B-cell lymphoma-2; C3: caspase 3; CC3: cleaved-caspase 3; DAPI: 4',6-diamidino-2-phenylindole; DM: diabetes mellitus; DAB: diaminobenzidine; GSH-PX: glutathione-peroxidase; HFHS: high-fat/high-sucrose; IPGTT: intraperitoneal glucose tolerance test; IPITT: intraperitoneal insulin tolerance test; iNOS: inducible nitric oxide synthase; MDA: malondialdehyde; MTT: 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; NO: nitric oxide; PA: palmitic acid; PDX1: pancreatic duodenal homeobox-1; PDT: photodynamic therapy; PDD: photodynamic diagnosis; T-AOC: total-antioxidant-capacity.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Key New Drug Creation and Manufacturing Program of Ministry of Science and Technology (No. 2013ZX09103003004), the Fundamental Research Funds for the Central Universities (No. 2412018QD011), the Grant of Jilin Province Science & Technology Committee (No. 20180520105JH, No. 20180101242JC; No. 20170414028GH, No. 20150204038YY and No. 20150309003YY), the Grant of Jilin Province Development and Reform Commission (No. 2015Y057), the Grant of Changchun City Science and Technology Bureau (No. 17YJ003) and the Grant of Changchun Industry and Information Technology Committee, China (No. 2017(342)).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xiong X, Wang X, Li B. et al. Pancreatic islet-specific overexpression of Reg3β protein induced the expression of pro-islet genes and protected the mice against streptozotocin-induced diabetes mellitus. Am J Physiol Endocrinol Metab. 2011;300:E669-80
2. Ganugula R, Arora M, Jaisamut P. et al. Nano-curcumin safely prevents streptozotocin-induced inflammation and apoptosis in pancreatic beta cells for effective management of Type 1 diabetes mellitus. Br. J. Pharmacol. 2017;174:2074-2084
3. Pagliuca FW, Melton DA. How to make a functional-cell. Development. 2013;140:2472-2483
4. Butler AE, Janson J, Bonner-Weir S. et al. βeta-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102-110
5. Cernea S, Dobreanu M. Diabetes and beta cell function: from mechanisms to evaluation and clinical implications. Biochem Med. 2013;23:266-280
6. Taniguchi S, Kang L, Kimura T. et al. Hydrogen sulphide protects mouse pancreatic β-cells from cell death induced by oxidative stress, but not by endoplasmic reticulum stress. Br. J. Pharmacol. 2011;162:1171-1178
7. Del Guerra S, Lupi R, Marselli L. et al. Functional and Molecular Defects of Pancreatic Islets in Human Type 2 Diabetes. Diabetes. 2005;54:727-735
8. Kawahito S, Kitahata H, Oshita S. Problems associated with glucose toxicity: role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009;15:4137-4142
9. McKinnon CM, Docherty K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of β-cell identity and function. Diabetologia. 2001;44:1203-1214
10. Fujimoto K, Polonsky KS. Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes Metab. 2009;11(Suppl 4):S30-S37
11. Fujimoto K, Chen Y, Polonsky KS. Targeting cyclophilin D and the mitochondrial permeability transition enhances beta-cell survival and prevents diabetes in Pdx1 deficiency. Proc Natl Acad Sci USA. 2010;107:10214-10219
12. Johnson JD, Ahmed NT, Luciani DS. et al. Increased islet apoptosis in Pdx1+/- mice. J Clin Invest. 2003;111:1147-60
13. Hong SW, Lee J, Park SE. et al. Repression of sterol regulatory element-binding protein 1-c is involved in the protective effects of exendin-4 in pancreatic β-cell line. Mol Cell Endocrinol. 2012;362:242-52
14. Youl E, Bardy G, Magous R. et al. Quercetin potentiates insulin secretion and protects INS-1 pancreatic β-cells against oxidative damage via the ERK1/2 pathway. Br. J. Pharmacol. 2010;161:799-814
