Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(8):1630-1636. doi:10.7150/ijbs.33772 This issue Cite
Research Paper
A Synthetic Light-switchable System based on CRISPR Cas13a Regulates the Expression of LncRNA MALAT1 and Affects the Malignant Phenotype of Bladder Cancer Cells
Fuming Qi1, Bo Tan1, Fujun Ma1, Bo Zhu1, Li Zhang2, Xiaoyun Liu1, Honglei Li3, Jinhui Yang1, Bo Cheng1 ![]()
1. Urology and Andrology Department, Shengli OilFiled Central Hospital, Dongying, 257034, Shandong, China
2. Burn and Plastic surgery Department, Shengli OilFiled Central Hospital, Dongying, 257034, Shandong, China
3. Medical Department, Shengli OilFiled Central Hospital, Dongying, 257034, Shandong, China
Received 2019-2-2; Accepted 2019-4-10; Published 2019-6-4
Abstract

DNA sequences drive their various functions through post-transcriptional processes, using mRNA or lncRNA (long non-coding RNA), and this accommodates the gene network by using various RNA types. However, the tools necessary to regulate RNA molecules are few. Likewise, RNA knockdown techniques that can be artificially controlled have not been extensively explored. Here, we investigated a novel light-inducible synthetic system based on CRISPR-Cas13a that can be used for RNA knockdown and binding in cancer cells. Based on the techniques of synthetic molecular biology, we constructed a light sensor, which efficiently induced Cas13a protein expression after blue light illumination. We also chose a lncRNA, Metastasis-associated Lung Adenocarcinoma Transcript 1 (MALAT1), as the functional target and detected it in bladder cancer 5637 and T24 cells in order to demonstrate the application of our synthetic system. Fluorescence reporter assays and real-time quantitative PCR (qRT-PCR) were used to detect the expression of the target gene. Phenotypic experiments were also used to test the effects of our synthetic system in bladder cancers. The results showed that our synthetic light-switchable system could inhibit the expression of MALAT1, and the fluorescence activity of enhanced green fluorescent protein. Our novel system provides a new technique to study RNA functions in gene networks and for precise tumor treatments.
Keywords: Cas13a, cancer cell, light inducible, RNA, synthetic
Introduction
The ability to regulate and manipulate target genes is important both in biomedical research and clinical treatments.[1] With the continuous discovery of gene functions and signal pathways, it is increasingly important to construct artificial synthetic devices or systems to transform or manipulate the original signal pathway and the level of gene expression.[2, 3] Over the past decade, various artificial synthetic technologies have been exploited to regulate specific functions in cells, which have improved the prospects for clinical treatments and biological research. Synthetic biological devices and techniques have greatly enriched the fields of biotechnology research and clinical gene therapy. However, the fidelity of genome editing has always been a controversial topic[4, 5], and it is important to correctly edit the original genes at the genome level in order to prevent the addition of incorrect genetic information into cells. In contrast, regulation at the post-transcriptional level does not alter the original genetic information. Therefore, new molecular tools to directly manipulate and regulate the product of gene expression, such as mRNA or lncRNA (long non-coding RNA) are necessary.
Previous studies have found that RNA has important biological functions in cell growth and development. However, the tools that directly regulate the levels of RNA are limited. Traditionally, RNA interference[6] was used to knock down the target RNAs, but off-target effects[7] are still a serious problem with this process. The effect on RNA in the nucleus is even less efficacious and specific. With increasing advancements in CRISPR technology, the toolkits for RNA manipulations have greatly expanded. Zhang et al.[8] developed a RNA toolkit based on CRISPR Cas13a in human cells, which was more efficient and more specific for RNA knockdown and binding than traditional RNA interference methods. Xie et al,[9] designed a synthetic system that targeted oncogenic microRNAs, which inhibited the malignant phenotypes of bladder cancer cells. However, there are still limitations to the synthetic procedures. A challenge to the use of this methodology is its lack of safety, reversibility, and spatiotemporal control.
