Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(8):1733-1742. doi:10.7150/ijbs.35138 This issue Cite
Research Paper
Inhibition of super enhancer downregulates the expression of KLF5 in basal-like breast cancers
Chuan-Huizi Chen1,2, Nong Yang3, Yongchang Zhang3, Jiancheng Ding4, Wenjuan Zhang4, Rong Liu1, Wen Liu4 ![]() , Ceshi Chen1,5,6
, Ceshi Chen1,5,6 ![]()
1. Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences & Yunnan Province, Kunming Institute of Zoology, Kunming, Yunnan, 650223, China
2. School of Chinese Materia Medica, Yunnan University of Chinese Medicine, Kunming, Yunnan, 650500, China
3. Department of Medical Oncology, Lung Cancer and Gastrointestinal Unit, Hunan Cancer Hospital/The Affiliated Cancer Hospital of Xiangya School of Medicine, Changsha, Hunan, 410013, China
4. School of Pharmaceutical Science, Fujian Provincial Key Laboratory of Innovative Drug Target Research, Xiamen University, Xiang'an South Road, Xiamen, Fujian, 361102, China
5. Institute for Stem Cell and Regeneration, Chinese Academy of Sciences, Beichen West Road, Chaoyang District, Beijing, 100101, China
6. Affiliated Cancer Hospital & Institute of Guangzhou Medical University, Guangzhou, China, 510095.
Received 2019-3-22; Accepted 2019-4-27; Published 2019-6-10
Abstract

The transcription factor KLF5 (Krüpple-like factor 5) is highly expressed in basal-like breast cancer (BLBC), which promotes cell proliferation, survival, migration and stemness, serving as a potential therapeutic target. In the current study, a super-enhancer (SE) was identified to be located downstream of the KLF5 gene in BLBC cell lines, HCC1806 and HCC1937. JQ-1, a BRD4 inhibitor, inhibits the expression and activity of KLF5 in both HCC1806 and HCC1937 cells in a time- and dose-dependent manner. Compound 870, an in-house BRD4 inhibitor, exhibited higher potency than JQ-1 to inhibit KLF5 and BLBC growth by arresting cells in G1 phase. Additionally, THZ1, a CDK7 inhibitor, also inhibits KLF5 and BLBC growth in a similar manner. Our findings suggested that KLF5 is regulated by SE, and modulation of SE could be an effective therapeutic strategy for treating BLBC.
Keywords: KLF5, Super-enhancer, BRD4, JQ-1, THZ1, BLBC
Introduction
Breast cancer is the most prevalent type of cancer among women worldwide [1]. According to molecular profiling, breast cancer can be divided into five subtypes, including luminal A, luminal B, HER-2 (Human Epidermal Growth Factor Receptor 2)-positive, basal-like breast tumors (BLBC) and normal-like [2]. BLBC is characterized by the lack of expression of the estrogen receptor (ER)α, progesterone receptor (PR) and HER2 and the positive expression of basal markers (cytokeratin 5/6, 14 and 17) [3]. As one of the main features of BLBC, basal cytokeratin expression is associated with poor clinical outcome, according to the study of a large pool of random patients with breast cancer [4, 5].
The transcription factor KLF5 (Krüpple-like factor 5) is highly expressed in BLBC and has been reported to promote breast cancer cell proliferation, survival, migration and stemness [6-9]. Our previous studies have shown that progesterone induces KLF5 transcription through PR in breast cancer cell lines [10], dexamethasone upregulates KLF5 transcription through the glucocorticoid receptor [11], and mithramycin A downregulates KLF5 expression by blocking the recruitment of Sp1 to the KLF5 gene promoter in BLBC [12]. A high expression level of KLF5 is a prognostic factor for poor clinical outcome in breast cancer [13]. Knockdown of KLF5 significantly attenuated BLBC tumor growth [14, 15]. However, the mechanism by which KLF5 is specifically highly expressed in BLBC is unclear.
Previous analysis of The Cancer Genome Atlas (TCGA) data showed that the amplification of SE activated the expression of KLF5 in seven types of tumors, including head and neck squamous cell carcinomas, esophageal carcinomas, cervical squamous cell carcinomas, lung squamous cell carcinomas, bladder carcinomas, stomach adenocarcinomas and colorectal adenocarcinomas [16, 17]. SE is composed of clusters of transcriptional enhancers, which are strongly enriched for the binding of MED1, BRD4, H3K27ac and other transcriptional coactivators [18, 19]. The first reported oncogenic SE was discovered in multiple myeloma in 2013[20]. Subsequently, oncogenic SEs were identified in various types of cancers, including small-cell lung cancer, breast cancer, neuroblastoma, head and neck squamous cell carcinoma, melanoma, gastric cancer, and esophageal carcinoma[21]. In general, SEs are located in proximity of master regulators and oncogenes, such as OCT4 and SOX2 in embryonic stem cells and c-MYC in cancer cells [20, 22]. Disrupting SE complexes is a new strategy for cancer treatment. For example, the expression of c-MYC can be selectively inhibited by JQ-1 (IC50 = 77/33 nM for BRD4 1/2 bromodomain) [23] by reducing BRD4 binding to the SEs that drives c-MYC expression [20, 24, 25]. In addition to BRD4, cyclin-dependent kinases (CDKs) are components of SE complexes [26]. A CDK7 covalent inhibitor THZ1 (IC50 = 3.2 nM) [27] was shown to inhibit SEs in triple-negative breast cancer [28] and esophageal squamous cell carcinoma [29].
