Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(9):1892-1904. doi:10.7150/ijbs.32429 This issue Cite
Research Paper
miR-671-5p Blocks The Progression Of Human Esophageal Squamous Cell Carcinoma By Suppressing FGFR2
Xiaoyan Li1,2,3*, Changjun Nie1,2,3*, Baoqing Tian1,2,3, Xuan Tan1,2,3, Wei Han1,2,3, Jiakang Wang4, Yuan Jin1,2,3, Yadan Li1,2,3, Xinyuan Guan5, An Hong1,2,3 ![]() , Xiaojia Chen1,2,3
, Xiaojia Chen1,2,3 ![]()
1. Institute of Biomedicine & Department of cell Biology, Jinan University, Guangzhou, Guangdong, 510632, P. R. China.
2. National Engineering Research Center of Genetic Medicine, Guangzhou, Guangdong, 510632, P. R. China.
3. Guangdong Provincial Key Laboratory of Bioengineering Medicine, Guangzhou, Guangdong, 510632, P. R. China.
4. Cancer Center of Guangzhou Medical University, Guangzhou, Guangdong, 510090, P. R. China.
5. Department of Clinical Oncology, University of Hong Kong, Hong Kong, China
*Both authors contributed to the work equally.
Received 2018-12-19; Accepted 2019-6-2; Published 2019-7-21
Abstract

Esophageal cancer is the eighth most common malignant tumor worldwide, of which esophageal squamous cell carcinoma (ESCC) is the dominant histological subtype. A drug shortage for ESCC therapy triggered us to explore the roles of fibroblast growth factor receptor 2 (FGFR2) and its upstream regulator miR-671-5p in ESCC progression. We compared the levels of FGFR2 and miR-671-5p between human ESCC tissues and their matched normal esophageal tissues and found an association between higher levels of FGFR2 and lower levels of miR-671-5p in ESCC tissues. High levels of FGFR2 resulted in the activation of the ERK and AKT pathways and a promotion of ESCC progression. High levels of miR-671-5p specifically reduced the expression of FGFR2 and suppressed ESCC progression in both in vitro and in vivo models. Therefore, suppressing FGFR2 and enhancing miR-671-5p expression may be the right approaches for ESCC therapy.
Keywords: FGFR2, miR-671-5p, ESCC, tumor progression
Introduction
Esophageal cancer is the eighth most frequent and the sixth most fatal cancer type worldwide, and is highly prevalent distributed in Asia and Africa [1]. According to its tissue origin, esophageal cancer is divided into esophageal adenocarcinoma and esophageal squamous cell carcinoma (ESCC), the dominant histological subtype [2]. The overall 5-year survival rate for patients with ESCC at advanced stages is only approximately 10-20% [3]. Therefore, understanding the mechanism of ESCC progression is very important for future therapy.
Fibroblast growth factor receptors (FGFRs) belong to a family of receptor tyrosine kinases, comprising of five members, FGFR1 to FGFR5, and are critical for not only normal development but also tumorigenesis [4-7]. FGFRs bind to their extracellular ligands fibroblast growth factors, which induces receptor dimerization in the cell membranes, leading to trans-auto-phosphorylation of the intracellular tyrosine residues. This phosphorylation results in the activation of the downstream signaling pathways, such as MAPK, PI3K-AKT and PLCγ, and plays key roles in cellular anti-apoptosis, angiogenesis, proliferation, and migration [8, 9]. Mutations or abnormal expression of FGFRs may result in constitutive dimerization of receptors and auto-activation of kinases, thereby inducing aberrant intracellular signaling [8]. FGFR2 is generally considered as an oncogene. For example, overexpression of FGFR2 promotes the proliferation and survival of gastric cancer cells through activating the downstream MAPK/ERK and PI3K/AKT signaling pathways [10], and FGFR2 enhances the phosphorylation activity of RSK2 and regulates the migration of human mammary epithelial cells [11]. Previous evidences have shown that FGFR2 is a potential therapeutic target, but the role of FGFR2 in ESCC progression has rarely been reported.
miRNA is a kind of small non-coding RNA, approximately 20-30 nucleotides in length. It inhibits translation or promotes the degradation of targeting mRNA by binding to its 3′-untranslated region (3′-UTR) to regulate gene expression [12-15]. It has been reported that several miRNAs activate or inhibit tumor progression [16]. For example, miR-3188 significantly inhibits the development of nasopharyngeal carcinoma by targeting mTOR to inactive the p-PI3K/p-AKT/p-mTOR pathway [17]; and the deficiency of miRNA-150 leads to inhibition of differentiation of intraepithelial lymphocytes [14]. miR-671-5p mentioned in this study, promotes the migration and proliferation of human glioblastoma multiforme by targeting CDR1-AS, CDR1, and VSNL1 [18], and suppresses the development of breast cancer via inhibiting epithelial-to-mesenchymal transition by targeting FOXM1 [19]. High levels of miR-671-5p and miR-193a-5p inhibit oncogene SMARCB1/INI1 expression in pediatric chordomas [20]. miR-206, miR-381, and miR-671-5p suppress the expression of SMARCB1 in epithelioid sarcoma [21]. However, the roles of miR-671-5p in ESCC progression are still unclear.
Here, we showed that suppressing the expression of FGFR2 led to significant decreases in ESCC xenograft size. Most of the human ESCC tissues displayed higher levels of FGFR2 and lower levels of miR-671-5p compared with their matched normal esophageal tissues. Furthermore, miR-671-5p targeting the FGFR2 3′-UTR was used to reduce the expression of FGFR2, which suppressed the proliferation, migration, invasion, and xenograft growth of ESCC cells.
Materials and Methods
Collection of normal esophageal and ESCC tissues and culture of human ESCC cell lines
Thirty-five pairs of ESCC tumor tissues and their matched normal esophageal tissues were collected for immunochemical analyses of FGFR2 protein as previously described [22], and six pairs of ESCC tumor tissues and their matched normal esophageal tissues were collected for analyses of miR-671-5p. All tissues were provided by the Cancer Center of Guangzhou Medical University, Guangzhou, China. None of the patients received prior chemo-radiotherapy. The relative intensities of FGFR2 in the tissue sections were unbiasedly estimated by researchers based on a scale from 0-10. A line of normal esophageal cells and eight lines of ESCC cells were maintained in RPMI medium (HyClone, USA) supplemented with 10% fetal bovine serum (FBS) at 37 °C in an incubator with a humidified atmosphere of 5% CO2. All procedures performed in studies involving human patients were in accordance with the ethical standards of the Institutional and/or National Research Committee and the Declaration of Helsinki and its later amendments, or comparable ethical standards. This study was in accordance with the ethics review regulations, and was reviewed and approved by the Human Subject Research Ethics Committee of Cancer Center of Guangzhou Medical University.