Small molecularly regulated synthetic systems have been successfully used in synthetic biology research. However, it was difficult to control the effects in the target organisms. Compared with molecular approaches, light is an ideal “switch” for artificial synthetic systems because of its reversibility and spatiotemporal resolution.[10] Therefore, we have used light as a switch to control artificial synthetic systems. We used an optimized light-switch system as a fundamental construct and inserted the RNA nuclease Cas13a DNA sequence into the output gene of the system.[11] Under the stimulation of blue light, the system quantitatively outputed Cas13a protein. After the expression of Cas13a in the cancer cells, it was specifically targeted for knockdown of either mRNA or lncRNA; thus, the levels of RNA were artificially manipulated using light. We choose lncRNA Metastasis-associated Lung Adenocarcinoma Transcript 1 (MALAT1), which has been confirmed to be highly expressed as an oncogene for human bladder cancer in previous works [12, 13] as the target RNA sequence.
Herein, we exploited a light-switchable synthetic system, which controlled the Cas13a nuclease activity to efficiently knockdown the level of MALAT1 and affect the malignant phenotype of bladder cancer cells. Light is noninvasive, reversible, and has high spatiotemporal resolution, which provides an ideal switch for safe biotechnological studies and clinical treatments.
Methods and Materials
Cell lines and cell culture
We purchased the bladder carcinoma cell lines (5637 and T24) from the America Type Culture Collection (ATCC, Manassas, VA, USA). Fetal bovine serum (FBS, 10%) and 1% antibiotics (100 µg/mL streptomycin and 100 U/mL penicillin) were added to RPMI-1640 medium (Gibco-BRL, Grand Island, NY, USA) and DMEM medium (Gibco-BRL). The 5637 cells were cultured in the above RPMI-1640 medium, in a 37°C humidified incubator with 5% CO2. The T24 cells were cultured in the above DMEM medium, in a 37°C humidified incubator with 5% CO2.
Construction of plasmids
The constructions of plasmids used standard biological methods. Briefly, we purchased the pcDNA3.1-EGFP vector from Addgene (#13031; Cambridge, MA, USA) and inserted the GAVPO or tGAVPO-NC sequences into the vector. The sequence of GAVPO consisted of Gal4, VVD, and VP64. We used an upstream activating sequence (UAS) of five repeat sequences and a minimal promoter to replace the CMV promoter and inserted the Cas13a gene sequence into a downstream site. Cas13a-MALAT1 sgRNA and NC vector were purchased from SyngenTech, Beijing, China (http://www.syngentech.com.cn/en/). The sequences are shown in a Supplementary table 1.
Cell transfections and blue light irradiation
The 5637 and T24 bladder cancer cells were cultured in 6-well plates, and after 24 hours, when the cells were 75%-90% confluent, pcDNA3.1-GAVPO and 5×UASg-Cas13a or negative control (NC) were transiently transfected at a ratio of 1:1 (1.25 μg for each plasmid in a 6-well plate) following the manufacturer's protocol using Lipofectamine® 3000 transfection reagent (Invitrogen, Carlsbad, CA, USA). Six hours after transfection, and in dark conditions, the cells were exposed to a 0.84W/m2, 460 nm (peak) blue light LED lamp, or were kept in the dark for 24-48 hours. The dose of light was controlled by a controller.
Total RNA extraction and real-time qPCR
Total RNA from cells irradiated by blue light was extracted by TRIzol reagent (Invitrogen) following the manufacture's protocol. After measuring the concentrations, RNAs were reverse transcribed to cDNA by using a Revertra Ace qPCR RT Kit (Toyobo, Osaka, Japan). Primers and cDNAs were mixed with SYBR Green Premix (Takara, Japan) on ice. Real-time qPCR (qRT-PCR) was performed in triplicate using the ABI PRISM 7000 Fluorescent Quantitative PCR system (Applied Biosystems, Foster City, CA, USA). Relative MALAT1 lncRNA expression level was calculated by the 2-ΔΔCt method. The primer sequences are shown in a Supplementary table 2.