The bromodomain (BRD) recognizes the acetyl-lysine residues of proteins [30]. There are eight families of BRD-containing proteins. The bromodomain and extra terminal domain (BET) family includes BRD2, BRD3, BRD4 and BRDT. BRD4 contains two BRDs, which endow BRD4 function in diverse biological processes. BRD4 can bind acetylated histones through the first BRD[31], and the binding can be blocked by JQ-1 by forming hydrogen bonds with asparagine (Asn) 140 in the acetyl-lysine binding cavity of BRD4 [23]. Since the anticancer effects of JQ-1 have been reported, a large number of novel molecules derived from JQ-1 have been developed. More than a dozen BET inhibitors are currently in clinical trials [32]. The Phase I clinical trial of OTX015, an analog of JQ-1, in triple negative breast cancer was completed in 2017.
In this study, we identified an SE located downstream of the KLF5 gene in BLBC. Both BRD4 and CDK7 inhibitors can inhibit KLF5 transcription in BLBC. Furthermore, a new BRD4 inhibitor, compound 870, was shown to inhibit KLF5 much more efficiently than JQ-1. Our discoveries thus provide new potential agents for BLBC treatment.
Materials and Methods
Cell lines, plasmids and inhibitors
All cell lines were purchased from the American Type Culture Collection. HCC1806 and HCC1937 were cultured in RPMI-1640 medium (Gibco, Grand Island, USA) supplemented with 10 % FBS (Gibco). All cell lines were cultured at 37 ℃ in a humidified atmosphere containing 5 % CO2. Plasmids lenti-KRAB-dCas9-blast and lentiGuide-Puro were gifts from professor Matthew Meyerson at Harvard University. JQ-1 (HY-13030) and THZ1 (HY-80013A) were purchased from MedChem Express (MCE, Monmouth Junction, USA).
Chromatin immunoprecipitation (ChIP) sequencing
ChIP-seq assays were performed as previously described [33]. Briefly, HCC1806 and HCC1937 cells were cross-linked with 1 % formaldehyde (for H3K27ac), or cross-linked with disuccinimidyl glutarate, followed by double cross-linked with 1 % formaldehyde (for BRD4). 125 mM glycine was used to quench the cross-linking. Chromatin DNA was sonicated using a Bioruptor Pico (Diagenode, Belgium), and immunoprecipitated with H3K27ac (Abcam, ab4729) or BRD4 (Bethyl, A301-985A50) antibodies. Purified DNA was subjected to high throughput sequencing (RiboBio, Guangzhou, China).
CRISPR/Cas9-mediated enhancer repression
First, HCC1806 cells were infected with lenti-KRAB-dCas9-blast and screened by 6 μg/mL blasticidin to obtain KRAB-expressing HCC1806 cells (HCC1806-KRAB). The sgRNAs were cloned into lentiGuide-Puro. Then, HCC1806-KRAB cells were infected with lentiGuide-sgRNAs and screened using 2 μg/mL puromycin. For sg-Mix, three sgRNA plasmids were equally used to package lentivirus, which was then used for cell infection. The sequences of sgRNA were as follows: sgCON: ATCGTTTCCGCTTAACGGCG, sgRNA1: TCGCACGAGAGTTCCAAAAC, sgRNA2: AGAAGCTTGTAGATACCCTG, sgRNA3: CTGCCCTCACATAAACACGT.
Quantitative PCR (qPCR)
The total mRNA was isolated using the reagent of TRIzol® (15596-026, Invitrogen) following the manufacturer's protocol. Complementary DNA synthesis was performed using the iScriptTM cDNA synthesis kit (1708891, Bio-Rad, USA). Quantitative PCR was carried out using the SYBR Green Select Master Mix system (4472908, Applied Biosystems, USA) with the ABI-7900HT system (Applied Biosystems). The sequences of the primers used for qPCR were as follows: GAPDH-F: 5'-GGTGAAGGTCGGAGTCAACG-3', GAPDH-R: 5'-TGGGTGGAATCATATTGGAACA-3'. KLF5-F: 5'-ACACCAGACCGCAGCTCCA-3', KLF5-R: 5'TCCATTGCTGCTGTCTGATTTGTAG-3'.
Western Blotting (WB)
Cell pellets were collected at the indicated time points, and their proteins were extracted in lysis buffer with a protease inhibitor cocktail (P8340, Sigma). The concentrations of the total protein were measured using DC™ Protein Assays (Bio-Rad). Equal amounts of protein extracts were analyzed by SDS-PAGE electrophoresis and blotted onto PVDF membranes. Membranes were blocked with 5 % nonfat dairy milk in PBS with 0.2 % Tween-20 and incubated with the following primary antibodies: KLF5, c-MYC (abcam, 32072), FGFBP (R&D, 1593), p21 (Cell Signaling Technology, 2947S), and GAPDH (Proteintech, 10494-1-AP). Following wash with PBS-T, the blots were incubated with HRP-conjugated secondary antibody (Jackson Immuno Research, USA) and developed using the ECL reagent (Thermo Fisher Scientific, CA, USA) on an ImageQuant LAS4000 Biomolecular Imager (GE Healthcare).