RNA extraction and quantification by quantitative real-time PCR
Total RNA was extracted from cells using RNAiso Plus, according to the manufacturer's protocol (Takara). The cDNA was synthesized using a reverse transcription kit (Takara). qPCR (Real-time Quantitative PCR Detecting System) was performed using CFX96 TouchTM Real Time PCR Detection System with SYBR Green Dye mix (Takara). The relative expression levels of mRNA or miRNAs were calculated by the comparative CT method and normalized to the levels of GAPDH or U6, respectively. The primer sequences are shown in Table 1.
Primers used in this study
| Primers | Sequences ( From 5' to 3' ) |
|---|---|
| miR-5585-RT | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAG ACCTGT |
| miR-5585-F | ACACTCCAGCTGGG CTGAATAGCTGGGACT |
| miR-4534-RT | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAG AGACCC |
| miR-4534-F | ACACTCCAGCTGGG GGATGGAGGAG |
| miR-5703-RT | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAG ACCTTC |
| miR-5703-F | ACACTCCAGCTGGG AGGAGAAGTCG |
| miR-6086-RT | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAG CTCTGC |
| miR-6086-F | ACACTCCAGCTGGG GGAGGTTGGGAAGG |
| miR-4485-RT | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAG TTAGGG |
| miR-4485-F | ACACTCCAGCTGGG TAACGGCCGCGGTA |
| miR-3141-RT | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAG TCCTCC |
| miR-3141-F | ACACTCCAGCTGGG GAGGGCGGGTGGA |
| miR-1973-RT | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAG TATGCT |
| miR-1973-F | ACACTCCAGCTGGG ACCGTGCAAAGGT |
| miR-3132-RT | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAG TCCTCT |
| miR-3132-F | ACACTCCAGCTGGG TGGGTAGAGAAGGAGCTC |
| miR-671-5p-RT | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAG CTCCAG |
| miR-671-5p-F | ACACTCCAGCTGGG AGGAAGCCCTGGAGGGG |
| U6-R | AACGCTTCACGAATTTGCGT |
| U6-F | CTCGCTTCGGCAGCACA |
| FGFR2-3'-UTR-R | TTGCGGCCGCGTCTTGTTAACATTAATATC |
| FGFR2-3'-UTR-F | CCGCTCGAGTCTTCAGGAGATGATTCTGT |
| FGFR2-M8-R | GCGTCTCCAACGCCAAAGAGTCTTTCGTATATTATCAAAAT |
| FGFR2-M8-F | CAGTGAATTTTGATAATATACGAAAGACTCTTTGGCGTTG |
| GAPDH-R | AAGTGGTCGTTGAGGGCAATG |
| GAPDH-F | CTGGGCTACACTGAGCACC |
| FGFR2-R | ACACTGCCGTTTATGTGTGGA |
| FGFR2-F | AGCCAACCTCTCGAACAGTAT |
Creation of stable cell lines using lentivirus system
We used ta lentivirus system to create stable cell lines with overexpression or knocked-down expression of FGFR2. The vector pCDH-FGFR2 was created by ligating the DNA fragment of the FGFR2 gene with the vector pCDH-CMV-MCS-EF1-GreenPuro (Cat. #CD513B, SBI, CA), digested by XhoI1 and BamH1, and co-transfected into 293T cells with the packaging vectors psPAX2 and pMD2G, using Lipofectamine 3000 reagent (Invitrogen, CA) according to the manufacturer's protocol. Stable cell lines were selected based on their resistance to puromycin. Following the same procedure, both shFGFR2-1 (shRNA-1, TRCN0000231053, BROAD) and shFGFR2-4 (shRNA-4, TCRN0000218493, BROAD) oligonucleotides were designed and cloned into the pLKO vector to create their respective stable cell lines.
Assays for cell proliferation and colony formation
ESCC cells (1.5×103 cells per well) were seeded in 96-well plates, and cultured for 1-7 days to achieve lentivirus-mediated overexpression or knock-down FGFR2, and for 1-4 days to test the impact of transient transfection with miR-671-5p mimics or miR-671-5p inhibitor, respectively. Subsequently, the rates of cells proliferation were measured using the cell Counting Kit-8 (CCK8) kit according to the manufacturer's protocol. For colonies formation assays, ESCC cells (1000 cells per well) were seeded in 6-well plates, and cultured for 7 days at 37 °C in an incubator with 5% CO2. The number of colonies formed in each well was counted after the cells in each well were fixed with 4% paraformaldehyde for 10 min, and then stained with crystal violet for 10 min.
Assays for cell migration and invasion
Assays for cell migration and invasion were performed using Transwell inserts (Costar, USA) and invasion using Matrigel-coated plates (BD Bioscience, USA), respectively. Approximately, 5×104 tumor cells in serum-free medium were placed in the upper chamber, and cells in medium with 20% FBS were placed in the lower chamber. Cells were incubated for 36h, fixed with 4% paraformaldehyde, and stained with crystal violet. The number of cells that attached on the underside of the filter was counted under a microscope.
Animal experiments
All experimental procedures were performed according to the institutional ethical guidelines approved by the Institutional Animal Care and Use Committee of Jinan University. Male 4-5 week-old BALB/c nude mice were subcutaneously injected with 3×106 KYSE180-CTL, KYSE180-shFGFR2-4, EC109-CTL, or EC109-FGFR2 cells to establish their respective ESCC xenografts. To test the impact of miR-671-5p, the same mice were subcutaneously injected with 3×106 KYSE180 cells to establish ESCC xenografts with a size of 1 cm in diameter, and then 1 nM miR-671-5p mimics and its negative control were injected into the intra-tumor spaces. Tumor growth in mice was monitored on daily base and tumor volume was calculated with the following equation: V=0.5xLxW2 [23].
Immunoblot analysis
Immunoblot analysis was performed as previously described [24, 25]. The following antibodies were used: anti- FGFR2, from Santa Cruz Biotechnology, anti-GAPDH from Sigma-Aldrich, and anti-AKT, anti-phosphor-AKT, anti-ERK, anti-phosphor-ERK, anti-cyclinD1, anti-cyclinB1, and anti-phosphor-FGFR from Cell Signaling Technology. Secondary antibodies were from Sigma-Aldrich. Densitometry (Bio-Rad Laboratories) was applied to analyze the intensities of the respective protein bands.
Prediction of miR-671-5p target genes and dual luciferase reporter assay
TargetScan, miRwalk, miRanda and miRDB softwares were used to predict the target genes of miR-671-5p and FGFR2-3′-UTR was identified as the target sequence. The 3′-UTR of human FGFR2 mRNA fragment was amplified by PCR using the listed primers (Table 1) and cloned into psi-CHECK-2 vector to create WT (Wild-type) vector. Site-Directed Mutagenesis of miR-671-5p binding site in FGFR2-3′-UTR (Mutation type, MT vector) was created using Gene Tailor Site-Directed Mutagenesis System (Invitrogen). For luciferase reporter assays, cells were transfected with WT or MT vector and treated with miR-671-5p mimics or its inhibitor for 48 hours. Luciferase activities were measured using the Dual-Luciferase Reporter kit (Promega, USA) and normalized to the activities of firefly luciferase.
miRNA transfection of Cells
miR-671-5p mimics (miR-671-5p) and its negative control (mimics Nc), and miR-671-5p inhibitor and its negative control (inhibitor Nc) were designed and synthesized by RiboBio (Guangzhou, China). miR-671-5p mimics and mimics Nc were transfected into KYSE180 cells and miR-671-5p inhibitor and inhibitor Nc were transfected into EC109 cells using Lipofectamine 3000 transfection reagent (Invitrogen) according to the manufacturer's protocol.
