Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(9):1993-2005. doi:10.7150/ijbs.34269 This issue Cite
Research Paper
Deletion of CD163 Exon 7 Confers Resistance to Highly Pathogenic Porcine Reproductive and Respiratory Viruses on Pigs
Haitao Wang1,2,*, Liangcai Shen1,2,*, Jingyao Chen1,2, Xiaojuan Liu1,2, Tan Tan1,2, Yiqing Hu1,2, Xiaofei Bai3, Yuexin Li1,2, Kegong Tian3,4, ![]() , Ning Li1,2,
, Ning Li1,2, ![]() , Xiaoxiang Hu1,2,
, Xiaoxiang Hu1,2, ![]()
1. State Key Laboratory for Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing, China.
2. National Engineering Laboratory for Animal Breeding, China Agricultural University, Beijing, China.
3. National Research Center for Veterinary Medicine, Luoyang, Henan Province, China.
4. College of Animal Science and Veterinary Medicine, Henan Agricultural University, Zhengzhou, Henan Province, China.
* Those authors contribute equally to the article.
Received 2019-2-20; Accepted 2019-5-19; Published 2019-7-25
Abstract

Porcine reproductive and respiratory syndrome (PRRS) caused by PRRS virus (PRRSV) is a severe infectious disease in the swine industry. PRRSV infection is mediated by porcine CD163 (pCD163). Scavenger receptor cysteine-rich domain 5 coded by exon 7 of pCD163 is essential for PRRSV infection. In this study, we generated CD163 exon 7 deleted (CD163E7D) pigs using CRISPR/Cas9 mediated homologous recombination and somatic cell nuclear transfer (SCNT). The deletion of exon 7 had no adverse effects on CD163-associated functions. Pigs were further challenged with a highly pathogenic PRRSV (HP-PRRSV) strain. The CD163E7D pigs exhibited mild clinical symptoms and had decreased viral loads in blood. All CD163E7D pigs survived the viral challenge, while all the WT pigs displayed severe symptoms, and 2 out of 6 WT pigs died during the challenge. Our results demonstrated that CD163 exon 7 deletion confers resistance to HP-PRRSV infection without impairing the biological functions of CD163.
Keywords: CD163, SRCR 5, Highly pathogenic PRRSV, CRISPR/Cas9, SCNT
Introduction
Porcine Reproductive and Respiratory Syndrome (PRRS) has caused significant economic losses in pig farming. Since its first report in 1987 in the United States [1], PRRS had spread across the world for decades [2]. The etiological agent of PRRS is Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) [3, 4], which causes reproductive and respiratory symptoms in different ages of pigs, especially in piglets and sows. Once pigs are infected, fever, inappetence, lethargy, and breathing difficulty are observed in fattening pigs, and late-term abortion and fetal mummification frequently occur to sows. The newborn piglets delivered by infected sows are weak and suffer from severe respiratory symptoms [5-7]. The large-scale reduction in pregnant sows and death of piglets caused a loss of more than 650 million dollars from 2005 to 2010 in the United States alone [8]. In China, the epidemic of PRRS also leads to a devastating loss in the pig industry since its first emergence in 2006 [9, 10].
PRRSV is a positive-stranded RNA virus belonging to the genus Arterivirus of the family Arteriviridae within the order Nidovirale [11, 12]. Genetically, there are two different PRRSV genotypes: European (EU genotype, type 1) and North American (NA genotype, type 2). The approximate 15kb PRRSV genome contains 11 open reading frame and encodes at least seven structural proteins, including nucleocapsid protein N, protein M, and protein E, as well as GP2a, GP3, GP4, and GP5 [13-15]. PRRSV can only infect pigs, and the host cells are specific swine macrophage subsets, especially porcine alveolar macrophages (PAMs) [16]. PRRSV can also infect other host cells in vitro, such as African green monkey kidney epithelial cell lines MA-104 and MARC-145 [17].
The process of viral infection includes viral adhesion, host cells endocytosis, and genome release. Three cellular factors, heparin sulphate (HS) [18], CD163 [19], and CD169 (Sn/cluster of differentiation 169) [20], involve in the binding, internalization, and uncoating of PRRSV respectively. Furthermore, studies have demonstrated that CD163 is the essential protein for PRRSV infection in vivo or in vitro [21-23].
CD163, also known as a hemoglobin/haptoglobin scavenger receptor or p155, belongs to the scavenger receptor cysteine-rich (SRCR) superfamily. CD163 was shown to bind the complex of hemoglobin and plasma protein haptoglobin (Hb-Hp) in early studies [24]. Further studies demonstrated CD163 involves in PRRSV infection. The extracellular structure of CD163 contains nine SRCR domains, which are anchored on the cell surface by a transmembrane segment and a short cytoplasmic domain [25]. The nine extracellular domains are separated by two proline-serine-threonine (PST) rich motifs located at the middle and the end of the extracellular region [26]. Among the extracellular domains, SRCR domain 5 (SRCR5) is essential for PRRSV infection [27]. Further structure-based mutation analysis revealed the important amino acid on SRCR5 for virus invasion in vitro [28].
Due to the high diversity of PRRSV, PRRS is difficult to control with traditional approaches [29, 30]. However, gene editing of CD163 made great progress in anti-PRRSV research. Although pigs with the complete ablation of CD163 are resistant to PRRSV infection challenge in vivo [31], more studies focus on the genomic engineering of CD163 exon 7, which encodes SRCR5, due to its essential role in PRRSV invasion, to avoid the potential harmful effects on CD163-associated biological functions. Pigs with CD163 exon7 replaced with the CD163L1 exon11 sequence (encoding human CD163L1 SRCR8) are resistant to type 1 PRRSV infection, but not to type 2 PRRSV infection [32]. Pigs lacking SRCR5, which were generated using CRISPR/Cas9 editing in zygotes, are resistant to PRRSV-1 in vivo [33]. However, it is unknown if the pigs lacking SRCR5 of CD163 are resistant to the type 2 HP-PRRSV in vivo. Here, we describe a different strategy, which used homologous recombination mediated by CRISPR/Cas9 and somatic cell nuclear transfer (SCNT), to generate CD163 exon 7 deleted (CD163E7D) pigs. The generated CD163E7D pigs were challenged with high doses of HP-PRRSV and were remarkably resistant to PRRSV in vivo. The PRRSV-infected CD163E7D pigs also inhibited PRRSV infection to the WT pigs housed with them. We also showed the deletion of exon 7 had no adverse effects on other biological functions associated with CD163 in vivo.
Materials and methods
Ethics statement of Animal Usage
In this study, all application institutional the care and use of animals were followed. The animal experiments were performed according to the Guide for the Care and Use for Laboratory Animals issued by the Ministry of Science and Technology. The experimental protocols were approved by the Committee on the Ethics of Animal Experiments of China Agricultural University (Permit Numbers: CAU20170830-3, CAU20171215-3). The experimental animals were maintained in an animal facility in which room temperature of 20-26°C, humidity 40%-60%, and a 9/15 h light/dark cycle were maintained, and HEPA-filtered air was provided. The pigs were fed with a pig diet and tap water. At the end of the challenge, the surviving pigs were euthanized by ketamine.
