Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(8):1676-1684. doi:10.7150/ijbs.35265 This issue Cite
Research Paper
Enhancement of oxaliplatin-induced colon cancer cell apoptosis by alantolactone, a natural product inducer of ROS
Peihai Cao1#, Yiqun Xia2#, Wei He1#, Tingting Zhang1#, Lin Hong1, Peisen Zheng1, Xin Shen1, Guang Liang1, Ri Cui1 ![]() , Peng Zou1
, Peng Zou1 ![]()
1. Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang 325035, China
2. Department of Digestive Diseases, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang 325035, China
# These authors contributed equally to this work
Received 2019-3-27; Accepted 2019-5-11; Published 2019-6-4
Abstract

Colon cancer is a malignant type of cancer with high prevalence and is one of the primary causes of cancer-related deaths. Oxaliplatin plays a significant role in the treatment of cancer, but the application of oxaliplatin is restricted due to its toxic side effects and drug resistance in clinical practice. Therefore, there is an urgent need for new strategies that can synergize with oxaliplatin for confronting colon cancer. Alantolactone (ALT), a natural sesquiterpene lactone, possesses antitumor properties in a number of cancer cell lines. In the present study, we investigated how ALT acts synergistically with oxaliplatin on human colorectal cancer HCT116 and RKO cells in vitro and in vivo. We observed that ALT strengthened the effect of oxaliplatin-induced growth restrain and apoptosis in HCT116 and RKO cells. It is through a mechanism concerning remarkable accumulation of intracellular reactive oxygen species (ROS) and activation of JNK and p38 MAPK signaling pathways. These changes ultimately induced apoptosis of HCT116 and RKO cells. Pretreatment of cells with the ROS reversal agent NAC significantly blocked the apoptosis induced by the combination treatment, and suppressed expression of JNK and p38 phosphorylation in HCT116 and RKO cells. In the xenograft model, the combination therapy displayed stronger antitumor activity compared with single agents. Immunohistochemistry of subsequent treatment tumors showed a significant decrease in proliferation as compared to either of the treatments alone. These results suggest that the combination treatment with ALT and oxaliplatin may become a potential therapeutic strategy for colon cancer.
Keywords: Reactive oxygen species, MAPK, Alantolactone, Oxaliplatin, Colon cancer
Introduction
Colon cancer remains one of the leading causes of cancer-related deaths in the world [1]. Currently, the treatment of colon cancer is mainly surgery and chemotherapy. Although chemotherapy has enhanced the survival rate of patients, resistance develops in nearly all patients with colon cancer and limits the therapeutic efficacies of many anti-carcinogens. These developments ultimately led to the failure of chemotherapy [2]. Oxaliplatin was the third generation of platinum antitumor drugs, which had the characteristics of powerful antitumor effects. It had been widely applied for the treatment of various types' tumors [3]. Paradoxically, high doses of oxaliplatin can cause strong side effects, such as gastrointestinal reactions, bone marrow suppression of nerve ending inflammation, and low doses can cause drug resistance [4]. Therefore, a novel approach to confronting colon cancer is needed [5].
Currently, drug combination therapies have become increasingly popular to reduce drug side effects and overcome chemoresistance [6]. Combination of oxaliplatin with conventional chemotherapy drugs such as capecitabine has improved the survival of colon cancer patients [7]. Alantolactone is a sesquiterpene lactone isolated from the roots of Inula helenium L, a traditional chinese herbal medicine which has been shown to possess strong anti-inflammatory and antitumor activities [8,9].
In this study, we investigated whether alantolactone can synergize to enhance the antitumor efficacy of oxaliplatin in colon cancer cells. We found that co-treatment of alantolactone and oxaliplatin in colon cancer cells induced stronger cell killing effect compared with either of these drugs alone, and further demonstrated that ROS accumulation was involved in the synergistic effect of alantolactone and oxaliplatin. Our studies elucidated the underlying mechanism of synergistic effect of alantolactone and oxaliplatin, and suggest that such a combinational treatment might potentially become a more effective regimen for the treatment of colon cancer.