Statistical analysis
Statistical analysis was performed using SPSS 19.0 statistical software. Data were expressed as means ± standard deviation (SD). The statistical significances of differences between two independent groups was tested by Student's t-test and expressed as *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Results
Levels of FGFR2 are elevated in ESCC tissues and cell lines
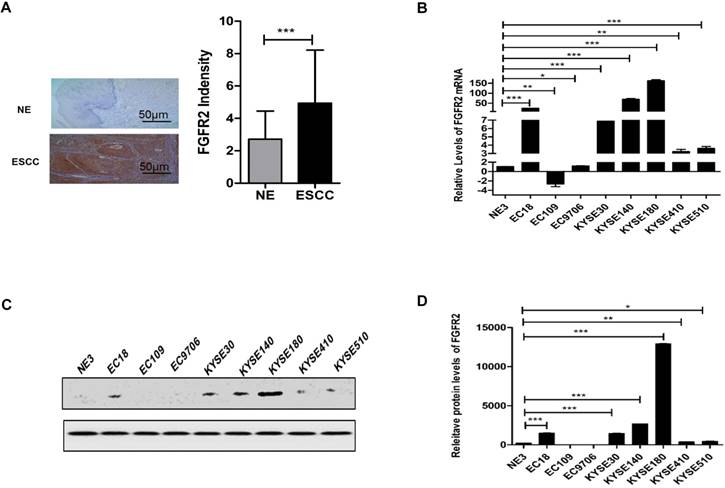
Previously, we reported that the expression levels of FGFRs between carcinoma and para-carcinoma cells in patients with digestive or reproductive system cancers are significantly different, and FGFR2 and FGFR4 are closely related to the susceptibility of digestive and reproductive system cancers [22]. We further evaluated the expression levels of FGFR2 in normal and ESCC tissues from 35 pairs of human patients by immunohistochemistry. We found that the levels of FGFR2 in ESCC tissues were significantly higher than those in normal tissues (Fig. 1A) although there was no significant difference between different groups with different clinical parameters (Table 2). We also examined the levels of FGFR2 mRNA in a panel of ESCC cell lines by qPCR assays, and found that majority of the ESCC cell lines, except EC109 had significantly higher levels of FGFR2 mRNA than the immortalized NE3 cell line derived from normal human esophageal tissues (Fig. 1B-D). Therefore, our results suggested that ESCC tissues and cell lines express higher levels of FGFR2.
Expression of FGFR2 in normal and ESCC tissues and cells. A. Representative images and a quantification analysis showing the levels of FGFR2 in 35 pairs of normal esophageal tissues (NE) and esophageal squamous cell carcinoma (ESCC) tissues as detected by immunohistochemical staining. Bar=50μm. B. A plot showing the relative levels of FGFR2 mRNA in normal esophageal cell line NE3 and several ESCC cell lines. Data shown are the means and standard deviations (Mean±S.D.) of at least three repeats. The significances of difference between ESCC cell lines and NE3 are analyzed using Student's T-test. Here and later, * indicates p < 0.05; ** p < 0.01; and *** p < 0.001.C,D Western blot showed the FGFR2 expression levels in NE3 and several ESCC Cell lines.
Relationship between FGFR2 expression and clinical pathological parameter of ESCC and NE.
| Characteristics | Total number | FGFR2 Expression level | P-Value | ||
|---|---|---|---|---|---|
| ESCC | NE | ESCC | NE | ||
| Age | 0.99 | 0.63 | |||
| <60 | 15 | 4.93±3.91 | 2.80±1.70 | ||
| >60 | 20 | 4.95±2.80 | 2.65±1.87 | ||
| Gender | 0.62 | 0.03* | |||
| Female | 6 | 4.33±2.50 | 1.51±1.51 | ||
| Male | 29 | 5.07±3.43 | 3.00±1.67 | ||
| Pathology grade | 0.98 | 0.33 | |||
| I+II | 14 | 4.92±3.38 | 3.07±1.85 | ||
| II-III+III | 21 | 4.95±3.27 | 2.48±1.66 | ||
| Lymph node metastasis | 0.66 | 0.59 | |||
| Negative | 13 | 4.62±3.31 | 2.92±1.97 | ||
| Positive | 22 | 5.14±3.31 | 2.59±1.62 | ||
FGFR2 promotes proliferation, colony formation, migration, invasion, and tumorigenesis of ESCC cells
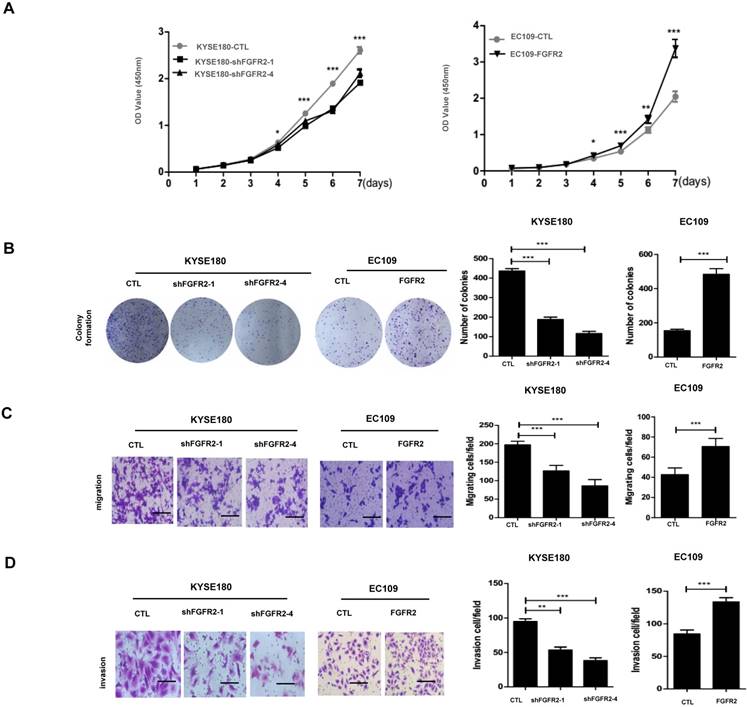
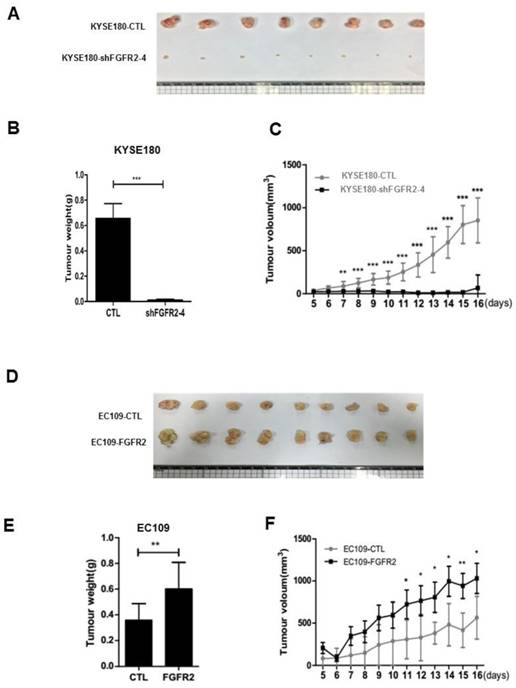
We selected KYSE180 cells with the highest levels of FGFR2 mRNA and EC109 cells with the lowest levels of FGFR2 to manipulate the expression levels of FGFR2. KYSE180 and EC109 cells were transfected with two types of shFGFR2 to silence and increase the expression of FGFR2, respectively. Two KYSE180 cell lines transfected with shFGFR2 displayed a reduction, while EC109 cells overexpressing FGFR2 displayed an increase in the rates of cell proliferation, colony formation, migration, and invasion compared with their respective controls (Fig. 2). We further investigated the effect of FGFR2 on ESCC progression in vivo using a xenograft tumor nude mouse model. Results showed that KYSE180 cells infected with shFGFR2 developed dramatically smaller tumors, while EC109 cells overexpressing FGFR2 developed significantly larger tumors as compared with their respective controls (Fig. 3). Therefore, our findings indicate that FGFR2 may act as an oncogene to promote ESCC progression.