Vectors, Cells, and Viruses
The px330-501 vector was modified from the original pX330 vector (Addgene, plasmid #42230) in State Key Laboratory for Agrobiotechnology, China Agricultural University. The homologous recombination vector was constructed using pGKneo (provided by Dr. Chen Jinyao, China Agricultural University) as the backbone. The homologous arms containing the sequences of CD163 were synthesized and inserted into pGKneo by restriction enzymes (New England Biolabs). Porcine fetal fibroblasts (PFFs) were obtained from 30-day-old Large White fetuses and maintained at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Gibco, 11995-065) supplemented with 15% fetal bovine serum (FBS) (Gibco, 10099-141). The HP-PRRSV used in this study is strain JXA1 with GenBank Accession Number EF1122445, which was a generous gift of Professor Kegong Tian, National Research Center for Veterinary Medicine. Viral particles were prepared according to the method reported by Zhang et al. [34] and stored at -80°C until use. Marc145 cells were from the stocks in State Key Laboratory for Agrobiotechnology, China Agricultural University, and cultured with DMEM plus 5-10% FBS. The Marc145 cells were used for determining the viral titers of serum separated from the challenged pigs using the TCID50 method as reported by Zhang et al. [35].
Cell transfection and selection
Electrotransfection of the vectors was applied according to the protocol provided by Amaxa Basic Nucleofector Kit (Lonza, VPI-1002). In brief, five micrograms of the px330-501 and homologous recombination vectors were mixed at a 1:2 ratio and added to 150μl electroporation medium. Approximate 1×104 trypsinized PFFs were added to the medium cocktail, mixed, and transferred into a Lonza cuvette for electroporation. Then, all the cells were divided into multiple 10-cm plates with G418 (600 μg/mL, Promega) selection medium, and incubated for 9-12 days. The resistant clones were picked up with cloning cylinders. Healthy clones were sub-cultured and subject to genotyping. 30% sub-cultured cells from each colony were used for genomic DNA extraction. The forward primer E7F (5'-TTCTCCCTCACCGAAATGCT-3') and reverse primer E7R (5'-GCAGTGACGGAACAATCTCC-3') were used to amplify the exon7 fragment. The forward primer KZBF (5'-CTCCCAAGTGTCTTCCCTGATGCT-3') and reverse primer KZBR (5'-CAAGATGGATTGCACGCAGGTTCTC-3') were used to amplify a 7513bp fragment from the edited genome locus, which contains a part of the drug-selection marker, 5' arms, and the sequences before 5' arms in the WT genome. The primers designed for amplification of the 3' homological arm were KYBF (5'- GAGAACCTGCGTGCAATCCATCTTG-3') and KYBR (5'-TCTGACTCTGGTTCCCTGCAAACAG-3'). The expected PCR product size is 6020bp. Homozygous CD163 exon7 null clones were expanded and cryopreserved for SCNT.
Off-target analysis of sgRNA
Potential off-target sites (OTSs) were predicted on website http://crispr.genome-engineering.org/. The predicted OTSs were aligned to the whole pig genome on website http://asia.ensembl.org/index.html. The OTSs with less than 6 mismatches to the sgRNA sequence and located within a gene were picked up for further analysis. PCR amplification (primers were listed Table S1) and Sanger sequencing were performed to determine whether any point mutations exist.
Generation of CD163E7D pigs by SCNT
The homozygous CD163 exon 7 null cells were trypsinized and kept at 4°C as the donor cells. SCNT was performed as the previous study [36]. In brief, cumulus-oocyte complexes (COCs) were aspirated and cultured in the maturation medium at 38.5°C for 40-44h. The mature COCs were digested with hyaluronidase (Sigma, H4273) for less than 5 minutes. The denudated oocytes that had released the polar bodies were enucleated by aspirating the cytoplasm adjacent to the first polar body with a 20μm glass pipette. Subsequently, a single donor cell was injected into the perivitelline space between the zona and cytoplasm of the enucleated oocytes. The reconstructed embryos were fused and activated with two successive dendritic cell pulses at 150V for 100ms using an electrofusion instrument (BLS, CF-150B). Then the embryos were cultured in porcine zygote medium at 38.5°C overnight and transferred into the oviduct of estrous recipient gilts. We monitored the pregnancy status of surrogates monthly until delivery. The piglets were delivered naturally. The ear skin of the newborn piglets was collected, and the genomic DNA was isolated for genotyping. The primers E7F/E7R were used to amplify the exon7 fragment. To detect whether the drug-selection gene was excised in the pigs, the forward primer 169F (5'-TGAATTGCCTCTCAGTCTG-3') and reverse primer 172R (5'-CATGATAGGAGTAAGCCAG-3') were used to amplify the whole fragment between homologous arms. Next, total RNA in the lungs of piglets was isolated with TRIzol (Invitrogen, 15596026) for RT-PCR. Primers ZLBF (5'-TCAGTGCCTGCTTGGTCACTAG-3') and ZLBR (5'-CCGTTCATCTGCTTTCAGGCAAG-3') were used to amplify CD163 transcript. The tissue samples of stillborn piglets were collected for further analysis.
Western blot
Tissues were collected and lysed by immunoprecipitation (IP) lysis buffer (Biyotime, P0013). Protein concentrations were quantified with a BCA kit (Beyotime, P0010). An equal amount of total protein from each sample was separated on 10% sodium dodecyl-sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane (Millipore). Then membranes were blocked with 5% non-fat milk and incubated with a mouse anti-pig CD163 monoclonal antibody as the primary antibody. After washing with TBST, HRP-conjugated anti-mouse IgG was used as the secondary antibody. β-actin was blotted as a loading control. Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific) was used to visualize the signals, and the images were captured with a camera (Tanon, 5200).
ELISA
PRRSV-specific antibody was detected using IDEXX PRRS X3 Ab Test (IDEXX Laboratories) according to the protocol of manufacturer. Serum samples were considered positive for PRRSV antibody if the S/P ratio was greater than 0.4. The amount of Hp in serum was measured using a pig ELISA kit (Cloud-Clone Crop Inc.) according to the manufacturer's instructions. Each sample was detected in triplicate.
Viral Challenge
The in vivo infection of the experimental pigs with HP-PRRSV was performed at National Research Center for Veterinary Medicine in Luoyang, Henan province. All the tested pigs were female, 8 weeks old, and were transferred to the challenge facility to acclimate 5 days before the challenge. Pigs were divided into four groups in two separate rooms: the challenged CD163E7D pigs (n=6) and 6 non-challenged WT pigs were maintained in one room, while the challenged WT pigs (n=6) and another 6 non-challenged WT pigs were housed in the other room. Before the challenge, the pigs were confirmed to be negative for PRRSV antibody in the serum and free of PRRSV infection. The rectal temperatures of all the pigs were normal, and there were no any observable clinical symptoms, and all the pigs were also energetic and ate actively. The pigs were challenged with the HP-PRRSV strain JXA1 by intramuscular injection of 106.5 TCID50 of viruses per animal. During the intramuscular injection, the unchallenged pigs that would be housed with the challenged pigs were separated from the challenged pigs temporally and transferred back several hours later. To avoid the leakage of virus from the needles, they were left to stay in the muscle for seconds after injection, alcohol wipes were immediately used for disinfection and were hold for several minutes at the injection sites after pulling out of the needles. Infected pigs were monitored for the next 21 days. The rectal temperature was measured twice daily, and clinical symptoms including respiratory distress, lameness, diarrhea, inappetence, and fever were recorded and scored as previously described [37, 38]. In short, the pigs were scored 0 for normal, 1 for mild inappetence or decreased demeanor, 2 for sneezing/coughing, diarrhea, or rubefaction, 4 for severe respiratory distress, lameness, blue ears, or stopping feed intake. Blood samples were collected on days 0, 3, 5, 7, 10, 14, 17, 21 and allowed to clot. The serum was separated and preserved at -80 °C. The surviving pigs were sacrificed by euthanasia and their tissues were collected for necropsy.
Immunohistochemistry Staining
Tissues, including lung, tonsil, spleen, lymph node of the different challenged groups, were collected and subject to immunohistochemistry to detect PRRSV as described previously [39]. The primary antibody was a mouse monoclonal antibody against PRRSV nucleocapsid protein. The slides were visualized using a microscope.