Cell apoptosis analysis by flow cytometry
After blue light illumination or darkness for 48 hours, the cells were harvested by using EDTA-free trypsin, and washed with 1× phosphate-buffered saline (PBS), and 2×105-2×106 cells were resuspended in ice-cold 1× Annexin V Binding Buffer (100 μL). Annexin V-FITC and propidium iodide (PI) were added to the cell suspension following the manufacturer's protocol. Cell samples were detected by a Beckman EPICS XL-4 system (Beckman, West Sacramento, CA, USA). Living cells vs apoptotic cells were identified from the flow cytometry graphs.
Transwell® assays
Cells motility assays used Transwell® chambers. After 6-8 hours of transfection, the cells were illuminated under blue light or kept in the dark for 48 hours. Then, approximately 5×104 T24 cells or 1.5×105 5637 cells were mixed with 200 μL serum-free medium and placed into the upper uncoated chambers (24-well plate, 8 μm; Corning Corning, NY, USA). At the same time, 500 μL medium with 10% FBS and 1% antibiotics was added to the lower (basolateral) compartment. Cells were then cultured at 37°C with 5% CO2 for 24 hours. Transwell® inserts were then placed in 4% paraformaldehyde for 40 min, and then placed in 0.1% Crystal Violet for 20 min. The inserts were washed with double-distilled H2O (ddH2O) at least three times. Invaded cells were observed using an inverted microscope and imaged. Each Transwell® insert with the invaded cells was then placed in 1 mL of 33% acetic acid for 15 min, and 100 μL of 33% acetic acid was added into each well. The absorbance was detected by a microplate reader (570 nm; Bio-Rad, Hercules, CA, USA). All the experiments were repeated at least three times.
Statistical analysis
All experimental assays were performed in triplicate. The data are presented as the mean ± standard deviation (SD). The statistical analysis of data was processed by Student's t-tests or ANOVAs using SPSS software, version 21.0 (IBM Corp. Armonk, NY, USA). According to the statistical definition, a value of P less than 0.05 was considered statistically significant.
Results
Construction of a light-switchable synthetic system
The light-switchable synthetic system consisted of a genome anchor with a light sensor and gene effector (Fig. 1A). Gal4 is a transcription activator, which specifically recognizes and binds to the UAS. Vivid (VVD) is a light/oxygen voltage domain, which forms a dimer under blue light. To create the genome light sensor anchor, we removed the dimerization domain of Gal4 and replaced it with VVD. Furthermore, recombinant Gal4 protein was fused with P65 protein, which formed the optimized P65 activation domain (GAVPO) (Fig. 1Aa). We also replaced the CMV promoter of the EGFP gene with a five repeat UAS of the Gal4 sequence (5×UASg) (Fig. 1Ab). Upon blue light irradiation, the fusion protein of GAVPO dimerized and bound to the sequence of 5×UASg, which activated the expression of downstream genes. In contrast, the protein does not bind in the dark, and transcriptional activation does not occur. We assessed the GAVPO-induced activity using an EGFP plasmid assay in human 5637 cells (Fig. 1B, Fig. 1C; p<0.001). This system effectively controlled gene expression in eukaryotic cells. Compared with the dark and NC groups, the light-switchable system showed increased induction of gene expression of 180-200-fold when exposed to blue light (0.84 W/m2, 24 hours) [11].