Cell proliferation inhibition assays
HCC1806 and HCC1937 cells were exposed to gradient concentrations of JQ-1, compound 870, or THZ1 for 72 h. The cell viability was assessed using sulforhodamine B (SRB) [9] with a microplate reader (Epoch, Bio-Tek) at a wavelength of 560 nm.
Cell cycle analysis
Cells were seeded in 6-well plates, cultured overnight and treated with JQ-1 or 870 for 24 h. Following treatment, the cells were fixed with 70 % ethanol, treated with 40 μg/ml RNase A, stained with 10 μg/ml propidium iodide and analyzed by flow cytometry (BD LSR Fortessa) for DNA synthesis and cell cycle status. The percentages of cells in the G1, S and G2/M cell cycle phases were analyzed using the FlowJo software.
DNA synthesis-based cell proliferation assay
DNA synthesis was detected with Click-iT® EdU Imaging Kits (Invitrogen) following the manufacturer's protocol. The cells were treated with different concentrations of JQ-1 or 870 and then labeled with EdU, which was followed by fixation and DNA staining. The cell images were collected using fluorescence microscopy. For each sample, five random fields were counted. The percentage of EdU-positive cells (%) was calculated as the EdU-positive cell number/the total cell number.
Stable overexpression of KLF5
The KLF5 cDNA was amplified and cloned into the pCDH-puro-3×Flag-3×HA lentiviral vector using the forward primer 5'-ATTCGGATCCATGGCTACAAGGGTGCTGAG-3' and the reverse primer 5'-ATGGCTCGAGTCAGTTCTGGTGCCTCTTCA-3'. Lentiviruses were packaged according to published protocols [34]. The cell lines were infected with lentiviruses, followed by selection with 1 μg/ml puromycin.
Statistical analysis
All data in this study were analyzed using the GraphPad Prism software (version 6.0) and SPSS software (version 20.0). The differences between two samples were analyzed using the unpaired two-tailed Student's t test. P values less than 0.05 were considered statistically significant.
Results
Super-enhancer drives KLF5 transcription in BLBC
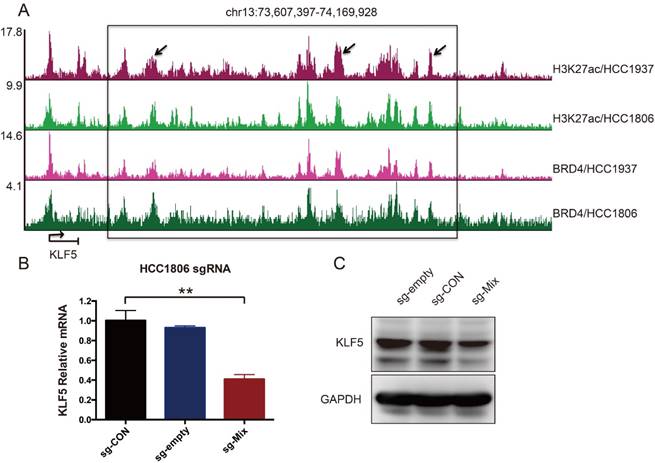
It has been reported that amplified SEs activate KLF5 expression and three individual enhancers within the SEs played dominant roles [17]. Although the KLF5 SE is not amplified in breast cancer, whether it controls KLF5 transcription in BLBC is unknown. To test this, H3K27ac and BRD4 ChIP-seq was performed in two BLBC cell lines, as both are markers of SEs [17, 20]. A cluster of peaks was identified downstream of the KLF5 gene, indicating the existence of SEs (Figure 1A). We used the CRISPR-mediated repression system to inhibit these enhancers. This system can recruit KRAB-dCas9 (the Krüpple-associated box (KRAB) transcriptional repressor domain fused to inactivated Cas9 (dCas9)) using a short guide RNA (sgRNA). We designed three sgRNAs to target three individual enhancers. Compared with sg-CON and sg-empty, sg-Mix (combine three sgRNAs) repressed 59 % of KLF5 mRNA expression (Figure 1B). Additionally, sg-Mix reduced the KLF5 protein level to a similar extent (Figure 1C). These results indicate that KLF5 transcription may be regulated by SEs in BLBC.
A super-enhancer located downstream of the KLF5 gene positively regulates KLF5 gene transcription in BLBC cell lines. A. The genome browser view of H3K27ac- and BRD4-binding profile around the KLF5 gene in HCC1937 and HCC1806 cell lines. B. The KLF5 mRNA level was downregulated by the KRAB-dCas9-mediated repression of the enhancers in the HCC1806 cell line guided by sgRNAs. The sg-Mix stands for the mixture of three sgRNAs target the enhancers as the arrow is directed in panel A. The error bars represent the SD of experimental triplicates. **, P<0.01, t-test. C. The KLF5 protein level was downregulated by the KRAB-dCas9-mediated repression of the enhancers in the HCC1806 cell line guided by sgRNAs. GAPDH was used as a loading control.