FGFR2 promotes ESCC progression in vitro. A. Plots showing the relative densities of KYSE180 cells treated with random shRNA (CTL), shFGFR2-1 or shFGFR2-4, and EC109 transfected with control plasmid (CTL) or plasmid expressing FGFR2. B-D. Representative images and plots showing the relative levels of colony formation (B), migration (C) and invasion (D) in the same cell lines as described in panel A. Bar=100μm.
FGFR2 promotes the ESCC progression in vivo. A-F. Images (A,D) and plots of weights (B,E) and volumes (C,F) of tumors formed in BALB/c nude mice injected of KYSE180 cells treated with random shRNA (CTL), shFGFR2-1 or shFGFR2-4 (A-C), or EC109 transfected with control plasmid (CTL) or plasmid expressing FGFR2 (D-F).
FGFR2 activates the ERK and AKT signaling pathways and alters cell cycle
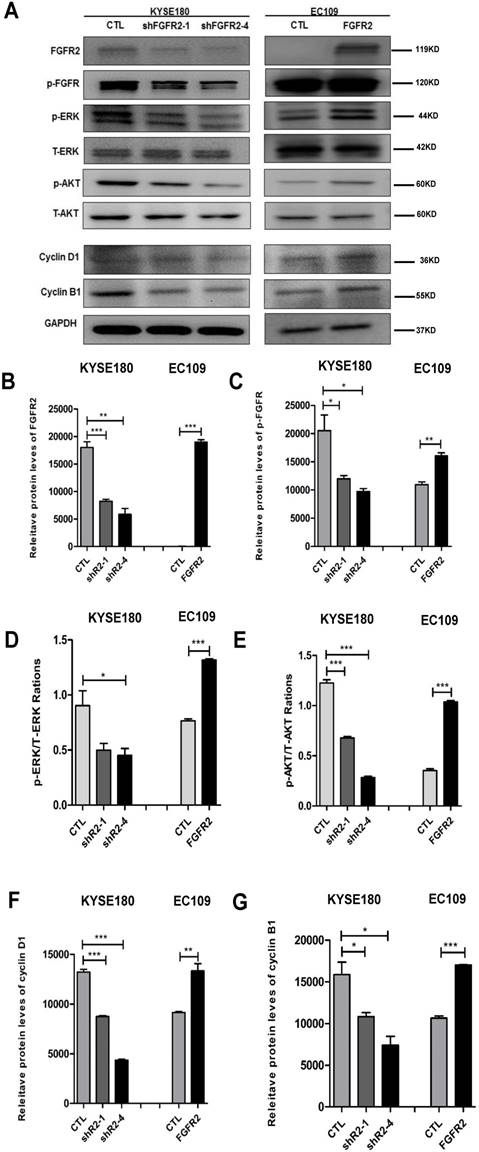
As expected, all cell lines expressed the expected levels of FGFR2 proteins (Fig. 4 A, B). The intensities of FGFR2 signals represented by the levels of phosphorylated FGFR were correspondingly altered (Fig. 4A, C). It has been reported that FGFR2 may impact tumorigenesis through ERK and AKT [26, 27]. ERK is a classical signaling molecule involved in the MAPK signaling pathway, which is well-known to enhance cell differentiation, proliferation and motility [28, 29], while AKT is a key molecule connecting many pathways to influence cell survivals [30]. Here, suppression of FGFR2 led to a decrease in the levels of the phosphorylated ERK (Fig. 4A, D) or AKT (Fig. 4A, E), while overexpression of FGFR2 led to an enhancement of these levels. In addition, cyclin D1 expression has been frequently reported to be elevated in a variety of tumors and may contribute to tumorigenesis [31]; while cyclin B1 is a regulatory protein involved in cell mitosis [32]. The levels of cyclin D1 and cyclin B1 were reduced in cells lines expressing lower levels of FGFR2 and increased in cells overexpressing FGFR2 (Fig. 4A,F,G). Therefore, FGFR2 may enhance cell proliferation through accelerating the cell cycle.
FGFR2 activates the MAPK-ERK and PI3K-AKT pathways. A-E. Representative immunoblots (A) and plots of relative levels of FGFR2 (B) and p-FGFR proteins (C), ratios of p-ERK to ERK (D), ratios of p-AKT to AKT (E), relative levels of cyclin D1 (F) and cyclin B1 proteins (G). GAPDH serves as loading control.
