Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(10):2087-2099. doi:10.7150/ijbs.27231 This issue Cite
Research Paper
Ciliary IFT80 regulates dental pulp stem cells differentiation by FGF/FGFR1 and Hh/BMP2 signaling
Xue Yuan1, Min Liu2, Xu Cao3, Shuying Yang1,2 ![]()
1. Department of Oral Biology, School of Dental Medicine, University of Buffalo, State University of New York, Buffalo, NY, United States
2. Department of Anatomy & Cell Biology, School of Dental Medicine, University of Pennsylvania, PA, United States
3. Department of Orthopedic Surgery, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Received 2018-5-13; Accepted 2019-2-19; Published 2019-8-6
Abstract

Primary cilia and intraflagellar transport (IFT) proteins control a wide variety of processes during development and tissue homeostasis. However, their potential roles in the regulation of stem cell differentiation and tooth development remain elusive. Here, we uncovered the critical roles of ciliary IFT80 in cilia formation and differentiation of dental pulp stem cells (DPSCs). IFT80-deficient DPSCs showed reduced fibroblast growth factor receptor 1 (FGFR1) expression, leading to the disruption of FGF2-FGFR1 signaling. We found, during DPSC differentiation, FGF2-FGFR1 signaling induces stress fiber rearrangement to promote cilia elongation, meanwhile stimulates PI3K-AKT signaling to aid Hh/bone morphogenetic protein 2 (BMP2) signaling activation. These signaling pathways and their coupling were disrupted in IFT80-deficient DPSCs, causing impaired differentiation. Our findings revealed a novel mechanism that ciliary protein regulates the odontogenic differentiation of DPSCs through FGF/FGFR1 and Hh/BMP2 signaling.
Keywords: Primary cilia, intraflagellar transport, dental pulp stem cells, fibroblast growth factor signaling, hedgehog signaling, bone morphogenetic protein
Introduction
Stem cells, capable of self-renewal and differentiation, are essential for tissue development and homeostasis [1]. Tooth development, like many other organs' formation, requires several types of stem cells, which are controlled by different signaling pathways.
Despite our extensive knowledge in tooth formation, very little is known about the stem cells in dental pulp, until Shi, Gronthos, and coworkers isolated dental pulp stem cells (DPSCs) from human dental pulp and defined their properties of clonogenic, self-renewing, rapidly proliferative, and multi-lineage differentiation ability [2, 3]. DPSCs give arise to odontoblasts, which are the columnar polarized cells located at the outer edges of dental pulp. Odontoblasts express dentin matrix protein 1 (DMP1) and dentin sialophosphoprotein (DSPP) and produce dentin. Although DPSCs and bone marrow-derived MSCs (BMMSCs) share similarities in the morphology and flow cytometry profiles, DPSCs are more efficient than BMMSCs in producing mineral in vitro [4]. Recently, DPSCs were identified and isolated from mouse incisors [5, 6], providing an opportunity to determine the signaling pathways that govern DPSC self-renewal and differentiation. To date, the understanding of the maintenance and differentiation of DPSCs are still elusive. Fibroblast growth factor (FGF) signaling was reported to play an essential role in dentinogenesis, and several studies supported that FGF2 promotes the pulp cell proliferation [7-10] and early differentiation [7]. However, it was not clear whether FGF2 signaling has a direct function in DPSCs.
Primary cilia are highly conserved microtubule-based organelles that are present on almost all vertebrate cells. Primary cilia were observed in both embryonic and adult stem cells, including DPSCs [11-13], and recent studies suggested that primary cilia regulate stem cell maintenance and lineage determination [12, 14-16]. However, none of them examined the roles of IFT proteins in DPSC self-renewal and differentiation.
Formation and function of primary cilia require intraflagellar transport (IFT) proteins along with other ciliary proteins [17, 18]. There are 22 identified IFT proteins that form two complexes, IFT-A and IFT-B [19, 20]. Mutation of those proteins usually causes cilia defects, and a wide range of diseases called ciliopathies. These disorders target multiple organs, of which bone and tooth are common ones [21-23]. IFT80 belongs to IFT-B complex and mutations of IFT80 have been reported in human Jeune asphyxiating thoracic dystrophy (JATD) and short rib polydactyly type III (SRPIII). Previously, we reported that IFT80 promotes osteogenesis [24] and deletion of IFT80 in osteoblast precursor cells leads to decreased bone mass with impaired osteoblast differentiation through regulating Hh signaling pathway [25]. In this study, we further revealed that deletion of ciliary IFT80 impairs cilia formation and DPSC differentiation via disrupting FGF/FGFR1 signaling and the coupling of FGF2 and Hh/BMP2 signaling.
Results
Primary cilia elongate during odontogenesis
Although primary cilia have been visualized on odontoblasts, it is largely unknown whether primary cilia are presented on pulp cells. We stained mouse molar sections with cilia marker acetylated α-tubulin and found that most of the pulp cells displayed primary cilia (Fig. 1A). To examine the functions of primary cilia in dental pulp cells, we isolated primary DPSCs from mouse incisors (Fig. S1A) and characterized the phenotype of DPSCs by cell morphology (Fig. S1B), cell surface markers (Fig. S1C), colony forming ability (Fig. S1D), proliferation rate (Fig. S1E), and multi-lineage differentiation ability (Fig. S1F).