The BRD4 inhibitor JQ-1 and Compound 870 inhibit KLF5 transcription in BLBC
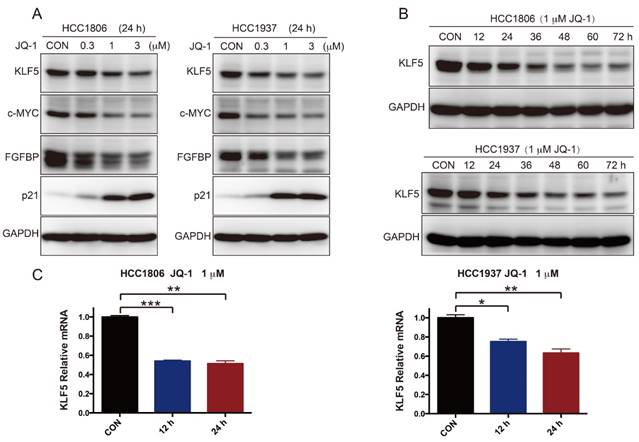
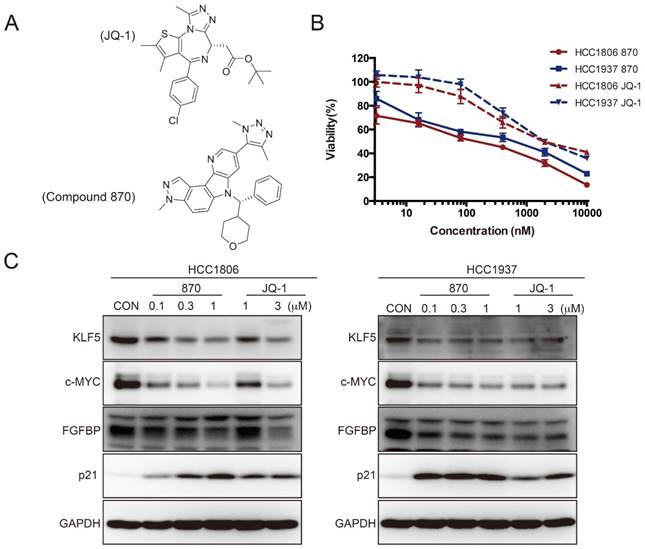
To further test whether SEs regulate KLF5 transcription in BLBC, we used the BRD4 inhibitor JQ-1 to treat BLBC cell lines (HCC1806 and HCC1937) and examined the KLF5 and c-MYC expression. As expected, JQ-1 reduced the c-MYC and KLF5 protein levels at 1-3 μM, and the inhibition was dose-dependent (Figure 2A). FGFBP is a positive target gene of KLF5 [8], while p21 is a negative target of KLF5 [35]. In agreement with the KLF5 expression change, JQ-1 inhibited FGFBP protein expression and induced p21 protein accumulation. Moreover, we traced the KLF5 protein expression changes after the cells were treated with 1 μM JQ-1. Immunoblotting analysis showed that JQ-1 reduced the KLF5 protein levels in a time-dependent manner in both the HCC1806 and HCC1937 cells (Figure 2B). Accordingly, the levels of KLF5 mRNA decreased after the cells were treated with 1 μM JQ-1 for 12 h or 24 h (Figure 2C). Due to its short half-life, JQ-1 failed to enter clinical trials [36]. However, many BET inhibitors have been reported [21, 37]. Compound 870 is a novel and highly potent BRD4 inhibitor developed in house recently. Its IC50 value is about 1 nM determined by a FRET (Fluorescence resonance energy transfer) assay [38]. Compound 870 was designed based on the structure of effective reported BET inhibitor (Figure 3A). We tested the growth inhibition rate of compound 870 in two BLBC cell lines. Compared with JQ-1, compound 870 showed more potent cytotoxicity. Importantly, compound 870 reduced the levels of KLF5 and c-MYC with approximately 10-fold higher efficiency compared with JQ-1 (Figure 3C). Taken together, these data suggested that JQ-1 and Compound 870, presumably through targeting BRD4 on SE of KLF5, could inhibit the expression of KLF5.
JQ-1 inhibits KLF5 expression in a dose- and time-dependent manner. A. HCC1806 and HCC1937 cells were treated with JQ-1 for 24 h. The protein level was detected by immunoblotting. c-MYC was used as a positive control. FGFBP and p21 are KLF5 target genes. B. The KLF5 protein levels were gradually decreased after HCC1806 and HCC1937 cells were exposed to JQ-1 for indicated time. GAPDH was used as a loading control. C. JQ-1 decreased the KLF5 mRNA levels in HCC1806 and HCC1937 cells. The mRNA levels of KLF5 were examined by qPCR. The error bars represent the SD of experimental triplicates. *, P<0.05, **, P<0.01, ***, P<0.001, t-test.
Compound 870 is more effective than JQ-1 in terms of its ability to inhibit KLF5 expression and BLBC growth. A. The structures of compound 870 and JQ-1. B. Compound 870 and JQ-1 decreased the cell viability of BLBC cells, as measured by the SRB assay. HCC1806 and HCC1937 cells were treated with the indicated compound 870 and JQ-1 for 72 h. The error bars represent the SD of experimental triplicates. C. Compound 870 and JQ-1 regulate KLF5 and its target gene expression in BLBC cells. HCC1806 and HCC1937 cells were exposed to the indicated compound 870 and JQ-1 for 24 h. The protein levels were detected by immunoblotting. c-MYC was used as a positive control.