Materials and methods
Reagents
Oxaliplatin and alantolactone were obtained from Aladdin (Shanghai, China). N-Acetyl-L-Cysteine (NAC) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against primary antibodies including p-p38 (sc-9211), p38 (sc-9212), p-JNK (sc-4668), JNK (sc-9252) and HRP-conjugated secondary antibodies (7074) were obtained from Cell Signaling Technology (Danvers, MA). Ki-67 (ab15580) was obtained from Abcam (Cambridge, England). Fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit I and Propidium Iodide (PI) were purchased from BD Pharmingen (Franklin Lakes, NJ). Reactive oxygen species probe 2′, 7′- dichlorodihydrofluorescin diacetate (DCFH-DA) was purchased from Thermo Fisher (Carlsbad, CA, USA). 3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyl-2 tetrazolium bromide (MTT) was obtained from Beyotime Biotech (Nantong, China).
Colon cancer cell lines
Human colon cancer cells, HCT116 and RKO were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). All cell types were cultured in McCoy's 5A medium (Gibco, Eggenstein, Germany) or Minimun Essential medium Alpha (Gibco, Eggenstein, Germany) supplemented with 10% heat-inactivated FBS (Gibco, Eggenstein, Germany), 100 units/mL of penicillin and 100 mg/mL streptomycin in a 5% CO2 atmosphere at 37℃.
Cell viability assay
To measure the cell proliferation activity of alantolactone and oxaliplatin against HCT116 and RKO cancer cells, 8 × 103 cells were plated per well onto 96-well plates. Cells were treated with the chemical agents as indicated in the figure legends. DMSO was use as vehicle. 3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyl-2 tetrazolium bromide (MTT) solution was added to each well and the cells were incubated at 37 °C with 5% CO2 for 4 h. Absorbance at 490 nm was then measured with a microplate reader. Combination index (CI) was calculated using CalcuSyn 2.0 software. CI is a quantitative determination of the degree of drug interaction and calculated over a range of FA levels from growth inhibition percentages. The CI > 1.0 indicates antagonism, CI = 1.0 indicates additive effect and CI < 1.0 could be considered as synergism.
Measurement of reactive oxygen species generation in cultured cells
Cellular ROS contents were measured by flow cytometry. Briefly, 5 × 105 cells were plated on 60-mm dishes, allowed to attach overnight, and then treated with oxaliplatin (60 or 80 μM) and alantolactone (10 μM) for 2 h. NAC pretreatment, where indicated, was carried out for 2 h. Cells were stained with 10 μM DCFH-DA at 37°C for 30 min in the dark. DCF fluorescence was analyzed using flow cytometry.
Cell apoptosis analysis
Apoptosis was determined using an apoptosis detection Kit (BD Biosciences, USA). HCT116 and RKO cells treated with oxaliplatin (60 or 80 μM) and alantolactone (10 μM) for 24 h. The treated cells (as described above) were simultaneously incubated with fluorescein-labeled Annexin V and PI. Annexin V-binding buffer was then added to the mixture before fluorescence was measured on a FACSCalibur (BD Biosciences, MD, USA).
Western blotting analysis
Lysates from cells and tumour tissues were prepared and protein concentrations were determined using the Bradford assay (Bio-Rad, CA, USA). Proteins were separated by 10% SDS-PAGE and transferred to PVDF transfer membranes. The blots were blocked at room temperature with freshly prepared 5% nonfat milk in TBST for 2 h. Then incubated with specific primary antibodies overnight at 4°C. HRP-conjugated secondary antibodies and ECL substrate (Bio-Rad, CA, USA) were used for detection.
Xenograft experiment
All animals were handled according to the Institutional Animal Care and Use Committee (IACUC) guidelines, Wenzhou Medical University. Five-week-old female athymic BALB/c mice (18-22 g; total n=20) were used for the experiment. The animals were kept in cages at a constant room temperature with a 12/12-hr light/dark cycle and fed a standard rodent diet. HCT116 cells (5 × 106) in 0.1 mL PBS was inject subcutaneously into the right flank of each mouse. The mice were divided into four experimental groups on randomization and blinding with five mice in each group. The mice were treated by intraperitoneal (i. p.) injection of alantolactone, oxaliplatin, or a combination of alantolactone and oxaliplatin once per day. The tumor volumes were determined by measuring length (l) and width (w) and calculating volume (V = 0.5 × l × w2) at the indicated time points. At the end of the experiment, mice were sacrifice and their tumors were isolated by surgery in a room separated from the other animals, then weighed for in vitro experiments.