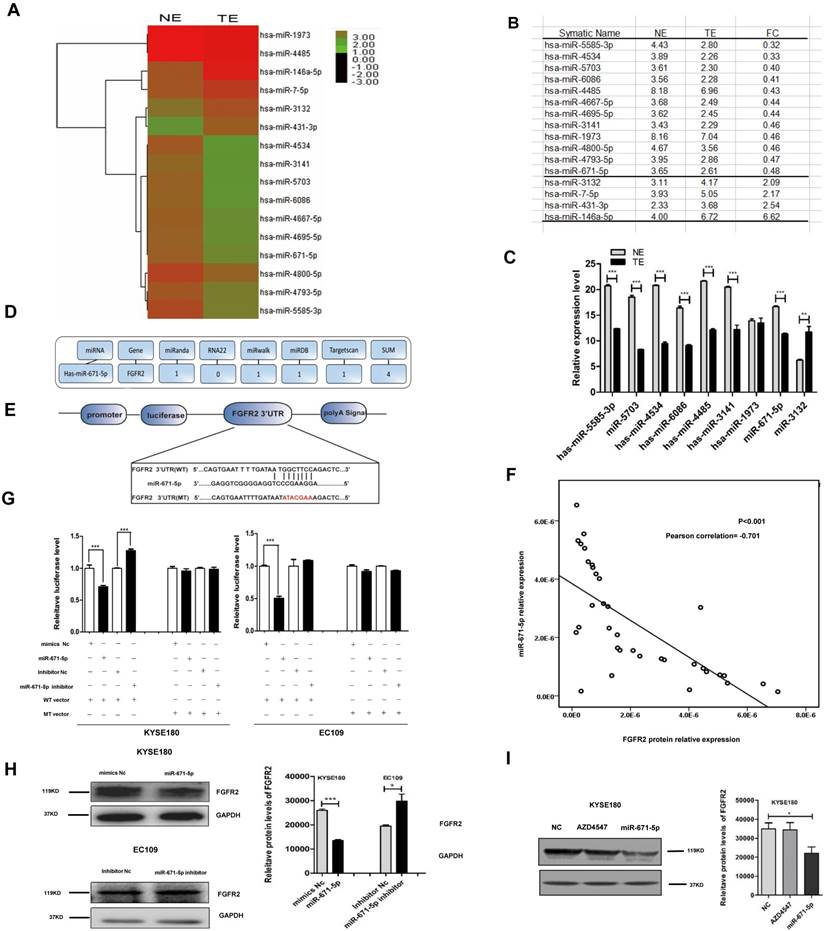
miR-671-5p suppresses the levels of FGFR2 protein in ESCC tissues
To investigate how FGFR2 expression is regulated in ESCC, we collected six pairs of FGFR2-positive ESCC tissues and FGFR2-negative normal tissues and performed, miRNA microarray analysis (Fig. 5A). We found that the levels of twelve miRNAs were reduced, while the levels of four miRNAs were increased in tumor tissues compared with those in normal tissues (Fig. 5A, B). The reduction in levels of 12 miRNAs was further confirmed by qPCR (Fig. 5C). Next, to predict the target genes of these miRNAs, multiple databases including miRnada, RNA22, miRwalk, miRDB, and TargetScan were used. We found that miR-671-5p was the only potential miRNA that specifically targets the 3′-UTR of FGFR2 (Fig. 5D, E). We further performed bivariate correlation analysis of 35 ESCC tissues as described above, and found a significantly negative correlation between the levels of FGFR2 protein and miR-671-5p (Fig. 5F). We treated KYSE180 and EC109 cells were treated with either the wild-type or mutant luciferase reporter of miR-671-5p (Fig. 5E) as well as with negative or specific mimics and inhibitors, as described earlier. We found that transfection with miR-671-5p mimics suppressed the levels of FGFR2, while transfection with miR-671-5p inhibitor enhanced its levels in KYSE180 cells treated with wild-type vector. Furthermore with miR-671-5p mimics suppressed the levels of FGFR2, but treatment with miR-671-5p inhibitor did not affect its levels in EC109 cells treated with wild-type vector (Fig. 5G). KYSE180 cells treated with miR-671-5p mimics displayed reduced the levels of FGFR2 protein, while EC109 cells treated with miR-671-5p inhibitor showed increased levels of this protein (Fig. 5H). At the same time, we used AZD4547 as a control to confirm that miR-671 was targeting the FGFR2-3'-UTR furtherly (Fig. 5I). Therefore, our result showed that miR-671-5p suppresses the expression of FGFR2.
miR-671-5p suppresses the expression of FGFR2 in ESCC. A,B. A plot (A) and a table (B) showing the relative levels of miRNAs in normal esophageal squamous tissue (NE) and ESCC tumor tissue (TE) as detected with miRNA microarrays. FC, fold of change. C. Plots of relative expression levels of miRNAs as verified by qPCR. U6 serves as internal control. D. A diagram showing the procedure to use different Bioinformatics websites to predict whether miR-671-5p targets to FGFR2-3'-UTR. E. A diagram showing the binding site and sequence of miR-671-5p targeting to FGFR2-3'UTR and the sequences inserted in wild-type (WT) and mutant (MT) reporter vectors. F. A bivariate correlation analysis of the level of FGFR2 and miR-671-5p. G. Plots showing the impact of miR-671-5p on the relative luciferase levels as detected by a dual fluorescence reporter system. KYSE180 and EC109 cells were transfected with WT or MT luciferase reporter vector. H. Representative immunoblots (left) and plots (right) showing levels of FGFR2 protein in KYSE180 and EC109 cells transfected with miR-671-5p. Nc, negative control. I. Representative immunoblots (left) and plots (right) showing levels of FGFR2 protein in KYSE180 and EC109 cells transfected with AZD4547 or miR-671-5p.
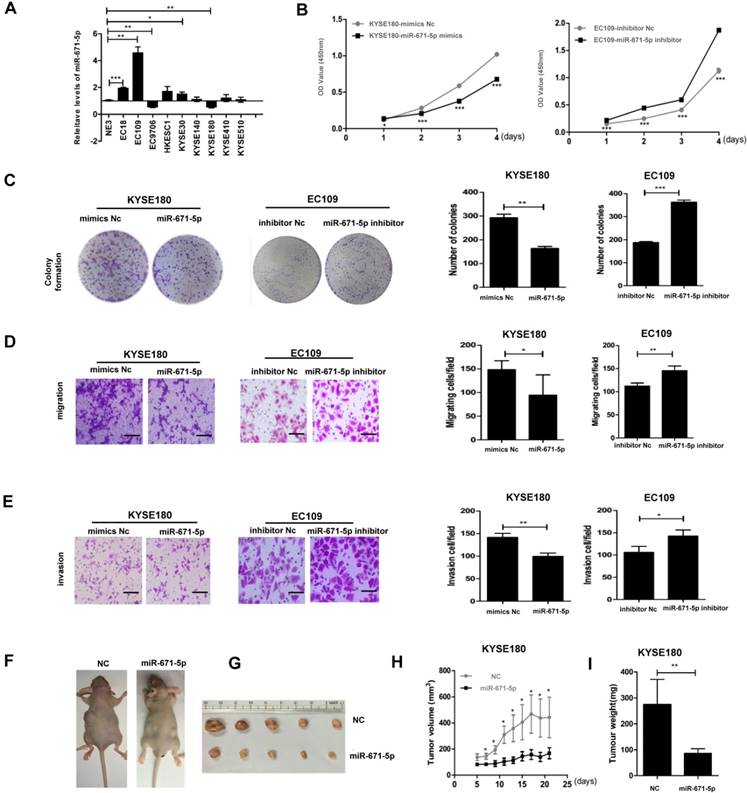
miR-671-5p suppresses proliferation, colony formation, migration, invasion, and tumorigenesis of ESCC cells
We screened the levels of miR-671-5p in the same panel of ESCC cell lines as described in Figure. 1B and found that EC109 cells with the lowest levels of FGFR2 displayed the highest levels of miR-671-5p, while KYSE180 cells with the highest levels of FGFR2 exhibited the lowest levels of miR-671-5p (Fig. 6A). The rates of cell proliferation, colony formation, migration, and invasion were reduced in KYSE180 cells treated with miR-671-5p mimics, but increased in EC109 cells treated with miR-671-5p inhibitor (Fig. 6B-E). Mice injected with KYSE180 cells developed tumors of a size of approximately 1 cm in diameters and then injected with miR-671-5p mimics or negative control mimics daily for 7 days. Treatment with miR-671-5p mimics significantly suppressed the development of tumors compared to treatment with mimics Nc (Fig. 6F-I). Therefore, our findings suggested that miR-671-5p suppresses the rates of proliferation, colony formation, migration, invasion, and tumorigenesis of ESCC cells specifically through regulating the expression of FGFR2.