Results
Generation of CD163 exon 7 deleted PFFs
To achieve CD163 exon7 biallelic deletions in PFFs, the CD163E7D homologous recombination vector was constructed as a donor plasmid. The donor vector was constructed by modifying the pKGneo vector and contains two homologous arms. A drug-selection cassette and an Oct4-Cre element were flanked by two loxP sites between the arms for excision of the drug-selection gene. A DTA (diphtheria toxin A-chain) element was put outside the homologous arm to avoid random integration. The donor vector was used as a template to repair the DNA double-strand break generated by sgRNA/Cas9 via homologous recombination (Fig. 1A). The 6255bp left arm and 999bp right arm are located upstream and downstream of exon 7, respectively. The left arm was cloned into the Sac II and Not I sites of pGKneo vector, and the right arm was cloned to the Sal I and Asc I sites of pGKneo vector.
Generation of CD163 exon 7 deleted PFFs. (A) Strategy for generation of CD163 exon 7 deleted allele. The WT CD163 exon 7 locus and donor vector are shown. The donor vector is composed of two homologous arms (pink and amber lines) and a drug-selection element (green line) flanked by two loxP sites (black triangles). The Cas9 enzyme makes a double-stranded break at the site targeted by 501-sgRNA. After homologous recombination repair mediated by the homologous arms, CD163 exon 7 will be replaced by the drug-selection cassette, resulting in an exon 7 deleted allele. The excision of the drug-selection gene is mediated by the Oct4-Cre element during the early stage of embryonic development, and a loxP sequence will be left in intron 6 of CD163. (B) Identification of exon 7 deletion in PFFs. E7F/E7R primers were used to amplify PFF colony DNA. (C) PCR amplification of the two homologous arms. The PCR products for the left (left) and right arms (right) are expected to be 7513bp and 6020bp in size, respectively. The result from the control homologous recombination vector was negative.
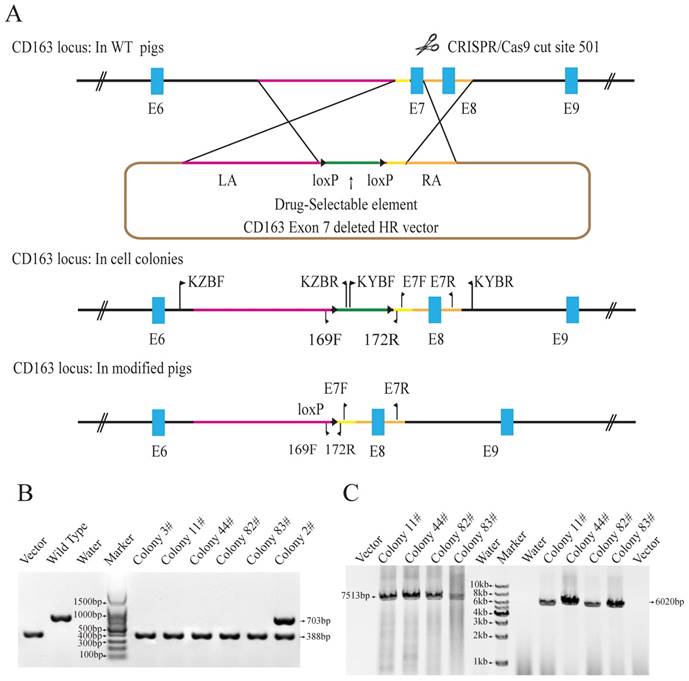
The Cas9 vector pX330-501 was mixed with the donor vector and transfected into porcine fetal fibroblasts. 62 colonies were screened using three pairs of primers to detect the recombination events. Primers E7F/R, which amplify the exon 7 sequences, were used to determine whether the exon 7 was deleted. Primers KZF/R and KYF/R were used to detect the recombination event by amplifying the fragments spanning the homologous arms and their adjacent genomic sequences. Our results showed that E7F/R primers amplified a 388bp product from the CD163 E7 deleted allele and a 703bp from the WT allele (Fig. 1B). KZF/R primers amplified a fragment of 7513bp containing the 5' homologous recombination arm and the drug-selection marker, and KYF/R primers amplified a fragment of 6020bp containing the right homologous arm (Fig. 1C). After PCR amplification and Sanger sequencing analysis of the products, a total of 5 colonies were demonstrated to carry the CD163 exon7 biallelic deletions.
Due to RNA-based recognition, the off-target effect of CRISPR/Cas9 system must be concerned. To check whether off-target genome modifications occurred, 7 potential off-target sites, which have less than 6 mismatches with the guide RNA sequence, were determined and screened (Table 1). All the seven possible off-target sites were examined by PCR and Sanger sequencing. The results showed no off-target mutations occurred in the genomes of all the 5 colonies. The results of PCR and Sanger sequencing of colony 11# were showed in Fig. S1. Two of the CD163E7D colonies were used as donor cells for SCNT.
Off-target analysis of colony #44.
| Sequencesa,b | Mism-atches | Gene | Position in genome | Resultsc | |
|---|---|---|---|---|---|
| Target site | GGAACTACAGTGCGGCACTGTGG | - | CD163 | - | - |
| Off site1 | GGGCCTGCATTGCGGCACTGAGG | 4MMs | SLC17A5 | chr1:92337898-92337920 | no off-target occurred |
| Off site2 | GGGGCTGCAGGGCGGCACTGAGG | 4MMs | KCNK12 | chr3:92999538-92999560 | no off-target occurred |
| Off site3 | AGAACCACAGTGCGACACTGAGG | 3MMs | ANKH | chr16:4129263-4129285 | no off-target occurred |
| Off site4 | GAAACGACAGTGCGACACTGAGG | 3MMs | APBA2 | chr1:144181060-144181082 | no off-target occurred |
| Off site5 | GGACGTGCAGTGAGGCACTGTGG | 4MMs | MEGF11 | chr1:164139020-164139042 | no off-target occurred |
| Off Site6 | TGAATACAAGTGCGGCACTGGGG | 5MMs | ACVR2A | chr15:4249480-4249502 | no off-target occurred |
| Off site7 | GAAGTCCCTGTGCGGCACTGTGG | 6MMs | DNAJC3 | chr11:65260383-65260405 | no off-target occurred |
a The nucleotides mismatched with 501-sgRNA were labeled red.
b The PAMs were labeled blue.
c No mutations were detected at these sites.