Immunohistochemistry
Harvested tumor tissues were fixed in 10% formalin at room temperature. Samples were dehydrated using ethanol gradient, and processed and embedded in paraffin. Parraffin embedded tissues were section at 5 μm thickness. Tumor tissue sections were stain for Ki-67 using primary antibodies at 1:200 dilution. HRP-conjugated secondary antibodies and diaminobenzidine (DAB) was used for detection.
Statistical analysis
All experiments were assayed in triplicate. Statistical analysis was perform using GraphPad Prism 5.0 software. Student's t-test and two-way ANOVA test were used to analyze the differences between data sets. A p value <0.05 was considered statistically significant.
Results
Combination of oxaliplatin and ALT inhibited the proliferation of colon cancer cells in a cooperative approach
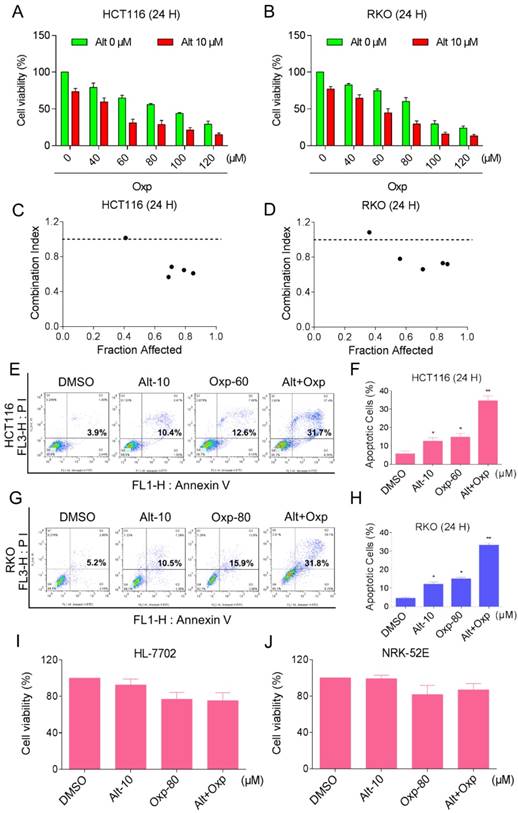
To determine whether the combination therapy with oxaliplatin and ALT is superior to monotherapy with either agent in HCT116 and RKO cells. An MTT assay was used to verify the inhibition of oxaliplatin or ALT on cell proliferation. As shown in Figures 1A and 1B, treatment with different concentrations of oxaliplatin increased from 40 to 120 µM, cell growth inhibition increased in a dose-dependent manner in HCT116 and RKO cells. Interestingly, combination of oxaliplatin and ALT significantly increased growth-inhibitory effect on HCT116 or RKO cells compared with the agent alone. To further comfirm the cytotoxic effect of the combination of oxaliplatin with ALT was synergism or antagonism. We evaluated combination index (CI) values using CalcuSyn 2.0 software. As shown in Figure 1C and 1D, analyses of the CI suggested that most of the data points were revealed a synergistic effect when FA values were 50% or greater. Next, the percentage of apoptotic cells was testified using the Annexin V-FITC/PI double staining method. The results showed that the combined treatment increased the proportion of apoptotic cells dramatically as compared with control cells or cells treated with the agent alone (Figure 1E-1H). By contrast, ALT did not increase the cytotoxicity of oxaliplatin in normal HL-7702 and NRK-52E cells, and the combined treatment has little effect on normal HL-7702 and NRK-52E cells (Figure 1I-1J). These results suggested that oxaliplatin and ALT possess a synergistic effect against colon cancer cells.