15. Quan X, Zhang L, Li Y. et al. TCF2 attenuates FFA-induced damage in islet β-cells by regulating production of insulin and ROS. Int J Mol Sci. 2014;15:13317-32
16. Wijesekara N, Krishnamurthy M, Bhattacharjee A. et al. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J Biol Chem. 2010;285:33623-31
17. Kwak HJ, Yang D, Hwang Y. et al. Baicalein protects rat insulinoma INS-1 cells from palmitate-induced lipotoxicity by inducing HO-1. PLoS One. 2017;12:e0176432
18. Costes S, Broca C, Bertrand G. et al. ERK1/2 control phosphorylation and protein level of cAMP-responsive element-binding protein: a key role in glucose-mediated pancreatic beta-cell survival. Diabetes. 2006;55:2220-30
19. Ahn M, Yoder SM, Wang Z. et al. The p21-activated kinase (PAK1) is involved in diet-induced beta cell mass expansion and survival in mice and human islets. Diabetologia. 2016;59:2145-55
20. Rys P, Wojciechowski P, Siejka S. et al. A comparison of biphasic insulin aspart and insulin glargine administered with oral antidiabetic drugs in type 2 diabetes mellitus-a systematic review and meta-analysis. Int. J. Clin. Pract. 2014;68:304-313
21. Cavarga I, Brezáni P, Cekanová-Figurová M. et al. Photodynamic therapy of murine fibrosarcoma with topical and systemic administration of hypericin. Phytomedicine. 2001;8:325-330
22. Jendželovská Z, Jendželovský R, Kuchárová B. et al. Hypericin in the light and in the dark: two sides of the same coin. Front. Plant Sci. 2016;7:560
23. Chang Y, Wang SJ. Hypericin, the active component of st. john's wort, inhibits glutamate release in the rat cerebrocortical synaptosomes via a mitogen-activated protein kinase-dependent pathway. Eur. J. Pharmacol. 2010;634:53-61
24. Shi XL, Ren YZ, Wu J. Intermittent high glucose enhances apoptosis in ins-1 cells. Exp Diabetes Res. 2015;2011:1687-5214
25. Sun Y, Zhang L, Gu HF. et al. Peroxisome Proliferator-Activated Receptor-α Regulates the Expression of Pancreatic/Duodenal Homeobox-1 in Rat Insulinoma (INS-1) Cells and Ameliorates Glucose-Induced Insulin Secretion Impaired by Palmitate. Endocrinology. 2008;149:662-671
26. Jiang Y, Huang W, Wang J. et al. Metformin plays a dual role in min6 pancreatic β cell function through ampk-dependent autophagy. Int J Biol Sci. 2014;10:268-277
27. Ai HH, Zhou ZL, Sun LG. et al. 20(S)-25-methoxyl-dammarane-3β, 12β, 20-triol negatively regulates activation of STAT3 and ERK pathways and exhibits anti-cancer effects in HepG2 cells. Apoptosis. 2017;1:1-15
28. Lee SJ, Choi HN, Kang MJ. et al. Chamnamul [pimpinella brachycarpa (kom.) nakai] ameliorates hyperglycemia and improves antioxidant status in mice fed a high-fat, high-sucrose diet. Nutr Res Pract. 2013;7:446-452
29. Hu SW, Wang JF, Xu H. et al. Fucosylated chondroitin sulphate from sea cucumber inhibits high-fat-sucrose diet-induced apoptosis in mouse pancreatic islets via down-regulating mitochondrial signaling pathway. J Funct Foods. 2014;7:517-526
30. Yang L, Yao D, Yang H. et al. Puerarin protects pancreatic β-cells in obese diabetic mice via activation of GLP-1R signaling. Mol Endocrinol. 2016;30:361-71
31. Benetti E, Mastrocola R, Chiazza F. et al. Effects of vitamin D on insulin resistance and myosteatosis in diet-induced obese mice. Plos One. 2018;13:e0189707
32. Sinha K, Das J, Pal PB. et al. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157-80
33. Mandrup-Poulsen T. Apoptotic signal transduction pathways in diabetes. Biochem Pharmacol. 2003;66:1433-1440