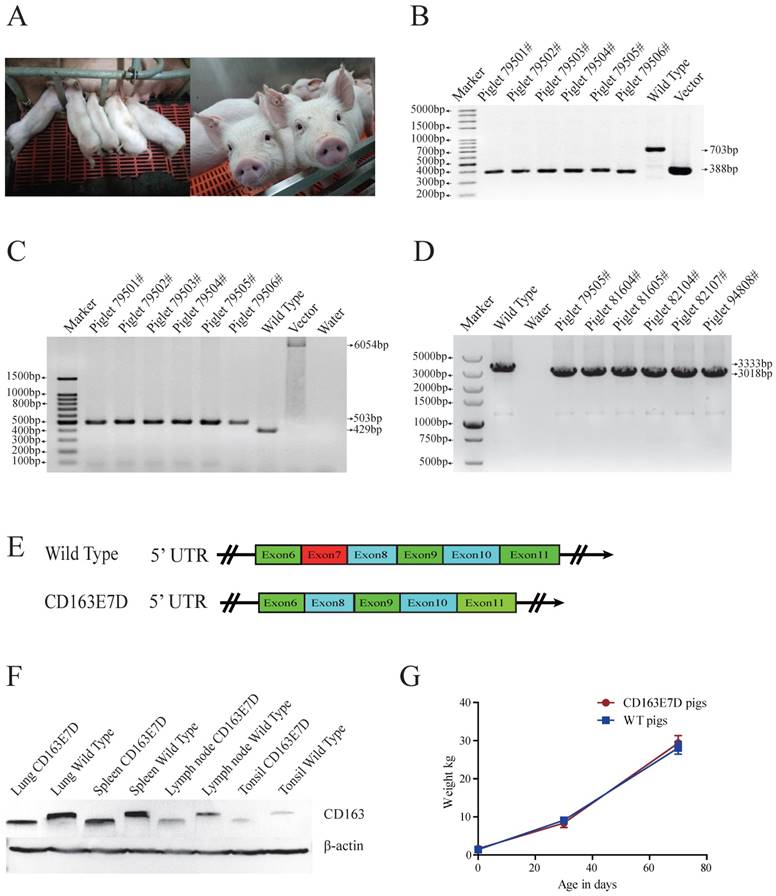
Generation of CD163 exon7 deleted pigs
The biallelic mutant cells derived from colonies 11# and 44# were chosen as donor cells for SCNT. Cells from the single colony or the pool of these two colonies were used for nuclear transfer. A total of 2192 reconstructed embryos were transferred to 6 surrogate mothers, and four surrogate sows were successfully pregnant. 26 piglets were delivered naturally (Table 2, Fig. 2A), including 4 stillborn piglets, 2 weak piglets died in 15 days, and 20 healthy piglets. The same PCR primers used for detecting the exon 7 deletion in PFFs were used for genotyping the piglets (Fig. 2B), and the exon 7 deletion was confirmed in the cloned animals. As the Oct4-Cre element was designed in the vector for the excision of the drug-selection gene in the early stage of embryonic development, primers 169F/172R were used for amplification of the whole fragment between homologous arms. As expected, PCR products of CD163E7D pigs were 502bp (Fig. 2C), and Sanger sequencing analysis showed only one loxP sequence was left in intron 6 of CD163, confirming the drug-selection gene was excised in the pigs. Furthermore, to detect the expression of CD163, the piglet lung RNA was extracted for RT-PCR. Primers ZLBF/R were used to amplify CD163 cDNA. The Sanger sequencing results of the PCR products showed the exon 7 of CD163 cDNA had been deleted, and the transcript of CD163E7D pigs was 315bp shorter than the WT transcript (Fig. 2D and E). The CD163 cDNA sequence of CD163E7D pigs and its predicted amino acid sequence were showed in Fig. S2. As a result, Western blot showed that the CD163 protein of CD163E7D pigs was 13.4kDa smaller than that of WT pigs in the lungs, tonsils, spleens, and lymph nodes (Fig. 2F). The weights of CD163E7D pigs and WT pigs were recorded, and the growth curves showed the CD163E7D pigs had similar growth rate as the WT pigs (Fig. 2G). There were no significant differences in weights between the 2 groups on their birthdays, 30 days, and 70 days. These results demonstrated that the CD163E7D pigs expressed CD163 only lacking SRCR5, and the CD163E7D pigs grew normally.
Somatic cell nuclear transfer results of CD163 exon7 deleted pig.
| Recipient ID | Donor colonies | No. of embryos transferred | Day of estrus | Piglets status |
|---|---|---|---|---|
| 20303 | 11# | 390 | 1 | 5 live piglets, 1 dead piglets |
| 10603 | 11# | 400 | 1 | No piglet |
| 21007 | 11# , 44# | 310 | 1 | 4 live piglets (one died at day 5), 1 dead piglet |
| 39003 | 11# , 44# | 396 | 1 | 6 live piglets (one died at day 13), 1 dead piglet |
| 10112 | 11# , 44# | 346 | 1 | No piglet |
| 33003 | 44# | 350 | 1 | 7 live piglets, 1 dead piglet |
Generation of CD163 exon7 deleted pigs via SCNT. (A) Represented pictures of 10-day-old (left) and 8-week-old (right) CD163E7D piglets. (B) Genotyping of the genome-modified pigs (#79501-#79506) by PCR using primers E7F/R. (C) PCR products of primers 169F/172R demonstrated the absence of the drug-selection gene in the pigs. The PCR product from CD163E7D pigs was 503bp, and the products of the control homologous recombination vectors were 6054bp. (D) RT-PCR proved that the transcripts of CD163 in the lung of the CD163E7D and WT pigs were 3018bp and 3333bp, respectively. (E) A schematic view of the CD163 transcripts in CD163E7D and WT pigs. (F) Western blot shows that the CD163E7D pigs express a smaller CD163 protein in the tissues compared with the WT pigs. (G) Growth curves of the CD163E7D pigs (n=4, 94801#-94804#) and WT pigs (n=15).
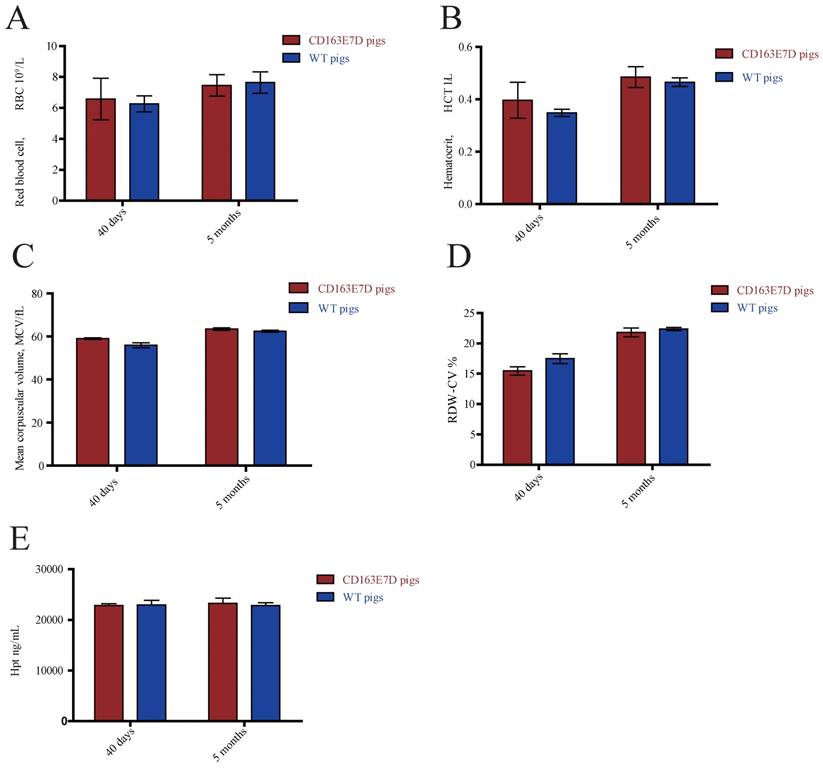
CD163 exon 7 deletion does not influence its normal biological functions
The biological functions of WT CD163 include prompting erythroblast growth and mediating the removal of hemoglobin from the blood. To determine whether the deletion of exon 7 influenced its normal functions, blood samples of 6-week-old and 5-month-old pigs were collected and subjected to routine blood tests. As shown in Fig. 3A, B, C and D, no significant differences were observed in red blood cell counts, blood hematocrit levels, red blood cell distribution width, or mean corpuscular volumes between the WT and CD163E7D pigs. Moreover, free hemoglobin binds to haptoglobin to form a Hb-Hp complex, which interacts with the SRCR3 domain of CD163. Thus, the serum of above pigs was separated to detect the Hp levels by ELISA (Fig. 3E). The results showed there were no differences in Hp levels between the WT and CD163E7D pigs. These results demonstrated that the CD163 exon 7 deletion had no adverse effects on its normal biological functions.