Combination of oxaliplatin and ALT inhibited the proliferation of colon cancer cells in a cooperative approach. (A-B) HCT116 or RKO cell lines were incubated with increasing doses of oxaliplatin (40-120 μM) and ALT (10 μM). The cytotoxic effect of oxaliplatin and alantolactone was measured by MTT assay. (C-D) The CI values were precisely calculated by the compusyn 2.0 software in a series of FA levels from growth inhibition percentages. A CI < 1.0 was regarded as synergy. (E-H) The induction of apoptosis as assessed by Annexin V/PI staining following combined treatment with oxaliplatin and ALT, and the percentage of apoptotic cells in the treatment groups was calculated. (I-J) HL-7702 or NRK-52E cells were treated with oxaliplatin (80 μM) or ALT (10 μM) alone or their combination (80 μM oxaliplatin and 10 μM ALT) at the indicated doses. At 24 h after treatment, the cell viability was determined by MTT assay. Data represent similar results from three independent experiments (* p< 0.05, ** p < 0.01).
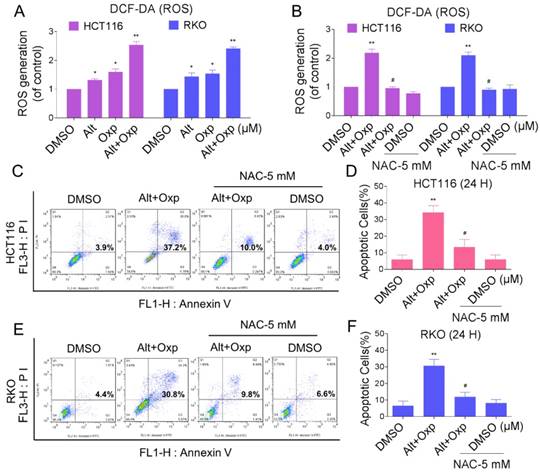
Elevation in ROS levels was a pivotal event in apoptosis induced by the combination of oxaliplatin and ALT
Reactive oxygen species (ROS) are the products of the natural metabolism of oxygen in cells, and they serve important roles in cell signaling and homeostasis. Nevertheless, cancer cells are more sensitive to increases in intracellular ROS levels than normal cells [10,11]. Elevated ROS levels have been confirmed as mechanisms behind the special events of numerous chemotherapeutic agents [12]. To demonstrate whether ROS was involved in the synergistic effect, we detected the intracellular ROS levels after oxaliplatin and ALT co-treatment. As shown in Figure 2A, treatment of cells with oxaliplatin or ALT alone both induced ROS generation, but the combined treatment resulted in significant increases in ROS levels. Next, we investigated whether superabundant generation of ROS was involved in the combined treatment-induced apoptosis. NAC is frequently used as a precursor of glutathione (GSH) and interact directly with ROS as a radical scavenger. As shown in Figure 2B, pretreatment of both cells with NAC markedly reversed the combined treatment-induced ROS accumulation. Moreover, the combined treatment-induced apoptosis was also blocked by NAC (Figure 2C-2F). These data suggested that ROS generation plays a vital role in the synergistic effect of oxaliplatin and ALT.
Elevation in ROS levels was a pivotal event in apoptosis induced by the combination of oxaliplatin and ALT. (A) HCT116 cells were treated with oxaliplatin (60 μM) or ALT (10 μM) alone or their combination (60 μM and 10 μM ALT) at the indicated doses. RKO cells were treated with oxaliplatin (80 μM) or ALT (10 μM) alone or their combination (80 μM oxaliplatin and 10 μM ALT) at the indicated doses. At 2 h after treatment, intracellular ROS levels were measured by flow cytometry. (B) HCT116 or RKO cells were pretreated with 5 mM NAC for 2 h before the combined treatment. At 2 h after treatment, intracellular ROS levels were measured by flow cytometry. (C-F) HCT116 or RKO cells were pretreated with 5 mM NAC for 2 h before exposure to oxaliplatin and ALT combination for 24 h. Percentage of cell apoptosis was determined by flow cytometry. The percentage of apoptotic cells in the treatment groups was calculated. Data represent similar results from three independent experiments (* p< 0.05, ** p < 0.01).