Construction of the light-switchable synthetic system in cancer cells. (A) A schematic of the light-switchable synthetic system. (B-C) The expression of EGFP after blue light illumination. Compared with the dark group or negative control group (NC), the expression of EGFP was significantly increased in 5637 cells after 6-8 hours of transient transfection; cells were illuminated by blue light (0.84 W/m2, 24 hours) or remained in the dark for 24 hours. (D) GAVPO and 5×UASg-Cas13a-EGFP sgRNA or negative control vector were co-transfected into the 5637 cells stably expressing EGFP. After blue light illumination (0.84W/m2) or after being in the dark for 48 hours, the fluorescence intensity of the 5637 cells was significantly decreased compared with that of the dark group or negative control group. (E) Relative expression of EGFP was measured by real-time qPCR. (F-G) Relative expression of MALAT1 as measured by real-time qPCR. (H) The expression level of MALAT1 was dynamically detected by real-time qPCR as the blue light illumination time was prolonged. All results are shown as the mean ± SD (*p<0.05, **p<0.01). EGFP, enhanced green fluorescent protein.
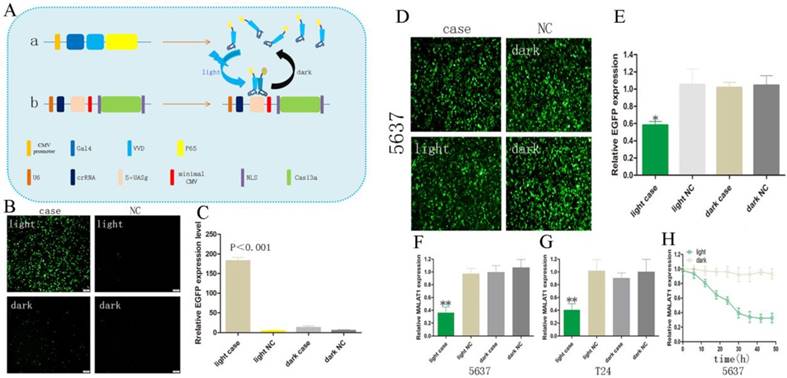
RNA knockdown by the light-switchable synthetic system
We performed an RNA knockdown assay in order to measure the efficiency of RNA knockdown using the synthetic light-switchable system. Both the GAVPO and 5×UASg-Cas13a vector or the negative control group plasmids were transiently transfected into 5637 cell lines (stably expressing EGFP protein). After the blue light illumination (0.84 W/m2) or in the dark for 48 hours, the fluorescence intensity of the 5637 cells was significantly decreased after transfection with the light-switchable synthetic system (Fig. 1D, E; p<0.05). Therefore, it was possible to manipulate RNA expression levels by using a light-switchable system. We then redesigned the sgRNA to target lncRNAs. MALAT1 was significantly decreased after transfection with the light-switchable synthetic system.
The effects of blue light illumination times on RNA expression
MALAT1 lncRNA is highly expressed in bladder cancer 5637 and T24 cells.[12, 13] This indicated that lncRNA MALAT1 could inhibit cell apoptosis by antagonizing miR-125b. Hence, we chose the 5637 and T24 cell lines to test our light-switchable system and compared the results with the dark group and negative control group. The expression levels of MALAT1 lncRNA were significant decreased under blue light illumination (Fig. 1F, G; p<0.01). We also detected the expression level of MALAT1 in order to explore the regulatory effects of the light-switchable system on knockdown of MALAT1. The data demonstrated that the expression level of MALAT1 decreased as the blue light illumination time was increased (Fig. 1H).