CD163 exon 7 deletion does not influence its normal biological functions. The blood samples of CD163E7D (n=5) and WT pigs (n=5) were collected for the test at the ages of 40 days and 5 months. (A) The red blood counts of the CD163E7D and WT pigs. (B) The hematocrit level of the CD163E7D and WT pigs. (C) The mean corpuscular volumes of the CD163E7D and WT pigs. (D) The red blood cell distribution width of the CD163E7D and WT pigs. (E) The serum Hp concentrations of the CD163E7D and WT pigs. All data are presented as the mean±SD.
CD163 exon7 deleted pigs are resistant to HP-PRRSV
To test whether the CD163 exon 7 deletion can protect pigs from the viral challenge in vivo, 8-week old CD163E7D pigs and WT Large White pigs were challenged with a highly pathogenic PRRSV strain, JXA1 (Table S2). The serum of all the tested pigs was negative for protein N and PRRSV-specific antibody before the viral challenge (Fig. S3 and S4). The viral challenge had lasted for 21 days, and clinical characteristics, including rectal temperatures, overall body condition, and respiratory disorders, were recorded daily until the animal death or the end of the challenge.
During the course of the disease, WT pigs started to show mild symptoms, such as fever and sternutation, at 2-5 days post inoculation (DPI). Then the signs continued and worsened gradually: the pigs exhibited respiratory disorders, lameness, diarrhea, and inappetence at 10-14 DPI. The most severe pigs presented with the blueing of ears and extremities, respiratory failure, ataxia, muscle paralysis, nystagmus, and other neurological symptoms. Some of the WT pigs died at this time, and other pigs recuperated and survived, but still exhibited some symptoms. Symptom scores were used to evaluate the disease course. The symptom scores of the challenged WT pigs and the pigs housed with them arose the earliest at 2 DPI, while the CD163E7D pigs only showed very mild symptoms at the later time. The symptoms of WT pigs became the most severe at 12 DPI, and some WT pigs started to die at 11 DPI (Fig. 4A). The surviving WT pigs still had mild symptoms at the end of the challenge. However, the mild signs of CD163E7D pigs disappeared at 16 DPI, and all the pigs were healthy without relapse at the end of the challenge (Fig. 4A). The WT pigs housed with the challenged CD163E7D pigs started to show symptoms the latest (Fig. 4A). One of them died at 20 DPI, and the others started to recover before the end of the challenge (Fig. 4A). The rectal temperature changes were similar to the symptom scores in all the groups of pigs. The challenged WT pigs and the pigs housed together with them had a fever as early as 2-4 DPI. Their rectal temperature increased to 41°C at 10-14 DPI. The surviving pigs recovered from fever at the end of the challenge. The challenged CD163E7D pigs had fevers later than WT pigs at 5 DPI, and their rectal temperatures were never higher than 41℃ during the entire process. The housed pigs had fever the latest, and they showed most severe fever between 13-19 DPI (Fig. 4B). At the same time, the surviving challenged WT pigs and their housed pigs had recovered from high fever. The results showed the symptoms and fever had been postponed, but not more severe in the WT pigs housed with the challenged CD163E7D pigs.
CD163 exon 7 deleted pigs are resistant to HP-PRRSV. (A) Symptom scores of the challenged WT and CD163E7D pigs, as well as the pigs housed with them, during the viral challenge. (B) Rectal temperature curves of the tested pigs during the viral challenge. (C) Survival curves of the tested pigs. (D) Macroscopic lesion of lungs, tonsils, spleen, lymph nodes, and kidney of the pigs of four severity categories (see Table 3). Only representative pictures were shown. Arrow1 indicates a lung hemorrhage and parenchymal lesion with secondary bacterial infection; arrows 2, 6, and 9 indicate lesions of lymph node hemorrhage; arrow 3 indicates a lesion of spleen infraction with secondary bacterial; arrow 4 indicates a lesion of kidney with blood spot; arrows 5 and 8 indicate lung hemorrhage lesions; arrow 7 indicates a lesion of spleen infraction.
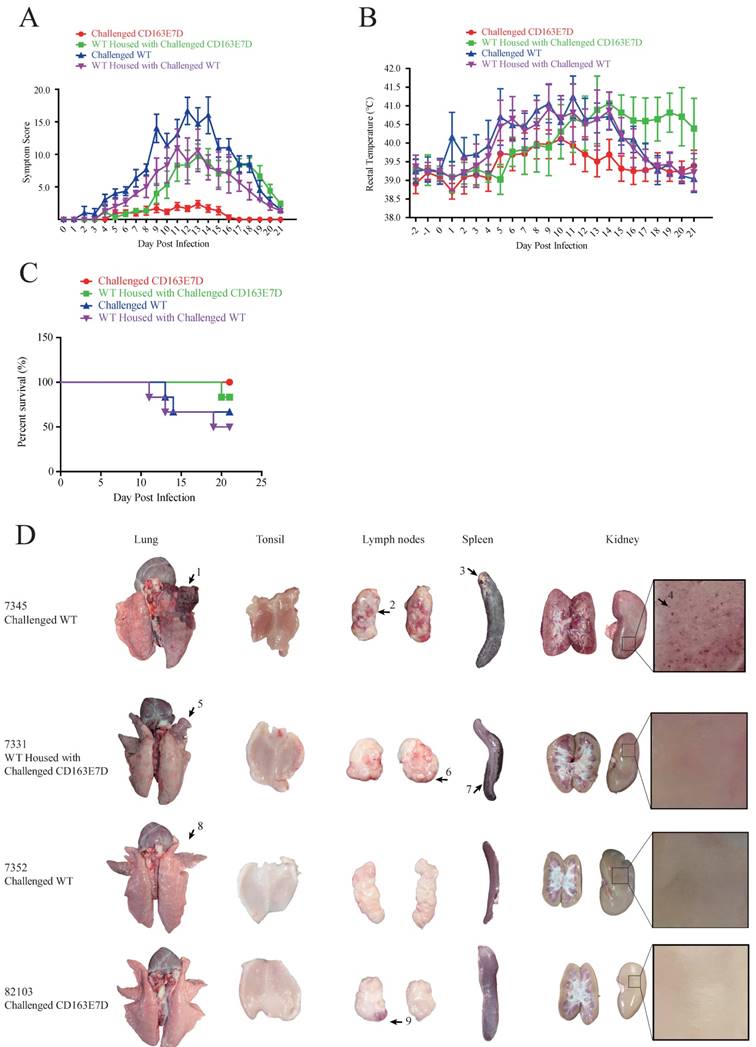
All the CD163E7D pigs survived the viral challenge, while 66.6% challenged WT pigs survived. The WT pigs housed with the challenged CD163E7D pigs had an 83.3% survival rate, while the survival rate of non-challenged WT pigs housed with challenged WT pigs was only 50% (Fig. 4C).
There were much less macroscopic lesions in CD163E7D pigs
After the viral challenge, the animals were euthanized, and the macroscopic lesions and pathology of their organs were examined. The CD163E7D pigs had almost no visible lesions in the tissues and organs. Only a slight lymph node hemorrhage was observed in CD163E7D pig #82103 (Fig. 4D), which showed the most severe symptoms in this group, other CD163E7D pigs had no visible lesions. WT pigs in other groups showed more serious lesions. Pulmonary parenchymal lesions, lung edema, lung hemarrhage, spleen infarct, renal hemorrhage, lymphatic hemorrhage, and secondary bacterial infection were common among the dead WT pigs. The detailed lesions in different treatment groups were listed in Table S3. The WT pigs housed with the challenged CD163E7D pigs had fewer lesions than those housed with challenged WT pigs (Table 3).