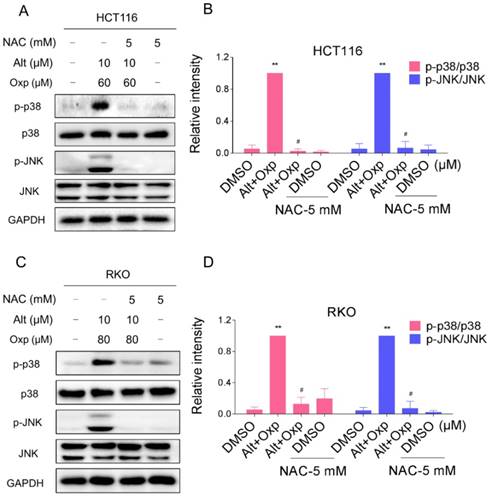
JNK and p38 MAPK signaling pathways activation is involved in the combined treatment-induced apoptosis of colon cancer cells
Excess ROS generation is known to activate JNK and p38 MAPK pathways. To investigate whether the JNK and p38 MAPK pathways is refer to the combined treatment-induced apoptosis, we assessed the expression of key proteins in related pathway in HCT116 and RKO cells. As shown in Figure 3A-3B, the combined treatment significantly activated JNK and p38 MAPK pathways in both cell lines. Furthermore, compared with oxaliplatin or ALT treatment alone, the combined treatment resulted in more significant increases in the phosphorylation levels of p38 and JNK in both HCT116 and RKO cells (Figure 3C-3D).
Oxaliplatin and ALT combination activated JNK and p38 MAPK signaling pathways. (A-B) HCT116 or RKO cells were treated with oxaliplatin and ALT combination for the indicated times, the protein levels of p-p38, p38, p-JNK and JNK were determined by western blot. (C-D) HCT116 or RKO cells were treated with oxaliplatin or ALT alone or their combination at the indicated doses. The protein levels of p-p38, p38, p-JNK and JNK were determined by western blot. Data represent similar results from three independent experiments (* p< 0.05, ** p < 0.01).

In order to identify the role of the JNK and p38 MAPK pathways in the combined treatment-induced cell apoptosis, the HCT116 and RKO cells were co-treated with oxaliplatin and ALT after pretreatment with JNK inhibitor SP600125 or p38 inhibitor BMS-582949. We found that pretreatment with SP600125 or BMS-582949 markedly reversed the combined treatment-induced phosphorylation of JNK or p38 in HCT116 and RKO cells (Figure 4A-4D). Moreover, the results in Figure 4E-4F showed that SP600125 or BMS-582949 can partially attenuate combined treatment-induced cell growth inhibition and apoptosis, suggesting that the activation of JNK and p38 signaling pathways is essential for the lethality of the combined treatment.
JNK and p38 MAPK signaling pathways activation is involved in the combined treatment-induced apoptosis of colon cancer cells. (A-B) HCT116 or RKO cells were pretreated with SP600125 for 2 h before exposure to oxaliplatin and ALT combination. The protein levels of p-JNK and JNK were determined by western blot. (C-D) HCT116 or RKO cells were pretreated with BMS-582949 for 2 h before exposure to oxaliplatin and ALT combination. The protein levels of p-p38 and p38 were determined by western blot. (E) HCT116 or RKO cells were pretreated with SP600125 or BMS-582949 for 2 h before exposure to oxaliplatin and ALT combination for 24 h, the cell viability was determined by MTT assay. (F) HCT116 or RKO cells were pretreated with SP600125 or BMS-582949 for 2 h before exposure to oxaliplatin and ALT combination for 24 h, percentage of cell apoptosis was determined by flow cytometry. The percentage of apoptotic cells in the treatment groups was calculated. Data represent similar results from three independent experiments (* p< 0.05, ** p < 0.01).

The combined treatment-induced activation of the JNK and p38 MAPK pathways is mediated by ROS generation in human colon cancer cells
The results mentioned above showed that ALT induced apoptosis in colon cancer cells through the generation of ROS. We next tested the relationship between ROS accumulation and activation of JNK and p38 signaling pathways. We observed that pretreatment with NAC markedly reversed the combined treatment-induced phosphorylation of JNK and p38 in both HCT116 and RKO cells (Figure 5A-5D). Taken together, these results suggested that the activation of p38 and JNK signaling pathways is due to accumulation of intracellular ROS in colon cancer cells.