JQ1 and Compound 870 induce G1 arrest in BLBC
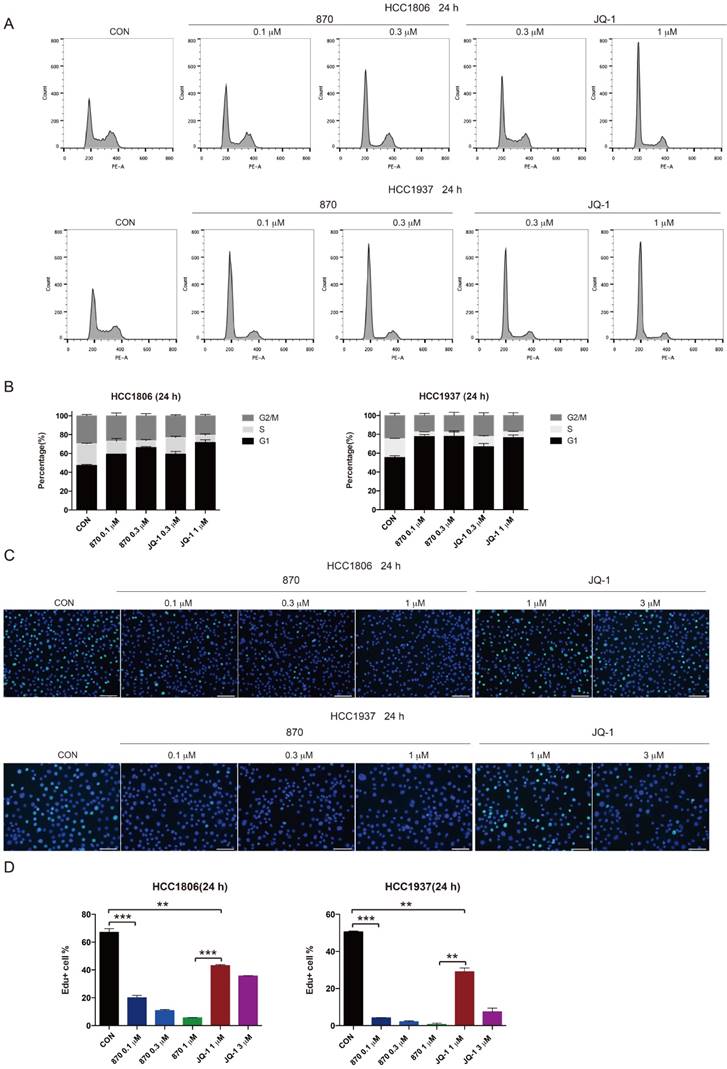
KLF5 is known to promote G1/S transition of cell cycle [39]. To test whether JQ-1 and compound 870 inhibit BLBC cell growth through regulating cell cycle progression, we treated BLBC cells with JQ-1 and found that the number of cells in the G1 phase increased and the number of cells in the S phase decreased. Consistently, compound 870 induced G1 arrest with more potent activity. Compared with JQ-1, compound 870 at similar concentration (0.3 μM) induced more cells in the G1 phase (Figure 4A-B). Furthermore, compound 870 inhibited DNA synthesis more potently than JQ-1, as measured by the Edu incorporation assay (Figure 4C-D).
Compound 870 shows more potent activity than JQ-1 in terms of its ability to induce G1 cell cycle arrest and inhibit DNA synthesis. A.HCC1806 and HCC1937 cells were treated with the indicated JQ-1 and compound 870 for 24 h. The number of cells in each phase of the cell cycle was analyzed by flow cytometry. B. Quantification of panel A. The error bars represent the SD of experimental triplicates. C. JQ-1 and compound 870 inhibited DNA synthesis as evaluated by an Edu incorporation assay. HCC1806 and HCC1937 cells were treated with JQ-1 or compound 870 for 24 h. The value of bar is 100 μm. D. Quantification of panel C. The error bars represent the SD of experimental triplicates. **, P<0.01, ***, P<0.001, t-test.
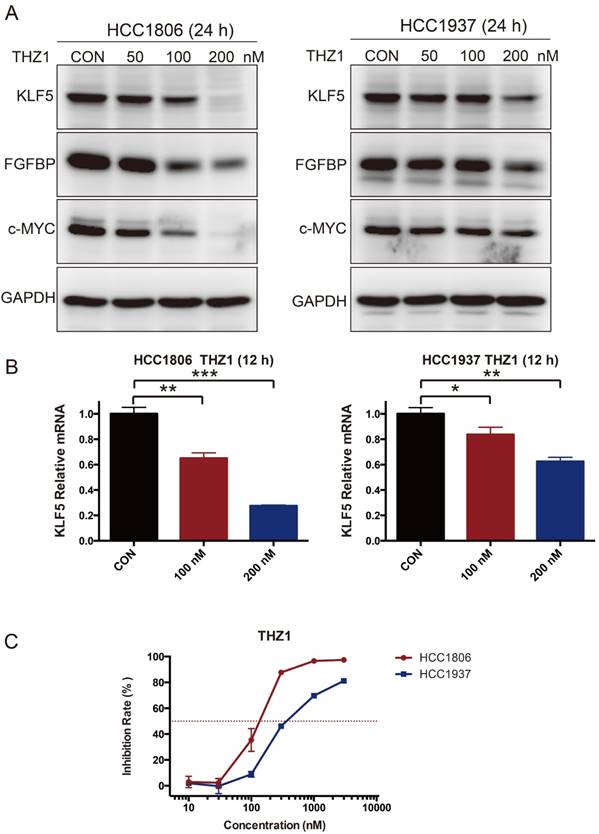
The CDK7 inhibitor THZ1 suppresses KLF5 transcription
As introduced earlier, THZ1 can inhibit SEs [28, 29]. We therefore tested whether THZ1 can also reduce the expression of KLF5. As expected, THZ1 inhibited the expression of KLF5, FGFBP and c-MYC (Fig 5A-B). THZ1 also strongly inhibited BLBC cell growth, as measured by the SRB assay (Fig 5C).