Macroscopic lesions of the challenged pigs and the pigs housed with thema.
| Groups | Lung hemorrhage | lung edema | Pulmonary parenchymal lesion | spleen infract | kidney with blood spot | secondary bacterial infection | lymph node hemorrhage | tonsil hemorrhage | Status before euthanasia |
|---|---|---|---|---|---|---|---|---|---|
| Challenged CD163E7D | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | All survived |
| WT housed with challenged CD163E7D | 1 | 1 | 6 | 4 | 1 | 1 | 6 | 1 | 1 died, 5 survived |
| Challenged WT | 2 | 2 | 5 | 3 | 2 | 1 | 6 | 4 | 2 died, 4 survived |
| WT housed with challenged WT | 3 | 3 | 6 | 3 | 3 | 1 | 6 | 3 | 3 died, 3 survived |
a The numbers of pigs that had the lesion in the group (n=6) were listed in the columns.
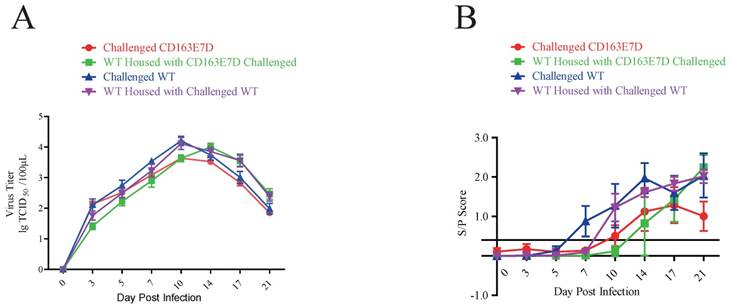
The CD163E7D pigs had less viral infection
Serum PRRSV viremia of the tested pigs was determined by TCID50 assay using Marc45 cells (Fig. 5A). A similar result to the symptom scores was observed. For WT pigs, the mean viremia titer increased to the peak steadily at 10-14 DPI after the challenge. However, the blood viral titers of CD163E7D pigs, which were never higher than the challenged WT pigs, increased more slowly and declined to a low level eventually. The rise of blood viral titer was delayed in the 2 non-challenged WT groups housed with either the challenged WT or CD163E7D pigs. The serum antibody levels were detected with ELISA. As a result, the challenged CD163E7D pigs had a lower PRRSV protein N antibody level than WT pigs, and the antibody levels of non-challenged pigs housed with challenged pigs were delayed compared with their challenged counterparts (Fig. 5B). Immunohistochemistry of PRRSV nucleocapsid protein also showed similar results: the viral protein signals were stronger and more intense in the lungs, spleens, tonsils and brains of most WT pigs, but positive signals were only observed in the tonsil of one CD163E7D pig (Table 4, Fig. S5).
The CD163E7D pigs had less viral infection. (A) Serum virus loads of all tested pigs determined by TCID50 assay. (B) The amounts of serum antibody against PRRSV nucleocapsid (N) protein of the tested pigs during the challenge.
Results of PRRSV immunohistochemistry staining.
| Group | Pig ID | Tonsila,b | Lung | Lymph Node | Spleen | Kidney | Brain |
|---|---|---|---|---|---|---|---|
| Challenged WT | 7345 (Died at 13 dpi) | + | + | + | - | - | + |
| 7498 (Died at 14 dpi) | + | + | - | - | - | - | |
| 7593 | + | - | - | - | - | - | |
| WT pigs housed with Challenged WT | 7434 (Died at 11 dpi) | - | + | - | - | - | - |
| 7399 (Died at 14 dpi) | + | + | - | - | - | + | |
| 7648 (Died at 19 dpi) | - | + | + | - | - | - | |
| Challenged CD163E7D | 81601 | - | - | - | - | - | - |
| 81602 | - | - | - | - | - | - | |
| 81603 | - | - | - | - | - | - | |
| 82101 | + | - | - | - | - | - | |
| 82102 | - | - | - | - | - | - | |
| 82103 | - | - | - | - | - | - | |
| WT pigs housed with challenged CD163E7D | 7589 | + | - | - | - | - | - |
| 7592 | + | - | - | - | - | - | |
| 7530 | ++ | + | ++ | - | - | - | |
| 7591 (Died at 20 dpi) | - | + | ++ | - | - | - |
a “+”, “++”: PRRSV positive.
b “-”: PRRSV negative.
Discussion
In this study, CD163 exon 7 precisely deleted pigs were generated with CRISPR/Cas9 mediated homologous recombination and somatic cells nuclear transfer. The genome editing of CD163 had no observable influences on the normal biological functions associated with the gene, such as erythroblast growth and Hb-Hp complex clearance. When the pigs were challenged with HP-PRRSV in vivo, the clinical signs appeared mild and lasted shortly in the CD163E7D pigs compared with the WT pigs. The WT pigs housed with the challenged CD163E7D pigs exhibited symptoms the latest and had a higher survival rate than the pigs housed with the challenged WT ones. Our results suggest that the CD163E7D pigs are resistant to HP-PRRSV infection without influencing its normal functions.
Genome editing of CD163 is not the only way to prevent PRRSV infection. However, traditional approaches based on vaccines or medicines were difficult to control PRRSV due to the diversity of the virus. Early studies primarily aimed at preventing PRRSV replication in cells to protect pigs against PRRSV. A study used RNA interference to silence viral gene expression in order to inhibit PRRSV replication in vitro [40]. Another study showed that adenoviruses carrying short-hairpin RNAs (shRNAs) against PRRSV genome could deliver the shRNA efficiently and inhibit PRRSV from spreading in vitro and in vivo [41]. PRRSV duplication was reduced in transgenic F1 pigs expressing PRRSV-specific siRNA compared with none-transgenic pigs, demonstrating the consistency of the RNAi-based approaches [42].
Different from preventing PRRSV replication, the genome-editing approach targets the PRRSV infection process mediated by the cell surface receptors of viral hosts. As an interacting protein of PRRSV, CD169 was considered to be an important determinant for PAM permissiveness of PRRSV infection. However, knockout of CD169 had not increased the pigs' resistance to PRRSV, and the CD169-/- pigs had the same disease course as the WT pigs after viral challenge [43]. In contrast, CD163-modified pigs from different origins were prominently resistant to genotype 1 and 2 PRRSVs [21]. CD163 consists of nine SRCR domains, and SRCR 5 was considered to be essential for PRRSV infection. To avoid the impairment of CD163 normal biological functions, genomic modifications of SRCR5 only are a better strategy than the knockout of entire CD163 gene. Thus far, substitution of SRCR5 with a homologous domain, missense point mutations in SRCR5, and knockout of CD163 SRCR 5 were used to obtain CD163 gene-editing pigs.
Recent studies showed that pigs in which SRCR5 was substituted with human homologous CD163L1 SRCR8 were resistant to the infection of genotype 1 PRRSV, but the animals were still permissive to the infection of genotype 2 viruses [32]. In Burkard's studies, SRCR 5 of CD163 was knocked out using two sgRNAs flanking exon 7 of CD163 in pig zygotes. presented resistance to both type 1 and type 2 PRRSV in vitro [44]. In their latest study, the CD163 SRCR5 null pigs were conferred resistant to the type 1 PRRSV, but the animal's resistance to type 2 viruses was not tested [33]. Using more sgRNAs for gene editing increased the possibility of off-targeting, and microinjection of sgRNAs might result in mosaicism. In the present study, we used CRISPR/Cas9 mediated homologous recombination to edit the CD163 exon 7 locus in porcine fetal fibroblasts and further used nuclear transfer to generate homozygous CD163 exon 7 deleted pigs. No off-target mutations were detected in the genome of the donor colonies. The viral challenge of the CD163E7D pigs with HP-PRRSV showed the pigs are resistant to the viruses.