Activation of the JNK and p38 MAPK pathways mediated by ROS accumulation. (A-B) HCT116 cells were pretreated with NAC (5 mM) for 2 h before exposure to oxaliplatin and ALT combination. The protein levels of p-p38, p38, p-JNK and JNK were determined by western blot. (C-D) RKO cells were pretreated with NAC (5 mM) for 2 h before exposure to oxaliplatin and ALT combination. The protein levels of p-p38, p38, p-JNK and JNK were determined by western blot. Data represent similar results from three independent experiments (* p< 0.05, ** p < 0.01).
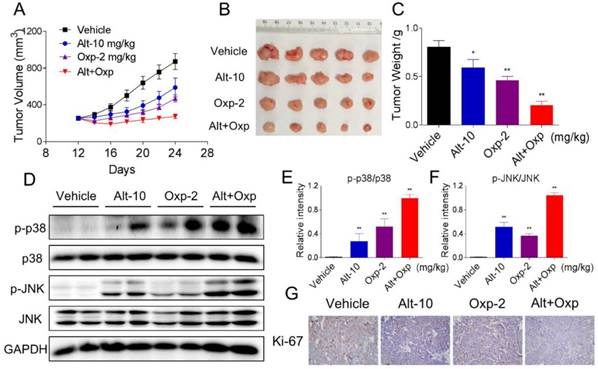
Oxaliplatin and ALT combination showed synergistic antitumor activity in human colon cancer HCT116 xenograft
Our last objective was to evaluate the in vivo effect of the combined treatment; we used a subcutaneous xenograft model of HCT116 cells in immunodeficient mice. After 13 days treatment, we found that 2 mg/kg oxaliplatin and 10 mg/kg ALT showed effective inhibition on the growth of HCT116 xenograft (Figure 6A-6C). However, the combined treatment exhibited stronger inhibitory effects on tumor volume and weight (Figure 6A-6C). Mechanistically, we found that the combined treatment resulted in significant increases in the phosphorylation levels of JNK and p38 (Figure 6D-6F). Moreover, the immunohistochemical staining assay was used to determine the expression of Ki-67. The expression level of Ki-67 was significantly decreased by the combined treatment (Figure 6G). Taken together, these results indicated that ALT could synergize the effect of oxaliplatin to inhibit tumor growth in vivo.
Combination of oxaliplatin and ALT showed synergistic antitumor activity on HCT116 tumor xenograft in nude mice. (A-B) Oxaliplatin and ALT combined treatment significantly decreased tumor volume and tumor weight (C) of HCT116 human colon cancer xenograft in nude mice. (D-F) The protein levels of p-p38, p38, p-JNK and JNK were determined by western blot. (G) The level of Ki-67 in tumor tissues was analyzed by immunohistochemical analysis.
Schematic illustration of the main findings of the present study.
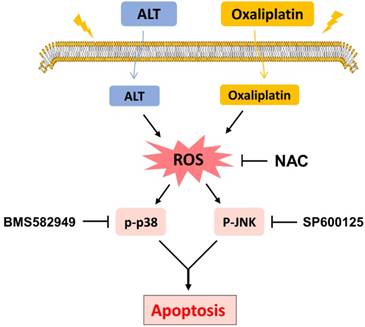
Discussion
During the past decades, the development of new chemotherapeutic drugs made a significant progress in cancer treatment. However, the application of conventional chemotherapeutic drugs is limited by toxicities and drug resistance [3,13]. Recently, the agents that are able to sensitize cancer cells to conventional antitumor drugs have received widespread attention. Natural products from diets and medicinal plants, in general, high efficiency and low toxicity, making them an ideal selection of “chemotherapy sensitizers” for cancer therapy [14,15]. In this study, we evaluated whether treatment of human colon cancer cells with ALT could act with oxaliplatin in a synergistic approach, aimed to provide preclinical evidence for agents to combine with oxaliplatin. Our results showed that the combination of oxaliplatin and ALT remarkably suppressed cell proliferation and induced the apoptosis of colon cancer cells. Moreover, oxaliplatin combined with ALT exhibited a synergistic inhibitory effect on tumor growth in vivo.