The effects of downregulation of MALAT1 on bladder cancer cell apoptosis and motility
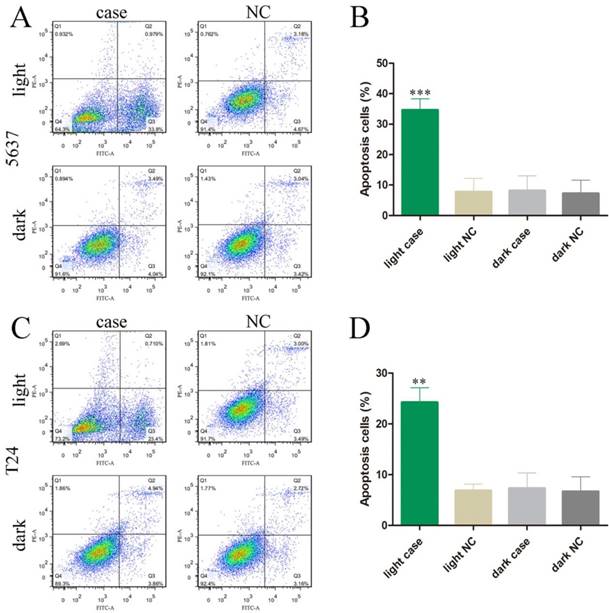
We also investigated the effects of MALAT1 knockdown on cell apoptosis and motility by using our light-switchable synthetic system in bladder cancer cells (Fig. 2). The bladder cancer cells were co-transfected with GAVPO and 5×UASg-Cas13a-MALAT1 sgRNA or negative control vector. After blue light sustainable illumination (0.84W/m2) or remaining in the dark for 48 hours, we used flow cytometry to measure apoptosis changes in bladder cancer cells (Fig. 3A-D; p<0.01).
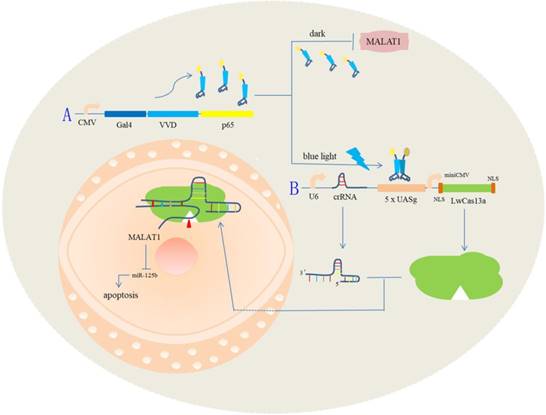
A schematic diagram of the light-switchable system based on CRISPR Cas13a and the effects on the malignant phenotype of bladder cancer cells by post-transcriptional regulation.
The effects of the light-switchable synthetic system on apoptosis in bladder cancer 5637 and T24 cells. (A-D) The apoptosis changes of cells were determined by flow cytometry assays, and representative images in 5637 and T24 cells are shown. Error bars illustrate the mean ± SD. NC, negative control, which are compared with the dark group or negative control group. One-sided t-test. (**p<0.01, ***p<0.001).
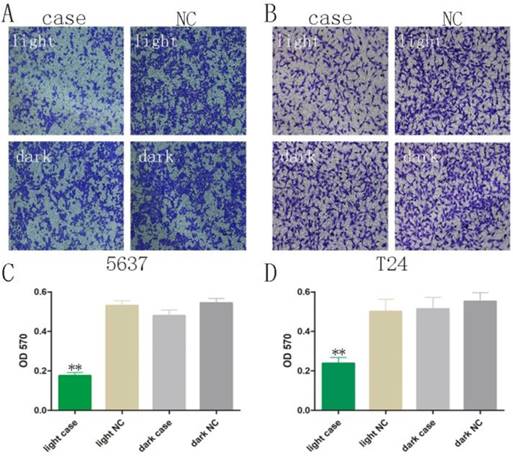
Decreased motility was also observed using Transwell® assays, after co-transfection of GAVPO and 5×UASg-Cas13a-MALAT1 sgRNA vs. negative control vector in 5637 and T24 bladder cancer cells. The results demonstrated that the light-switchable synthetic system downregulated MALAT1 lncRNA after blue light illumination and suppressed the migration of bladder cancer 5637 and T24 cells (Fig. 4A-D; p<0.01).