As a cell surface marker, CD163 was not only identified as a receptor of viruses, such as PRRSV and ASFV [45], but also involved in erythroblast adhesion, clearing of the Hb-Hp complexes, and potential anti-inflammatory activity [24]. The precise deletion of SRCR5 in pigs may have advantages over the CD163 knockout animals described previously, which were randomly introduced a premature stop codon in the exon 3 [21] or exon 7 [31]. The CD163 SRCR 3 domain plays a vital role in clearance of Hb-Hp complexes [46], and the SRCR 2 domain is an adhesion receptor for erythroblasts [47]. Thus, it is necessary to examine the functions of CD163 in SRCR5 null pigs. Our results showed the exon 7 deletion does not influence the normal functions of CD163. The effects of exon 7 deletion on animal growth and reproductivity need to be evaluated in future studies.
In the viral challenge experiment, although the symptoms of CD163E7D pigs (only transient fever were observed) were mild, the CD163E7D pigs were still infected by PRRSV as shown by the viremia titers. This observation is different from Burkard's study [33], in which no PRRSV infection was observed. We speculate that the different PRRSV strains used in the 2 studies are the primary reason. In Burkard's study, the subtype strains of PRRSV-1 BOR-57 were inoculated intranasally into the pigs. During the challenge, no respiratory or other severe symptoms were observed except decreased demeanor in only one WT pig. The fever of WT pigs lasted shorter than our study, and no pigs died in the challenge. However, in our study, the pigs were inoculated with type 2 HP-PRRSV by intramuscular injection. This strain was isolated from the infected pigs in China in 2006, and its nsp2 region had a unique 30-amino acid deletion. The WT pigs had sustained fever, respiratory symptoms, and behavioral abnormalities, and some pigs died because of the disease. Differences among the viral strains in infectivity are a possible reason for the various clinical signs. Moreover, in our previous study, SRCR5-substituted pigs were also generated and challenged with HP-PRRSV strains [48]. The results showed the PAMs from the modified CD163 were completely protected from the viral infection in vitro, but the pigs showed a transient viremia after the infection, and one pig died in the viral challenge. In vivo responses of the animals are more complicated than the cultured cells in vitro. Other types of monocytes/macrophages cells, such as bone marrow-derived macrophages, pulmonary interstitial or splenic macrophages, may also be infected by PRRSV in vivo [49]. Other unknown reasons may exist and need further studies. In our study, these cell populations and other factors may have contributed to the lower infection in the CD163E7D pigs, which requires further studies.
Furthermore, we also set up two pig groups that were housed with either the challenged WT pigs or the challenged CD173E7D pigs. Although one died at the end of challenge experiment, the WT pigs housed with the challenged CD163E7D pigs showed delayed and milder clinical signs, and the survival rate of this group was also higher compared with the WT pigs housed with the challenged WT pigs. We speculate that the failure of PRRSV infection into the host cells of the CD163E7D pigs reduced virus replication and inhibited viral spreading. The result demonstrates that the CD163E7D pigs can effectively suppress infection of PRRSV in a herd.
In conclusion, we generated CD163 exon 7 deleted pigs by CRISPR/Cas9 mediated homologous recombination and SCNT. In the viral challenge experiment of HP-PRRSV, the CD163E7D pigs had less viral infection and recovered rapidly and were able to reduce the mortality in the herd. Meanwhile, the deletion of CD163 exon 7 does not impair the biological functions associated with CD163.
Supplementary Material
Supplementary figures and tables.
Abbreviations
CRISPR: Clustered regularly interspaced short palindromic repeats; DC-SIGN: dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin; DMEM: Dulbecco's modified Eagle's medium; FBS: fetal bovine serum; DPI: day post inoculation; gRNA: guide RNA; Hb: Hemoglobin; Hp: haptoglobin; HP-PRRSV: highly pathogenic porcine reproductive and respiratory syndrome virus; HS: heparin sulphate; OTSs: off-target sites; PAM: protospacer adjacent motif; PAMs: porcine alveolar macrophages; PFFs: porcine fetal fibroblasts; PRRS: porcine reproductive and respiratory syndrome; PRRSV: porcine reproductive and respiratory syndrome virus; SCNT: somatic cell nuclear transfer; SN: sialoadhesin; SRCR: scavenger receptor cysteine-rich; WT: wild-type.
Acknowledgements
We thank Zhaoliang Liu for reading the early version of the manuscript and comments provision. We also thank the staffs of National Research Center for Veterinary Medicine for their help with the viral challenge experiment. This work was supported by National Transgenic Breeding Project of China (Project Grant No. 2016ZX08009003006 and No. 2011ZX08006001), the Open Research Program of State Key Laboratory for Agro-Biotechnology (Project Grant No. 2017SKLAB1-7) and the 111 Project (Project Grant No. B12008).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Keffaber K. Reproductive failure of unknown etiology. Am Assoc Swine Pract Newsl. 1989;1:1-9
2. Lunney JK, Benfield DA, Rowland RR. Porcine reproductive and respiratory syndrome virus: an update on an emerging and re-emerging viral disease of swine. Virus Res. 2010;154:1-6
3. Wensvoort G, Terpstra C, Pol JM. et al. Mystery swine disease in The Netherlands: the isolation of Lelystad virus. Vet Q. 1991;13:121-130
4. Collins JE, Benfield DA, Christianson WT. et al. Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J Vet Diagn Invest. 1992;4:117-126
5. Karniychuk UU, Saha D, Geldhof M. et al. Porcine reproductive and respiratory syndrome virus (PRRSV) causes apoptosis during its replication in fetal implantation sites. Microb Pathogenesis. 2011;51:194-202
6. Pejsak Z, Stadejek T, Markowska-Daniel I. Clinical signs and economic losses caused by porcine reproductive and respiratory syndrome virus in a large breeding farm. Vet Microbiol. 1997;55:317-322
7. Christianson WT, Joo HS. Porcine reproductive and respiratory syndrome: A review. J Swine Health Prod. 1994;2:10-28
8. Holtkamp DJ, Kliebenstein JB, Neumann EJ. et al. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod. 2013;21:72-84
9. Tian K, Yu X, Zhao T. et al. Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS One. 2007;2:e526
10. Tong GZ, Zhou YJ, Hao XF. et al. Highly pathogenic porcine reproductive and respiratory syndrome. China Emerg Infect Dis. 2007;13:1434-1436
11. Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol. 1997;142:629-633
12. Meulenberg JJ, Hulst MM, de Meijer EJ. et al. Lelystad virus belongs to a new virus family, comprising lactate dehydrogenase -elevating virus, equine arteritis virus, and simian hemorrhagic fever virus. Arch Virol Suppl. 1994;9:441-448
13. Meulenberg JJ, Petersen-den BA. Identification and characterization of a sixth structural protein of Lelystad virus: the glycoprotein GP2 encoded by ORF2 is incorporated in virus particles. Virology. 1996;225:44-51