miR-671-5p suppresses the progression of ESCC. A. A plot showing the relative levels of miR-671-5p as detected by qPCR in normal esophageal cell NE3 and several ESCC cell lines. B. Plots showing the relative densities of KYSE180 cells treated with negative control (Nc) or miR-671-5p mimics, and EC109 cells treated with Nc or miR-671-5p inhibitor. C-E. Representative images and plots showing the relative levels of colony formation (C), migration (D) and invasion (E) in the same cells as described in panel Bar=100μm B. F. Images showing the impact of miRNAs on the development of xenograft tumors originated from KYSE180 cells in mice. G Images showing the solid tumor tissues collected from ESCC xenograft mice. H,I. A plot showing the volumes (H) and weights of tumors collected from mice (I).
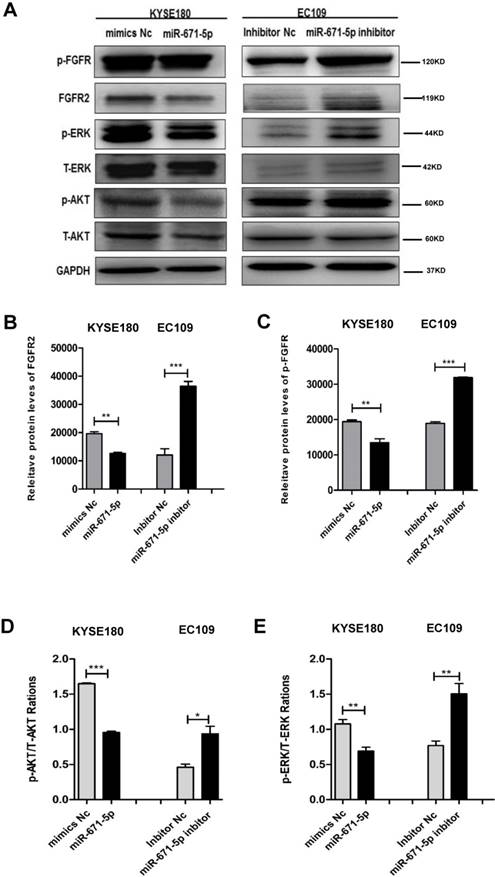
miR-671-5p suppresses ERK and AKT signaling pathway through the phosphorylation of FGFR2
To further decipher the mechanism by which miR-671-5p suppresses the tumorigenesis of ESCC cells, we tested the effect of miR-671-5p on the expression of FGFR2 and its downstream targets. KYSE180 cells treated with miR-671-5p mimics displayed reduced levels, while EC109 cells treated with miR-671-5p inhibitor showed increased levels of total FGFR2 (Fig. 7A, B), phosphorylated FGFR (Fig. 7A, C), phosphorylated AKT (Fig. 7A, D) and phosphorylated ERK (Fig. 7A, E). Therefore, miR-671-5p suppressed the progression of ESCC by suppressing the expression of FGFR2 via inhibiting the MAPK-ERK and PI3K-AKT pathways.
miR-671-5p inhibits the phosphorylation of FGFR2 and signals of MAPK-ERK and PI3K-AKT pathways in ESCC cells. A-E. Representative immunoblots (A), and plots of relative levels of FGFR2 (B) and p-FGFR proteins (C), and ratios of p-AKT to AKT (D) and ratios of p-ERK to ERK (E) in cells as described in Fig. 6B.
Discussion
Due to the shortage of precise therapeutic targets for ESCC, surgery is still the main line of treatment, although surgery alone has poor locoregional control and poor long-term outcome. Surgical treatment can achieve a five-year survival rate of approximately 10%-40% for patients with non-metastatic ESCC [33]. However, chemoradiotherapy is the most widely accepted non-surgical therapy, but is highly toxic and occasionally lethal to patients with ESCC [34]. Thus, targeted treatment is expected to have a greater potential for ESCC therapy [34].
The intracellular proteins SOX2 [35], AKT1 [36] and STAT3 [37] have been considered as potential therapeutic targets for ESCC, but are still not used for clinical application. The FGFR variant was reported to be involved in the development of various cancers [38]. High levels of FGFR2 have been associated with gastric cancer [39, 40], oral squamous cell carcinoma [41] and breast cancer [42, 43], and promote the proliferation, invasion, and metastases of tumor cells. We detected a similar trend. High levels of FGFR2 were detected in ESCC tissues, which promoted the migration, invasion, and proliferation of ESCC cells. Therefore, suppressing the expression of FGFR2 may be a potential strategy for ESCC therapy.
There are various methods to suppress the expression of FGFR2. Currently, the reported targeting molecules include inhibitors [44, 45], RNA aptamer [46-49], peptide mimetic [50], human antibody, siRNAs and synthetic miRNAs targeting the compounds of FGFRs [51-53] [26]. Drugs targeting FGFRs such as dovitinib, AZD4547, lucitanib, BGJ398, and JNJ-42756493 are currently in clinical trials, and may become a new line of treatment for patients with ESCC [54]. In addition, miRNAs also play important roles in various types of cancers [55, 56]. It has been showed that miR-494 [53], miR-381-3p [41] and miR-125b [57] directly target FGFR2 to regulate cancer progression. Here we reported that miR-671-5p specifically binds with the 3′-UTR of FGFR2 to suppress the expression of FGFR2, which inhibited the proliferation, migration, and invasion of ESCC cells. Since more and more evidences indicate that miRNAs may enhance chemotherapy efficacy for multiple human cancers [58, 59], miR-671-5p may potentially be utilized as a drug in combination with other treatments for ESCC therapy in the future.