THZ1 inhibits KLF5 expression and BLBC growth. A. HCC1806 and HCC1937 cells were treated with THZ1 for 24 h. The KLF5, FGFBP and c-MYC protein levels were detected by immunoblotting. THZ1 is a CDK7 inhibitor. B. THZ1 inhibited the KLF5 mRNA levels in HCC1806 and HCC1937 cells. The mRNA level of KLF5 was examined by qPCR after the cells were treated with 100 nM and 200 nM THZ1 for 12 h. The error bars represent the SD of experimental triplicates. *, P<0.05, **, P<0.01, ***, P<0.001, t-test. C. THZ1 inhibited HCC1806 and HCC1937 cell growth, as measured by the SRB assay. The cells were treated with THZ1 for 72 h.
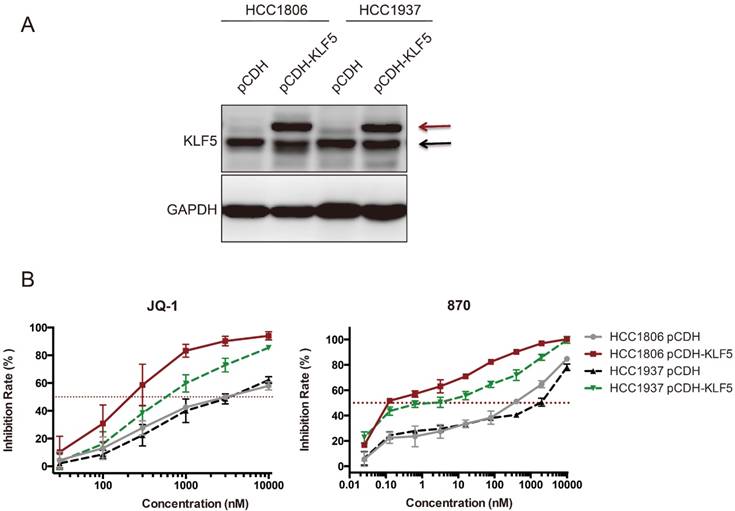
Ectopic KLF5 expression sensitizes HCC1806 and HCC1937 cells to JQ-1 and compound 870
It is well known that SEs regulates hundreds of target genes. Both KLF5 and c-MYC were downregulated by BRD4 and CDK7 inhibitors in BLBC. We wondered whether KLF5 overexpression could rescue the growth inhibition induced by BRD4 inhibitors. To our surprise, ectopic KLF5 expression in HCC1806 and HCC1937 cell lines did not abrogate the effects of JQ-1 or compound 870, but rather sensitized the BLBC cells to these BRD4 inhibitors (Fig 6). This phenomenon was not detected in other cell lines we tested, including MDA-MB-231, MDA-MB-468, Hs578T, and MCF10A (Data not shown).
Ectopic KLF5 expression sensitizes HCC1806 and HCC1937 cells to JQ-1 and compound 870. A. KLF5 was overexpressed in HCC1806 and HCC1937. The breast cell lines were infected with pCDH-3×Flag-KLF5 or pCDH vector control. The expression of KLF5 was detected by immunoblotting. The upper bands indicate the exogenous 3×Flag-KLF5. The lower bands indicate the endogenous KLF5 protein expression. B. KLF5 overexpression sensitized HCC1806 and HCC1937 cells to JQ-1 and compound 870. pCDH-3×Flag-KLF5 or pCDH vector control lentivirus-infected cells were plated in 96-well plates and treated with JQ-1 for 72 h. The cell inhibition rate was analyzed using the SRB assay. The error bars represent the SD of experimental triplicates.
Discussion
Recently, we reported that mithramycin A [12] and mifepristone [9] could inhibit KLF5 expression in triple-negative breast cancer cells. Gao Y et al. reported that curcumin [40] also promotes KLF5 degradation in bladder cancer cells. Metformin [41] was reported to increase the degradation of KLF5 in triple-negative breast cancer cells. However, the pharmacological efficacy of these compounds is weak. Here, we showed that KLF5 is regulated by SEs located downstream of the KLF5 gene based on BRD4- and H3K27ac-ChIP-seq analysis. The combinatorial effects of three sgRNAs targeting this SE inhibited the transcription of KLF5. Although BRD4 is a chromatin regulator expressed broadly in various cells and contributes widely to gene expression, the BRD4 inhibitor JQ-1 predominately inhibits SE-associated genes, such as c-MYC [20]. As expected, JQ-1 strongly inhibited KLF5 expression, both at the transcriptional and protein levels, in a more potent fashion than mithramycin A and mifepristone.