14. Wieringa R, de Vries AA, Rottier PJ. Formation of disulfide-linked complexes between the three minor envelope glycoproteins (GP2b, GP3, and GP4) of equine arteritis virus. J Virol. 2003;77:6216-6226
15. Van Nieuwstadt AP, Meulenberg JJ, van Essen-Zanbergen A. et al. Proteins encoded by open reading frames 3 and 4 of the genome of Lelystad virus (Arteriviridae) are structural proteins of the virion. J Virol. 1996;70:4767-4772
16. Duan X, Nauwynck HJ, Pensaert MB. Virus quantification and identification of cellular targets in the lungs and lymphoid tissues of pigs at different time intervals after inoculation with porcine reproductive and respiratory syndrome virus (PRRSV). Vet Microbiol. 1997;56:9-19
17. Kim HS, Kwang J, Yoon IJ. et al. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch Virol. 1993;133:477-483
18. Jusa ER, Inaba Y, Kouno M. et al. Effect of heparin on infection of cells by porcine reproductive and respiratory syndrome virus. Am J Vet Res. 1997;58:488-491
19. Calvert JG, Slade DE, Shields SL. et al. CD163 expression confers susceptibility to porcine reproductive and respiratory syndrome viruses. J Virol. 2007;81:7371-7379
20. Duan X, Nauwynck HJ, Favoreel HW. et al. Identification of a putative receptor for porcine reproductive and respiratory syndrome virus on porcine alveolar macrophages. J Virol. 1998;72:4520-4523
21. Whitworth KM, Rowland RR, Ewen CL. et al. Gene-edited pigs are protected from porcine reproductive and respiratory syndrome virus. Nat Biotechnol. 2016;34:20-22
22. Burkard C, Lillico SG, Reid E. et al. Precision engineering for PRRSV resistance in pigs: Macrophages from genome edited pigs lacking CD163 SRCR5 domain 463 are fully resistant to both PRRSV genotypes while maintaining biological function. PloS Pathog. 2017;13:e1006206
23. Van Gorp H, Van Breedam W, Delputte PL. et al. Sialoadhesin and CD163 join forces during entry of the porcine reproductive and respiratory syndrome virus. J Gen Virol. 2008;89:2943-2953
24. Kristiansen M, Graversen JH, Jacobsen C. et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409(6817):198-201
25. Law SK, Micklem KJ, Shaw JM. et al. A new macrophage differentiation antigen which is a member of the scavenger receptor superfamily. Eur J Immunol. 1993;23:2320-2325
26. Van Gorp H, Delputte PL, Nauwynck HJ. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol Immunol. 2010;47:1650-1660
27. Calvert JG, Slade DE, Shields SL. et al. CD163 expression confers susceptibility to porcine reproductive and respiratory syndrome viruses. J Virol. 2007;81:7371-7379
28. Ma H, Jiang L, Qiao S. The Crystal Structure of the Fifth Scavenger Receptor Cysteine-Rich Domain of Porcine CD163 Reveals an Important Residue Involved in Porcine Reproductive and Respiratory Syndrome Virus Infection. J Virol. 2016;91:JVI.01897-16
29. Chen J, Liu T, Zhu CG. et al. Genetic variation of Chinese PRRSV strains based on ORF5 sequence. Biochem Genet. 2006;44:425-35
30. Stadejek T, Oleksiewicz MB, Potapchuk D. et al. Porcine reproductive and respiratory syndrome virus strains of exceptional diversity in eastern Europe support the definition of new genetic subtypes. J Gen Virol. 2006;87:1835-41
31. Yang H, Zhang J, Zhang X. et al. CD163 knockout pigs are fully resistant to highly pathogenic porcine reproductive and respiratory syndrome virus. Antivir Res. 2018:151
32. Wells KD, Bardot R, Whitworth KM. et al. Replacement of Porcine CD163 Scavenger Receptor Cysteine-Rich Domain 5 with a CD163-Like Homolog Confers Resistance of Pigs to Genotype 1 but Not Genotype 2 Porcine Reproductive and Respiratory Syndrome Virus. J Virol. 2017:91
33. Burkard C, Opriessnig T, Mileham AJ. et al. Pigs lacking the scavenger receptor cysteine-rich domain 5 of CD163 are resistant to PRRSV-1 infection. J Virol. 2018;92:JVI.00415-18
34. Zhang Q, Guo XK, Gao L. et al. MicroRNA-23 inhibits PRRSV replication by directly targeting PRRSV RNA and possibly by upregulating type I interferons. Virology. 2014;450-451:182-195
35. Zhang W, Yang H, Kong X. et al. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat Med. 2005;11:56-62
36. Zhang Y, Pan D, Sun X. et al. Production of porcine cloned transgenic embryos expressing green fluorescent protein by somatic cell nuclear transfer. Sci China Ser C. 2006;49:164-71
37. Halbur PG, Paul PS, Meng XJ. et al. Comparative pathogenicity of nine US porcine reproductive and respiratory syndrome virus (PRRSV) isolates in a five-week-old cesarean-derived, colostrum-deprived pig model. J Vet Diagn Invest. 1996;8:11-20
38. Halbur P, Thanawongnuwech R, Brown G. et al. Efficacy of antimicrobial treatments and vaccination regimens for control of porcine reproductive and respiratory syndrome virus and Streptococcus suis coinfection of nursery pigs. J Clin Microbiol. 2000;38:1156-60
39. Zhang C, Guo L, Jia X. et al. Construction of a triple gene-deleted Chinese Pseudorabies virus variant and its efficacy study as a vaccine candidate on suckling piglets. Vaccine. 2015;33:2432-7
40. He YX, Hua RH Zhou YJ. et al. Interference of porcine reproductive and respiratory syndrome virus replication on MARC-145 cells using DNA-based short interfering RNAs. Antivir Res. 2007;74:83-91
41. Li G, Jiang P, Li Y. et al. Inhibition of porcine reproductive and respiratory syndrome virus replication by adenovirus-mediated RNA interference both in porcine alveolar macrophages and swine. Antivir Res. 2009;82:157-165
42. Li L, Li Q, Bao Y. et al. RNAi-based inhibition of porcine reproductive and respiratory syndrome virus replication in transgenic pigs. J Biotechnol. 2014;171:17-24
43. Prather RS, Rowland RR, Ewen C. et al. An intact sialoadhesin (Sn/SIGLEC1/CD169) is not required for attachment/internalization of the porcine reproductive and respiratory syndrome virus. J Virol. 2013;87:9538-9546
44. Burkard C, Lillico SG, Reid E. et al. Precision engineering for PRRSV resistance in pigs: Macrophages from genome edited pigs lacking CD163 SRCR5 domain are fully resistant to both PRRSV genotypes while maintaining biological function. PloS Pathog. 2017;13:e1006206
45. Sanchez EG, Quintas A, Perez-Nunez D. et al. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog. 2012;8:e1002754
46. Madsen M, Moller HJ, Nielsen MJ. et al. Molecular characterization of the haptoglobin center dot hemoglobin receptor CD163: ligand binding properties of the scavenger receptor cysteine-rich domain region. J Biol Chem. 2004;279:51561-7
47. Barbe E, Huitinga I, Dopp EA. et al. A novel bone marrow frozen section assay for studying hematopoietic interactions in situ: the role of stromal bone marrow macrophages in erythroblast binding. J Cell Sci. 1996;109:2937-45
48. Chen J, Wang H, Bai J. et al. Generation of Pigs Resistant to Highly Pathogenic-Porcine Reproductive and Respiratory Syndrome Virus through Gene Editing of CD163. Int J Biol Sci. 2019;15:481-492
49. Chaudhuri S, McKenna N, Balce D. et al. Infection of porcine bone marrow-derived macrophages by porcine respiratory and reproductive syndrome virus impairs phagosomal maturation. J Gen Virol. 2016;97:669-679
Author contact
![]() Corresponding authors: Ning Li (ninglcauedu.cn), Xiaoxiang Hu (huxxedu.cn) and Kegong Tian (tiankgnet).
Corresponding authors: Ning Li (ninglcauedu.cn), Xiaoxiang Hu (huxxedu.cn) and Kegong Tian (tiankgnet).