The bladder cancer cell migration was reversed upon activation of the light-switchable synthetic system. (A-B) The light-switchable synthetic system affected the mobility of 5637 and T24 bladder cancer cells by efficient knockdown of the MALAT1. Cell migration with co-transfection was inhibited under blue light illumination. (C-D) The cell migration was determined by enzyme-labelling assays. The results are shown as the mean ± SD. NC, negative control, compared with the dark group or negative control group. One-sided t-test (**p<0.01).
Discussion
Bladder cancer is considered one of the most common malignant tumors in the urinary system.[14] Studies have revealed that the occurrence of bladder cancer is related to genetic factors; upregulation of oncogenes may be an important factor that leads to changes in cellular biological functions.[15] lncRNA is considered to play an important and complicated role in cancer initiation, growth, and progression. Because lncRNA acts as a regulator, they can simultaneously regulate multiple protein signaling pathways and are the hub of gene regulatory networks. It is therefore critical to reduce the malignant phenotype of tumor cells by regulating the levels of lncRNA molecules. In this study, we spliced the cDNA of the Cas13a sequence to a light sensor recognition site to design a synthetic light-switchable system. In order to confirm the efficacy of our system, we used EGFP and oncogenic MALAT1 lncRNA as targets; the qRT-PCR results showed that the system caused knockdown of the expression level of EGFP in 5637 cells and also caused decreased expression of oncogenic MALAT1 lncRNA in bladder cancer cells (5637 and T24 cells) after blue light illumination. Additional phenotypic experiments illustrated that after blue light illumination, the light-switchable synthetic system also caused alteration in the phenotype of the bladder cancer cells.
Regulation of cellular gene networks or gene editing must be safe, have low toxicity, and show low background expression. Furthermore, interference with endogenous genes or proteins should also be minimized in order to reduce possible damaging crosstalk with the normal cellular signal systems. Therefore, a synthetic system should be simple and efficient. We used light as a switch for our system, which is harmless and reversible, and the effects on the cell can be removed immediately after the light source is turned off. Compared with small molecular switches and temperature control, a light switch is more convenient and easier to manipulate[16]. Light can control the multimerization state of light-sensitive proteins, which has been previously exploited in synthetic biology[17-19] and genome engineering[20].
RNA interference technology is not only a powerful tool for studying gene functions, but also is an important technology for clinical treatments. It provides efficacious clinical application value for cancer therapy by silencing of oncogenes. RNA targeting with CRISPR-Cas13a technology has been developed, and compared with traditional RNA interference, CRISPR-Cas13a technology was more efficient and more specific. We presented the first work that uses optical switches to regulate the activity of the Cas13a system. Compared to the dCas9 system, Cas13a specifically targets RNA molecules in cells, which is a special advantage.
However, achieving timely and accurate regulation of genes in order to ensure the safety of gene interference technology remains challenging. Our light-switchable synthetic system provides a possible novel technology to solve this problem; in particular, light was applied to control synthetic biological systems. The technology can also interfere with a variety of cellular RNA molecules (such as mRNA, lncRNA, and eRNA). However, there are still limitations that should be solved. For example, the occurrence and development of cancer often results from the activation of multiple oncogenes; therefore, regulation of a single target gene may not show efficacy in clinical treatments. Hence, simultaneous regulation of multiple RNA targets is an important direction for further research. Furthermore, studies have shown that it is feasible to simultaneously regulate multiple targets by designing crRNA arrays.[21] Therefore, optimizing the tandem crRNA arrays in our synthetic system should be done in the future.
In conclusion, we have exploited a novel light-switchable synthetic system, which manipulated RNA expression in cells. Moreover, the light-switchable synthetic system affected apoptosis and migration of bladder cancer cells by post-transcriptional regulation. Multi-target intervention could be an important direction in our future work. The results provide a new strategy to study the function of RNAs for use in biotechnology research and as clinical treatments. The key issue of current gene therapy is still the problem of gene delivery. So in the future, we plan to use viral vectors or nanomaterials to carry these light-control devices into tissue cells.