Since the first BRD inhibitor NP1 was reported in 2005 [42], the development of BRD inhibitors has thrived over the past decade. As the anti-cancer effects of the BRD4 inhibitor JQ-1 disrupting the SEs, the development of small-molecule inhibitors against SE complexes is a promising strategy to treat cancers. Inhibitors targeted the BET bromodomain are the most extensively studied small-molecules against the component of SEs. More than a dozen of BET inhibitors are in phase I/II clinical trials, including iBET762, OTX015 and CPI0610 [21]. However, no BRD inhibitor has been approved by the FDA yet [43]. There is an urgent need for more potent BET inhibitors. In this study, we investigated a new BRD4 inhibitor compound 870, which showed stronger activity than JQ-1 in terms of KLF5 and c-MYC inhibition. In addition, compound 870 was more potent than JQ-1 in G1 phase arrest, DNA synthesis, and cell growth inhibition.
Although KLF5 is dramatically downregulated by BRD4 inhibitors in BLBC, ectopic KLF5 overexpression did not abrogate the effects of JQ-1 but rather sensitized the BLBC cells to JQ-1 or compound 870 (Fig 6). This phenomenon is cell line specific because similar results were not detected in MDA-MB-231, MDA-MB-468, Hs578T, and MCF10A. A possible explanation for this is that the overexpressed KLF5 may interrupt the SE complexes through binding to components of SE, which makes the cells more sensitive to JQ-1. A previous work showed that ectopic MYC-N expression could not rescue the effects of the BRD4 inhibitor OTX015 in neuroblastoma [44]. The detailed mechanism requires further investigation in the future.
We previously demonstrated that knockdown of KLF5 inhibited HCC1806 and HCC1937 xenograft growth in immune-deficient mice [14, 15]. The in vivo anti-tumor effects of JQ-1 were evaluated thoroughly. In the HCC1806 xenograft model, JQ-1 inhibits tumor growth, has an anti-angiogenic effect, and decreases the expression of the hypoxia-regulated genes CA9 (carbonic anhydrase) and VEGF-A (vascular endothelial growth factor A) [45]. In addition, JQ-1 inhibits tumor growth of MDA-MB-231 and IDC50X xenografts [46]. For these reasons, we did not further assess the anti-tumor growth effects of JQ-1 in this study. Additionally, OTX015 suppresses tumor mass in MDA-MB-231 murine xenografts, and OTX015 decreases NANOG and OCT4 expression at the mRNA level in the MDA-MB-231, MDA-MB-468 and HCC1937 cell lines [47].
In summary, we showed that SEs regulate KLF5 in BLBC. Furthermore, the SE inhibitors JQ-1 and THZ1 inhibit KLF5 expression and BLBC growth. Finally, we demonstrated that a new BRD4 inhibitor compound 870 was more potent than JQ-1 in terms of its ability to inhibit KLF5 and BLBC.
Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16010405 to Chen, C), a Chinese Postdoctoral Science Foundation Grant (2015M582581 to Chen, C.H), the National Natural Science Foundation of China (81830087, U1602221, and 31771516 to Chen, C., 81772847 to Liu, R.), and the Yunnan Applied Basic Research Key Projects (2015FA027 to Liu, R.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: a cancer journal for clinicians. 2018;68:7-30
2. Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:2568-81
3. Shi J, Cao J, Zhou BP. Twist-BRD4 complex: potential drug target for basal-like breast cancer. Current pharmaceutical design. 2015;21:1256-61
4. Potemski P, Kusinska R, Watala C, Pluciennik E, Bednarek AK, Kordek R. Prognostic relevance of basal cytokeratin expression in operable breast cancer. Oncology. 2005;69:478-85
5. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z. et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:5367-74
6. Dong JT, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cellular and molecular life sciences: CMLS. 2009;66:2691-706
7. Jia L, Zhou Z, Liang H, Wu J, Shi P, Li F. et al. KLF5 promotes breast cancer proliferation, migration and invasion in part by upregulating the transcription of TNFAIP2. Oncogene. 2016;35:2040-51
8. Zheng HQ, Zhou Z, Huang J, Chaudhury L, Dong JT, Chen C. Kruppel-like factor 5 promotes breast cell proliferation partially through upregulating the transcription of fibroblast growth factor binding protein 1. Oncogene. 2009;28:3702-13
9. Liu R, Shi P, Nie Z, Liang H, Zhou Z, Chen W. et al. Mifepristone Suppresses Basal Triple-Negative Breast Cancer Stem Cells by Down-regulating KLF5 Expression. Theranostics. 2016;6:533-44
10. Liu R, Zhou Z, Zhao D, Chen C. The induction of KLF5 transcription factor by progesterone contributes to progesterone-induced breast cancer cell proliferation and dedifferentiation. Molecular endocrinology. 2011;25:1137-44
11. Li Z, Dong J, Zou T, Du C, Li S, Chen C. et al. Dexamethasone induces docetaxel and cisplatin resistance partially through up-regulating Kruppel-like factor 5 in triple-negative breast cancer. Oncotarget. 2017;8:11555-65
12. Liu R, Zhi X, Zhou Z, Zhang H, Yang R, Zou T. et al. Mithramycin A suppresses basal triple-negative breast cancer cell survival partially via down-regulating Kruppel-like factor 5 transcription by Sp1. Scientific reports. 2018;8:1138
13. Tong D, Czerwenka K, Heinze G, Ryffel M, Schuster E, Witt A. et al. Expression of KLF5 is a prognostic factor for disease-free survival and overall survival in patients with breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:2442-8