34. Nagy L, Márton J, Vida A. et al. Glycogen phosphorylase inhibition improves beta cell function. Br. J. Pharmacol. 2018;175:301-319
35. Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr. Rev. 2008;29:351-366
36. Rashid MA, Lee S, Tak E. et al. Carbonyl reductase 1 protects pancreatic beta-cells againstoxidative stress-induced apoptosis in glucotoxicity and glucolipotoxicity. Free Radic. Biol. Med. 2010;49:1522-1533
37. Muhammed SJ, Lundquist I, Salehi A. Pancreatic β-cell dysfunction, expression of iNOS and the effect of phosphodiesterase inhibitors in human pancreatic islets of type 2 diabetes. Diabetes Obes. Metab. 2012;14:1010-1019
38. Shimo N, Matsuoka TA, Miyatsuka T. et al. Short-term selective alleviation of glucotoxicity and lipotoxicity ameliorates the suppressed expression of key β-cell factors under diabetic conditions. Biochem Biophys Res Commun. 2015;467:948-954
39. Yang BT, Dayeh TA, Volkov PA. et al. Increased dna methylation and decreased expression of pdx-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol. 2012;26:1203-1212
40. Goff LM, Griffin BA, Lovegrove JA. et al. Ethnic differences in beta-cell function, dietary intake and expression of the metabolic syndrome among uk adults of south asian, black african-caribbean and white-european origin at high risk of metabolic syndrome. Diab. Vasc. Dis. Res. 2013;10:315-323
41. Servitja JM, Ferrer J. Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia. 2004;47:597-613
42. Tan Y, Ichikawa T, Li J. et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60:625-633
43. Krishnamurthy M, Liu S, Liu M. et al. Conditional knockout of the β1 integrin affects glucose metabolism and pdx-1 expression through erk1/2 signaling. Can. J. Diabetes. 2008;32:336-336
44. Yi J, Yang X, Zheng L. et al. Photoactivation of hypericin decreases the viability of rinm5f insulinoma cells through reduction in jnk/erk phosphorylation and elevation of caspase-9/caspase-3 cleavage and bax-to-bcl-2 ratio. Biosci Rep. 2015;35:pii e00195
45. Zhang Q, Li ZH, Li YY. et al. Hypericin-photodynamic therapy induces human umbilical vein endothelial cell apoptosis. Sci Rep. 2015;17:18398
46. Kascakova S, Refregiers M, Jancura D. et al. Fluorescence spectroscopic study of hypericin-photosensitized oxidation of low-density lipoproteins. Photochem Photobiol. 2005;81:1395-1403
47. Vandenbogaerde AL, Cuveele JF, Proot P. et al. Differential cytotoxic effects induced after photosensitization by hypericin. J Photochem Photobiol B. 1997;38:136-142
48. Miadokova E, Chalupa I, Vlckova V. et al. Genotoxicity and antigenotoxicity evaluation of non-photoactivated hypericin. Phytother Res. 2010;24:90-95
49. Jacobson JM, Feinman L, Liebes L. et al. Pharmacokinetics, safety, and antiviral effects of hypericin, a derivative of st. john's wort plant, in patients with chronic hepatitis c virus infection. Antimicrob Agents Chemother. 2001;45:517-24
50. Cott J M. In Vitro Receptor Binding and Enzyme Inhibition by Hypericum perforatum Extract. Pharmacopsychiatry. 1997;30(supplement 2):S108-S112
51. Gobbi M, Moia M, Pirona L. et al. In vitro binding studies with two hypericum perforatum extracts-hyperforin, hypericin and biapigenin-on 5-ht6, 5-ht7, gaba(a)/benzodiazepine, sigma, npy-y1/y2 receptors and dopamine transporters. Pharmacopsychiatry. 2001;34(supplement 1):S45-S48
Author contact
![]() Corresponding authors: Email: sunlg388edu.cn (Luguo Sun), liyx486edu.cn (Yuxin Li). Tel: 86-0431-8916-5922; Fax: 86-0431-8916-5917.
Corresponding authors: Email: sunlg388edu.cn (Luguo Sun), liyx486edu.cn (Yuxin Li). Tel: 86-0431-8916-5922; Fax: 86-0431-8916-5917.