Under normal physiological conditions, ROS production was tightly regulated, and ROS participate in more complex signaling networks. However, cellular damage resulting from excessive ROS production or reduced endogenous antioxidants [16]. Compared with normal cells, cancer cells usually generate and maintain higher levels of ROS. Elevated ROS levels render cancer cells more sensitive to agents that increase ROS generation [17,18]. Therefore, stimulation of ROS is a potential therapeutic strategy for cancer treatment. Numerous natural products exert antitumor effects on human cancer cells by inducing ROS generation [19,20]. Our study also revealed that ALT induced ROS production in colon cells, and the combined further enhanced ROS generation. Moreover, altitude of intracellular ROS levels seemed to be the primary mechanism of apoptosis induction via combination treatment because pretreatment with the ROS scavenging agent NAC almost completely reversed these changes.
The pathogenesis of cancer includes the transformation of signal transduction pathways. As well, the state change (excitation or inhibition) of abnormal signaling pathway molecules may be targets for cancer treatments. MAPK is a classical signal transduction pathway that accommodates a large amount of physiological processes and holds the post of crucial role in the induction of cellular damage [21]. The family of mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase, c-Jun N-terminal kinase and p38 MAPK pathways [22]. Activation of the MAPK signaling pathway has been shown to induce the phosphorylation of p38, activates transcription factors and accelerates apoptosis [23]. The p38 MAP kinases are a family of serine/threonine protein kinases that play important roles in cellular responses to external stress signals such as UV irradiation [24,25]. In present study, combined treatment of ALT and oxaliplatin resulted in further increasing in the expression of phosphorylated JNK and p38, suggesting that the combined treatment executed significant antitumor effects in a JNK/p38-dependent manner. In addition, pretreatment with BMS582949 or SP600125 partially reversed combination treatment-induced apoptosis, which confirmed the important role of the JNK and p38 MAPK pathway. Based on our results, in a xenotransplant mouse model and model cell, combined therapy remarkably activated in the p38 pathway than alone. What's more, pretreatment with NAC significantly blocked JNK and p38 MAPK activations and showed that ROS is an upstream regulator of JNK and p38 MAPK pathways. Our study revealed that ALT could potentiate oxaliplatin in vitro and in vivo.
In conclusion, the current study is the first to demonstrate that ALT synergized the antitumor effect of oxaliplatin by inducing ROS generation. These findings provide new insights into the molecular mechanisms by which ALT synergized with oxaliplatin, and suggest that such a combinatorial treatment might potentially become a more effective regimen for colon cancer.
Abbreviations
ROS: reactive oxygen species; DCFH-DA: 2',7'-dichlorodihydrofluorescin diacetate; JNK: c-Jun N terminal kinase; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NAC: N-Acetyl-L-Cysteine; PI: propidium iodide.
Acknowledgements
The research was supported by National Natural Science Foundation of China (81603153 and 81503107), Zhejiang Provincial Natural Science Foundation of China (LY16H310011).
Ethics approval and consent to participate
The experimental protocol was established according to the Guide for the Care and Use of Laboratory Animals, and was approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Stein A, Atanackovic D, Bokemeyer C. Current standards and new trends in the primary treatment of colorectal cancer. Eur. J. Cancer. 2011;47(Suppl 3):S312-4
2. Dallas N, Xia L, Fan F, Gray M, Gaur P, van Buren G, Samuel S, Kim M, Lim S, Ellis L. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69(5):1951-7
3. Noh SH, Park SR, Yang HK. et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(12):1389-96
4. Ramanathan RK, Clark JW, Kemeny NE, Lenz HJ, Gococo KO, Haller DG, Mitchell EP, Kardinal CG. Safety and toxicity analysis of oxaliplatin combined with fluorouracil or as a single agent in patients with previously treated advanced colorectal cancer. J. Clin. Oncol. 2003;21(15):2904-11