14. Zhao D, Zhi X, Zhou Z, Chen C. TAZ antagonizes the WWP1-mediated KLF5 degradation and promotes breast cell proliferation and tumorigenesis. Carcinogenesis. 2012;33:59-67
15. Qin J, Zhou Z, Chen W, Wang C, Zhang H, Ge G. et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nature communications. 2015;6:8471
16. Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD. et al. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nature genetics. 2016;48:176-82
17. Zhang X, Choi PS, Francis JM, Gao GF, Campbell JD, Ramachandran A. et al. Somatic Superenhancer Duplications and Hotspot Mutations Lead to Oncogenic Activation of the KLF5 Transcription Factor. Cancer discovery. 2018;8:108-25
18. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA. et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934-47
19. Pott S, Lieb JD. What are super-enhancers? Nature genetics. 2015;47:8-12
20. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320-34
21. Shin HY. Targeting Super-Enhancers for Disease Treatment and Diagnosis. Molecules and cells. 2018;41:506-14
22. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307-19
23. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O. et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067-73
24. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904-17
25. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA. et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16669-74
26. Sengupta S, George RE. Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends in cancer. 2017;3:269-81
27. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB. et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616-20
28. Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S. et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015;163:174-86
29. Jiang YY, Lin DC, Mayakonda A, Hazawa M, Ding LW, Chien WW. et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut. 2017;66:1358-68
30. Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology and gene transcription. Current opinion in drug discovery & development. 2009;12:659-65
31. Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q. et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer cell. 2014;25:210-25
32. Liu Z, Wang P, Chen H, Wold EA, Tian B, Brasier AR. et al. Drug Discovery Targeting Bromodomain-Containing Protein 4. Journal of medicinal chemistry. 2017;60:4533-58
33. Gao WW, Xiao RQ, Zhang WJ, Hu YR, Peng BL, Li WJ. et al. JMJD6 Licenses ERalpha-Dependent Enhancer and Coding Gene Activation by Modulating the Recruitment of the CARM1/MED12 Co-activator Complex. Molecular cell. 2018;70:340-57 e8
34. Bu W, Xin L, Toneff M, Li L, Li Y. Lentivirus vectors for stably introducing genes into mammary epithelial cells in vivo. Journal of mammary gland biology and neoplasia. 2009;14:401-4
35. Xia H, Wang C, Chen W, Zhang H, Chaudhury L, Zhou Z. et al. Kruppel-like factor 5 transcription factor promotes microsomal prostaglandin E2 synthase 1 gene transcription in breast cancer. The Journal of biological chemistry. 2013;288:26731-40
36. Moyer MW. First drugs found to inhibit elusive cancer target. Nature medicine. 2011;17:1325
37. Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Annals of oncology: official journal of the European Society for Medical Oncology. 2017;28:1776-87
38. Nenghui Wang. Carboline derivative serving as bromodomain inhibitor. WO2017/124936 A1. 2017
39. Chen C, Benjamin MS, Sun X, Otto KB, Guo P, Dong XY. et al. KLF5 promotes cell proliferation and tumorigenesis through gene regulation and the TSU-Pr1 human bladder cancer cell line. International journal of cancer. 2006;118:1346-55
40. Gao Y, Shi Q, Xu S, Du C, Liang L, Wu K. et al. Curcumin promotes KLF5 proteasome degradation through downregulating YAP/TAZ in bladder cancer cells. International journal of molecular sciences. 2014;15:15173-87
41. Shi P, Liu W, Tala, Wang H, Li F, Zhang H. et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell discovery. 2017;3:17010
42. Zeng L, Li J, Muller M, Yan S, Mujtaba S, Pan C. et al. Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. Journal of the American Chemical Society. 2005;127:2376-7
43. Pervaiz M, Mishra P, Gunther S. Bromodomain Drug Discovery - the Past, the Present, and the Future. Chemical record. 2018
44. Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F. et al. Targeting MYCN-Driven Transcription By BET-Bromodomain Inhibition. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:2470-81
45. da Motta LL, Ledaki I, Purshouse K, Haider S, De Bastiani MA, Baban D. et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene. 2017;36:122-32
46. Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM. et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413-7
47. Vazquez R, Riveiro ME, Astorgues-Xerri L, Odore E, Rezai K, Erba E. et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget. 2017;8:7598-613
Author contact
![]() Corresponding authors: Wen Liu, E-mail: w2liuedu.cn; Ceshi Chen, Tel: 86-871-5181944, E-mail: chenckiz.ac.cn
Corresponding authors: Wen Liu, E-mail: w2liuedu.cn; Ceshi Chen, Tel: 86-871-5181944, E-mail: chenckiz.ac.cn