Supplementary Material
Supplementary tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jusiak B, Cleto S, Perez-Pinera P, Lu TK. Engineering Synthetic Gene Circuits in Living Cells with CRISPR Technology. Trends Biotechnol. 2016;34:535-47
2. Liu Y, Zhan Y, Chen Z, He A, Li J, Wu H. et al. Directing cellular information flow via CRISPR signal conductors. Nature methods. 2016;13:938-44
3. Liu Y, Li J, Chen Z, Huang W, Cai Z. Synthesizing artificial devices that redirect cellular information at will. eLife. 2018:7
4. Ho P, Chen YY. Mammalian synthetic biology in the age of genome editing and personalized medicine. Current opinion in chemical biology. 2017;40:57-64
5. Jennifer Doudna. Tailoring the Genome. Trends in cancer. 2016;2:536-7
6. Root DE, Hacohen N, Hahn WC, Lander ES, Sabatini DM. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nature methods. 2006;3:715-9
7. Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M. et al. Expression profiling reveals off-target gene regulation by RNAi. Nature biotechnology. 2003;21:635-7
8. Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ. et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280-4
9. Xie H, Zhan H, Gao Q, Li J, Zhou Q, Chen Z. et al. Synthetic artificial "long non-coding RNAs" targeting oncogenic microRNAs and transcriptional factors inhibit malignant phenotypes of bladder cancer cells. Cancer letters. 2018;422:94-106
10. Muller K, Naumann S, Weber W, Zurbriggen MD. Optogenetics for gene expression in mammalian cells. Biological chemistry. 2015;396:145-52
11. Wang X, Chen X, Yang Y. Spatiotemporal control of gene expression by a light-switchable transgene system. Nature methods. 2012;9:266-9
12. Han Y, Liu Y, Nie L, Gui Y, Cai Z. Inducing cell proliferation inhibition, apoptosis, and motility reduction by silencing long noncoding ribonucleic acid metastasis-associated lung adenocarcinoma transcript 1 in urothelial carcinoma of the bladder. Urology. 2013;81:209 e1-7
13. Xie H, Liao X, Chen Z, Fang Y, He A, Zhong Y. et al. LncRNA MALAT1 Inhibits Apoptosis and Promotes Invasion by Antagonizing miR-125b in Bladder Cancer Cells. Journal of Cancer. 2017;8:3803-11
14. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
15. Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer letters. 2013;339:159-66
16. Nihongaki Y, Otabe T, Sato M. Emerging Approaches for Spatiotemporal Control of Targeted Genome with Inducible CRISPR-Cas9. Analytical chemistry. 2018;90:429-39
17. Nihongaki Y, Furuhata Y, Otabe T, Hasegawa S, Yoshimoto K, Sato M. CRISPR-Cas9-based photoactivatable transcription systems to induce neuronal differentiation. Nature methods. 2017;14:963-6
18. Zhou XX, Zou X, Chung HK, Gao Y, Liu Y, Qi LS. et al. A Single-Chain Photoswitchable CRISPR-Cas9 Architecture for Light-Inducible Gene Editing and Transcription. ACS chemical biology. 2018;13:443-8
19. Lin F, Dong L, Wang W, Liu Y, Huang W, Cai Z. An Efficient Light-Inducible P53 Expression System for Inhibiting Proliferation of Bladder Cancer Cell. International journal of biological sciences. 2016;12:1273-8
20. Hochrein L, Mitchell LA, Schulz K, Messerschmidt K, Mueller-Roeber B. L-SCRaMbLE as a tool for light-controlled Cre-mediated recombination in yeast. Nature communications. 2018;9:1931
21. Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM. et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nature biotechnology. 2017;35:31-4
Author contact
![]() Corresponding author: Bo Cheng, E-mail address: edwardchengbocom
Corresponding author: Bo Cheng, E-mail address: edwardchengbocom