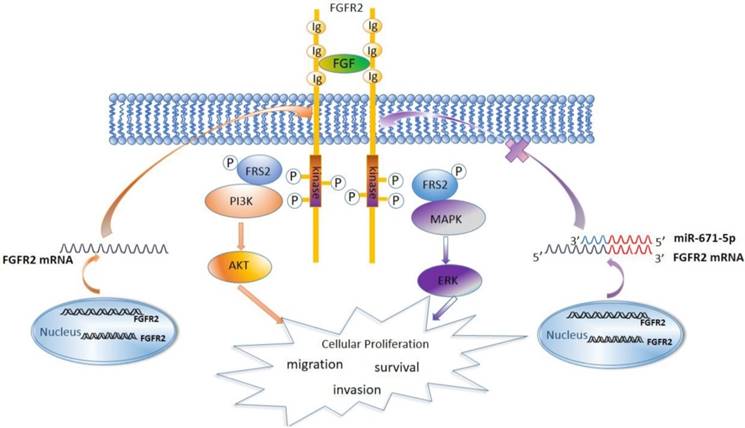
Previous studies have showed that the FGFR family members activate the MAPK, AKT, and PLCγ signaling pathways [27]. We found that FGFR2 similarly activated the MAPK-ERK and PI3K-AKT signaling pathways and increased the expression levels of the cell cycle-regulatory proteins cyclin D1 and cyclin B1. It has been reported that both cyclin D1 and cyclin B1 accelerate the cell cycle and are associated with malignance and proliferation of tumor cells [60-63]. Therefore, miR-671-5p suppresses the expression of FGFR2, inhibits the MAPK-ERK and PI3K-AKT signaling pathways to block the cell cycle, and inhibits the proliferation, invasion and migration of ESCC cells and progression of ESCC (Fig. 8).
A diagram showing the mechanism by which miR671-5p suppresses ESCC progression through FGFR2.
Abbreviations
ESCC: Esophageal Squamous Cell Carcinoma; ERK: extracellular regulated protein kinases; AKT: Protein kinase B; MAPK: mitogen-activated protein kinase; PI3K: Phosphoinositide 3-kinase; miRNA: MicroRNA; qPCR: Real-time Quantitative PCR Detecting System; STAT3: Signal transducer and activator of transcription 3; mTOR: mammalian target of rapamycin; p-AKT: phosphor-AKT; p-ERK: phosphor-ERK.
Acknowledgements
This study was supported by grants from the National Key Basic Research Program of China (2015CB553706), the National Nature Science Foundation of China (81472337 and 81473131), the Science & Technology Program of Guangdong Province (2016A020217012), and Science & Technology Program of Guangzhou City (2016201604030039). We thank Yuying Zhou and Shujun Guo from the Institute of Biomedicine of Jinan University for technical assistance.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
2. Gholipour M, Islami F, Roshandel G, Khoshnia M, Badakhshan A, Moradi A. et al. Esophageal Cancer in Golestan Province, Iran: A Review of Genetic Susceptibility and Environmental Risk Factors. Middle East journal of digestive diseases. 2016;8:249-66
3. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893-917
4. Lee PL, Johnson DE, Cousens LS, Fried VA, Williams LT. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science. 1989;245:57-60
5. Jaye M, Schlessinger J, Dionne CA. Fibroblast growth factor receptor tyrosine kinases: molecular analysis and signal transduction. Biochimica Et Biophysica Acta. 1992;1135:185-99
6. Givol D, Yayon A. Complexity of FGF receptors: genetic basis for structural diversity and functional specificity. Faseb Journal Official Publication of the Federation of American Societies for Experimental Biology. 1992;6:3362
7. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature reviews Cancer. 2010;10:116-29
8. Gallo LH, Nelson KN, Meyer AN, Donoghue DJ. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine & growth factor reviews. 2015;26:425-49
9. Ahmad I, Iwata T, Leung HY. Mechanisms of FGFR-mediated carcinogenesis. Biochim Biophys Acta. 2012;1823:850-60
10. Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A. et al. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008;68:2340-8
11. Czaplinska D, Turczyk L, Grudowska A, Mieszkowska M, Lipinska AD, Skladanowski AC. et al. Phosphorylation of RSK2 at Tyr529 by FGFR2-p38 enhances human mammary epithelial cells migration. Biochim Biophys Acta. 2014;1843:2461-70
12. Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642-55
13. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
14. Seo SH, Jang MS, Kim DJ, Kim SM, Oh SC, Jung CR. et al. MicroRNA-150 controls differentiation of intraepithelial lymphocytes through TGF-beta receptor II regulation. The Journal of allergy and clinical immunology. 2017
15. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
16. Song Y, Li L, Ou Y, Gao Z, Li E, Li X. et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91-5
17. Zhao M, Luo R, Liu Y, Gao L, Fu Z, Fu Q. et al. miR-3188 regulates nasopharyngeal carcinoma proliferation and chemosensitivity through a FOXO1-modulated positive feedback loop with mTOR-p-PI3K/AKT-c-JUN. Nat Commun. 2016:7
18. Barbagallo D, Condorelli A, Ragusa M, Salito L, Sammito M, Banelli B. et al. Dysregulated miR-671-5p/CDR1-AS/CDR1/VSNL1 axis is involved in glioblastoma multiforme. Oncotarget. 2016;7:4746
19. Tan X, Fu Y, Chen L, Lee W, Lai Y, Rezaei K. et al. miR-671-5p inhibits epithelial-to-mesenchymal transition by downregulating FOXM1 expression in breast cancer. Oncotarget. 2016;7:293
20. Malgulwar PB, Pathak P, Singh M, Kale SS, Suri V, Sarkar C. et al. Downregulation of SMARCB1/INI1 expression in pediatric chordomas correlates with upregulation of miR-671-5p and miR-193a-5p expressions. Brain tumor pathology. 2017
21. Papp G, Krausz T, Stricker TP, Szendroi M, Sapi Z. SMARCB1 expression in epithelioid sarcoma is regulated by miR-206, miR-381, and miR-671-5p on Both mRNA and protein levels. Genes, chromosomes & cancer. 2014;53:168-76
22. Wang J-k, Guo S-j, Tian B-q, Nie C-j, Wang H-l, Wang J-l. et al. Association between FGFRs and the susceptibility of digestive and reproductive system cancers in Chinese population. Molecular & Cellular Toxicology. 2017;13:379-85
23. Qin YR, Tang H, Xie F, Liu H, Zhu Y, Ai J. et al. Characterization of tumor-suppressive function of SOX6 in human esophageal squamous cell carcinoma. Clin Cancer Res. 2011;17:46-55
24. Wang X, Pan L, Feng Y, Wang Y, Han Q, Han L. et al. p300 plays a role in p16INK4a expression and cell cycle arrest. Oncogene. 2008;27:1894
25. Zhen Y, Liu Z, Yang H, Yu X, Wu Q, Hua S. et al. Tumor suppressor PDCD4 modulates miR-184-mediated direct suppression of C-MYC and BCL2 blocking cell growth and survival in nasopharyngeal carcinoma. Cell death & disease. 2013;4:e872
26. Katoh. FGFR2-related pathogenesis and FGFR2-targeted therapeutics (Review). International Journal of Molecular Medicine. 2009:23
27. Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine & growth factor reviews. 2005;16:139-49
28. Tanimura S, Takeda K. ERK signalling as a regulator of cell motility. Journal of biochemistry. 2017;162:145-54
29. Wellbrock C, Arozarena I. The Complexity of the ERK/MAP-Kinase Pathway and the Treatment of Melanoma Skin Cancer. Frontiers in cell and developmental biology. 2016;4:33
30. Downward J. PI 3-kinase, Akt and cell survival. Seminars in cell & developmental biology: Elsevier. 2004 p. 177-82