5. Thomas H, Coley H. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10(2):159-65
6. Demols A, Peeters M, Polus M, Marechal R, Gay F, Monsaert E, Hendlisz A, Van Laethem J. Gemcitabine and oxaliplatin (GEMOX) in gemcitabine refractory advanced pancreatic adenocarcinoma: a phase II study. Br. J. Cancer. 2006;94(4):481-5
7. Petrioli R, Francini E, Cherri S, Torre P, Fiaschi A, Miano S, Marrelli D, Rovello F, Francini G. Capecitabine Plus Oxaliplatin and Bevacizumab, Followed by Maintenance Treatment With Capecitabine and Bevacizumab for Patients Aged > 75 Years With Metastatic Colorectal Cancer. Clin Colorectal Cancer. 2018
8. Chun J, Choi RJ, Khan S, Lee DS, Kim YC, Nam YJ, Lee DU, Kim YS. Alantolactone suppresses inducible nitric oxide synthase and cyclooxygenase-2 expression by down-regulating NF-kappaB, MAPK and AP-1 via the MyD88 signaling pathway in LPS-activated RAW 264.7 cells. Int Immunopharmacol. 2012;14(4):375-83
9. Chun J, Li RJ, Cheng MS, Kim YS. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015;357(1):393-403
10. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48(6):749-62
11. Nogueira V, Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013;19(16):4309-14
12. Teppo H, Soini Y, Karihtala P. Reactive Oxygen Species-Mediated Mechanisms of Action of Targeted Cancer Therapy. Oxid Med Cell Longev. 2017;2017:1485283
13. Gamelin E, Gamelin L, Bossi L, Quasthoff S. Clinical aspects and molecular basis of oxaliplatin neurotoxicity: current management and development of preventive measures. Semin Oncol. 2002;29(5 Suppl 15):21-33
14. Fulda S, Debatin K. Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res. 2004;64(1):337-46
15. Banerjee S, Wang Z, Kong D, Sarkar F. 3,3'-Diindolylmethane enhances chemosensitivity of multiple chemotherapeutic agents in pancreatic cancer. Cancer Res. 2009;69(13):5592-600
16. Martin K, Barrett J. Reactive oxygen species as double-edged swords in cellular processes: low-dose cell signaling versus high-dose toxicity. Hum Exp Toxicol. 2002;21(2):71-5
17. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579-91
18. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931-47
19. Zou P, Xia Y, Ji J. et al. Piperlongumine as a direct TrxR1 inhibitor with suppressive activity against gastric cancer. Cancer Lett. 2016;375(1):114-126
20. Duan D, Zhang B, Yao J, Liu Y, Fang J. Shikonin targets cytosolic thioredoxin reductase to induce ROS-mediated apoptosis in human promyelocytic leukemia HL-60 cells. Free Radic Biol Med. 2014;70:182-93
21. Chun J, Kim Y. Platycodin D inhibits migration, invasion, and growth of MDA-MB-231 human breast cancer cells via suppression of EGFR-mediated Akt and MAPK pathways. Chem. Biol. Interact. 2013;205(3):212-21
22. Chun J, Choi R, Khan S, Lee D, Kim Y, Nam Y, Lee D, Kim Y. Alantolactone suppresses inducible nitric oxide synthase and cyclooxygenase-2 expression by down-regulating NF-κB, MAPK and AP-1 via the MyD88 signaling pathway in LPS-activated RAW 264.7 cells. Int. Immunopharmacol. 2012;14(4):375-83
23. Ghatan S, Larner S, Kinoshita Y, Hetman M, Patel L, Xia Z, Youle R, Morrison R. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J. Cell Biol. 2000;150(2):335-47
24. Raingeaud J, Gupta S, Rogers J, Dickens M, Han J, Ulevitch R, Davis R. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995;270(13):7420-6
25. Freshney N, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, Saklatvala J. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell. 1994;78(6):1039-49
Author contact
![]() Corresponding author: Peng Zou, Ph.D. Address: Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325035, China; Tel: +86-577-86699892; Fax: +86-577-86689982 E-mail: zoupengedu.cn Ri Cui, Ph.D. Address: Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325035, China; Tel: +86-577-86699892; Fax: +86-577-86689982 E-mail: wzmucuiricom
Corresponding author: Peng Zou, Ph.D. Address: Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325035, China; Tel: +86-577-86699892; Fax: +86-577-86689982 E-mail: zoupengedu.cn Ri Cui, Ph.D. Address: Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325035, China; Tel: +86-577-86699892; Fax: +86-577-86689982 E-mail: wzmucuiricom