31. Ewen ME, Lamb J. The activities of cyclin D1 that drive tumorigenesis. Trends in molecular medicine. 2004;10:158-62
32. Pines J, Hunter T. Human cyclin A is adenovirus E1A-associated protein p60 and behaves differently from cyclin B. Nature. 1990;346:760-3
33. Kaifi JT, Gusani NJ, Jiang Y, Mackley HB, Dye CE, Mathew A. et al. Multidisciplinary management of early and locally advanced esophageal cancer. Journal of clinical gastroenterology. 2011;45:391-9
34. Sasaki Y, Kato K. Chemoradiotherapy for esophageal squamous cell cancer. Japanese journal of clinical oncology. 2016;46:805-10
35. Liu Y, Xiong Z, Beasley A, D'Amico T, Chen XL. Personalized and targeted therapy of esophageal squamous cell carcinoma: an update. Annals of the New York Academy of Sciences. 2016;1381:66-73
36. Mao Y, Li L, Liu J, Wang L, Zhou Y. MiR-495 inhibits esophageal squamous cell carcinoma progression by targeting Akt1. Oncotarget. 2016;7:51223-36
37. Liu J, Mao Y, Zhang D, Hao S, Zhang Z, Li Z. et al. MiR-143 inhibits tumor cell proliferation and invasion by targeting STAT3 in esophageal squamous cell carcinoma. Cancer Lett. 2016;373:97-108
38. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179-85
39. Jang J, Kim HK, Bang H, Kim ST, Kim SY, Park SH. et al. Antitumor Effect of AZD4547 in a Fibroblast Growth Factor Receptor 2-Amplified Gastric Cancer Patient-Derived Cell Model. Translational oncology. 2017;10:469-75
40. Greer SU, Nadauld LD, Lau BT, Chen J, Wood-Bouwens C, Ford JM. et al. Linked read sequencing resolves complex genomic rearrangements in gastric cancer metastases. Genome medicine. 2017;9:57
41. Yang X, Ruan H, Hu X, Cao A, Song L. miR-381-3p suppresses the proliferation of oral squamous cell carcinoma cells by directly targeting FGFR2. Am J Cancer Res. 2017;7:913
42. Wang S, Ding Z. Fibroblast growth factor receptors in breast cancer. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2017;39:1010428317698370
43. Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R. et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29:2013-23
44. Mohammadi M, Froum S, Hamby JM, Schroeder MC, Panek RL, Lu GH. et al. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. The EMBO journal. 1998;17:5896-904
45. Nakamura K, Yashiro M, Matsuoka T, Tendo M, Shimizu T, Miwa A. et al. A novel molecular targeting compound as K-samII/FGF-R2 phosphorylation inhibitor, Ki23057, for Scirrhous gastric cancer. Gastroenterology. 2006;131:1530-41
46. Ireson CR, Kelland LR. Discovery and development of anticancer aptamers. Mol Cancer Ther. 2006;5:2957-62
47. Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nature biotechnology. 2005;23:1073-8
48. Ulrich H. DNA and RNA aptamers as modulators of protein function. Medicinal Chemistry. 2005;1:199-208
49. Lee HK, Choi YS, Park YA, Jeong S. Modulation of oncogenic transcription and alternative splicing by β-catenin and an RNA aptamer in colon cancer cells. Cancer Res. 2006;66:10560-6
50. Li S, Christensen C, Kiselyov VV, Køhler LB, Bock E, Berezin V. Fibroblast growth factor-derived peptides: functional agonists of the fibroblast growth factor receptor. Journal of neurochemistry. 2008;104:667-82
51. Katoh Y, Katoh M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA. International journal of molecular medicine. 2008;22:271-5
52. Katoh M. RNA technology targeted to the WNT signaling pathway. Cancer Biol Ther. 2008;7:275-7
53. Zhao X, Zhou Y, Chen YU, Yu F. miR-494 inhibits ovarian cancer cell proliferation and promotes apoptosis by targeting FGFR2. Oncol Lett. 2016;11:4245-51
54. Andre F, Cortes J. Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res Treat. 2015;150:1-8
55. Parikh A, Lee C, Joseph P, Marchini S, Baccarini A, Kolev V. et al. microRNA-181a has a critical role in ovarian cancer progression through the regulation of the epithelial-mesenchymal transition. Nat Commun. 2014:5
56. Guo SL, Ye H, Teng Y, Wang YL, Yang G, Li XB. et al. Akt-p53-miR-365-cyclin D1/cdc25A axis contributes to gastric tumorigenesis induced by PTEN deficiency. Nat Commun. 2013;4:2544
57. Xu N, Brodin P, Wei T, Meisgen F, Eidsmo L, Nagy N. et al. MiR-125b, a microRNA downregulated in psoriasis, modulates keratinocyte proliferation by targeting FGFR2. The Journal of investigative dermatology. 2011;131:1521-9
58. Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, Mendell JT. et al. Involvement of Human Micro-RNA in Growth and Response to Chemotherapy in Human Cholangiocarcinoma Cell Lines. Gastroenterology. 2006;130:2113-29
59. Sun L, Yao Y, Liu B, Lin Z, Lin L, Yang M. et al. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene. 2012;31:432-45
60. Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJPL, Lam EWF. et al. Cell Cycle Inhibition by FoxO Forkhead Transcription Factors Involves Downregulation of Cyclin D. Molecular and Cellular Biology. 2002;22:7842-52
61. Lehn S, Tobin NP, Berglund P, Nilsson K, Sims AH, Jirstrom K. et al. Down-regulation of the oncogene cyclin D1 increases migratory capacity in breast cancer and is linked to unfavorable prognostic features. The American journal of pathology. 2010;177:2886-97
62. Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny M. et al. P21-dependent G1arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. British journal of cancer. 2000;83:817-25
63. Song Y, Zhao C, Dong L, Fu M, Xue L, Huang Z. et al. Overexpression of cyclin B1 in human esophageal squamous cell carcinoma cells induces tumor cell invasive growth and metastasis. Carcinogenesis. 2008;29:307-15
Author contact
![]() Corresponding author: Xiaojia Chen and An Hong, Institute of Biomedicine & Department of cell Biology, Jinan University, National Engineering Research Center of Genetic Medicine, and Guangdong Provincial Key Laboratory of Bioengineering Medicine, Guangzhou, Guangdong, 510632, P. R. China. Tel: 020-85221983. E-mail for Xiaojia Chen: tchenxjedu.cn, An Hong: thaedu.cn
Corresponding author: Xiaojia Chen and An Hong, Institute of Biomedicine & Department of cell Biology, Jinan University, National Engineering Research Center of Genetic Medicine, and Guangdong Provincial Key Laboratory of Bioengineering Medicine, Guangzhou, Guangdong, 510632, P. R. China. Tel: 020-85221983. E-mail for Xiaojia Chen: tchenxjedu.cn, An Hong: thaedu.cn