Primary cilia in DPSCs and cilia elongation during differentiation. (A) Immunofluorescence analysis of primary cilia in molar sections. Primary cilia were stained with acetylated α-tubulin (green) antibody. DAPI was used for counterstaining. (B) Immunofluorescence analysis of primary cilia on cultured DPSCs. Primary cilia were stained with acetylated α-tubulin (green) antibody. DAPI staining was used as counterstaining. (C) SEM of primary cilia in DPSCs. (D) Immunofluorescence analysis of primary cilia in CiliaGFP;IFT80+/+ DPSCs during odontogenesis. White dot circle represents the nucleus. Scale bars represent 5 μm. (E) Calculated cilia length (n = 20 cells). (F) Calculated cilia percentage (n = 3 with at least 200 cells analyzed). (G) qPCR analysis of IFT80, IFT144 and IFT88 expression during DPSC odontogenesis. The expression level of target genes was normalized to GAPDH expression (n = 3, triplicates per group). Data are expressed as mean ± SEM; ns, not statistically significant; *p < 0.05; ***p < 0.00001.
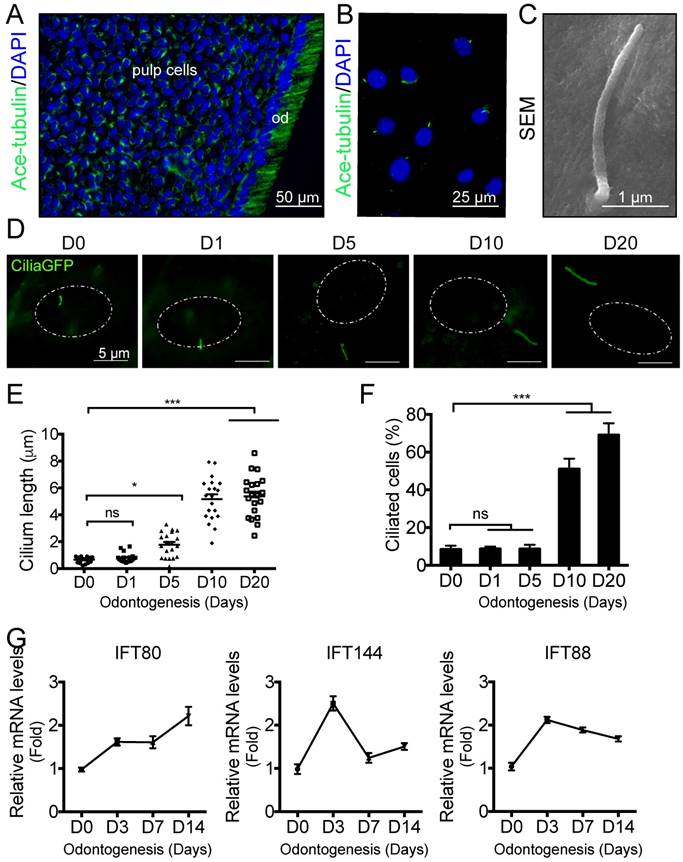
To visualize primary cilia, we isolated DPSCs from CMV;CiliaGFP mice, in which, primary cilia were labeled with GFP. Primary cilia were presented on DPSCs (Fig. 1B), and the average length is about 2 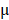 m based on SEM analysis (Fig. 1C). During in vitro induced odontogenesis, the length of primary cilia increased (Fig. 1D). Quantification showed that cilia length was significantly longer at day 5 after odontogenic differentiation compared with non-induced ones (Fig. 1E). At day 10, the cilia length reached a peak and any longer inducing time, e.g., 20 days, did not significantly change the cilia length (Fig.1E). We also quantified the percentage of ciliated cells. Before odontogenic inducing, the ciliated cell population was about 10%. This ratio remained the same with 5-day odontogenic induction (Fig. 1F). By day 10, the ciliated cell percentage reached to 50%, and this number increased to 70% by day 20 (Fig.1F). These data suggested that primary cilia might be involved in the odontogenic differentiation of DPSCs.
m based on SEM analysis (Fig. 1C). During in vitro induced odontogenesis, the length of primary cilia increased (Fig. 1D). Quantification showed that cilia length was significantly longer at day 5 after odontogenic differentiation compared with non-induced ones (Fig. 1E). At day 10, the cilia length reached a peak and any longer inducing time, e.g., 20 days, did not significantly change the cilia length (Fig.1E). We also quantified the percentage of ciliated cells. Before odontogenic inducing, the ciliated cell population was about 10%. This ratio remained the same with 5-day odontogenic induction (Fig. 1F). By day 10, the ciliated cell percentage reached to 50%, and this number increased to 70% by day 20 (Fig.1F). These data suggested that primary cilia might be involved in the odontogenic differentiation of DPSCs.
It is well established that IFT proteins are involved in primary cilia formation. We then asked whether the process of odontogenesis would change IFT protein expression. Using qPCR, we found that the expression of IFT80 was increased along with differentaition (Fig. 1G). However, not all the IFT proteins showed the same pattern. For example, both IFT144 and IFT80 showed the highest expression at day 3 but the expression decreased at a later stage (Fig.1G), suggesting IFT proteins may play different roles in odontogenic differentiation. In this study, we focused on the roles of IFT80 and primary cilia in the odontogenic differentiation of DPSCs.
Deletion of IFT80 in DPSCs causes cilia loss and odontogenesis defect
To further examine the role of IFT80 in DPSCs, we isolated primary DPSCs from IFT80f/f mice and then infected with adenovirus expressing Cre recombinase to delete IFT80 (named as IFT80d/d). Adenovirus expressing the green fluorescent protein (GFP) was used as the control, and the GFP-infected IFT80f/f DPSCs were still marked as IFT80f/f. Western blot confirmed Cre adenovirus transduction significantly reduced IFT80 expression in DPSCs (Fig. 2A).
Deletion of IFT80 disrupts cilia formation and odontogenic differentiation. (A) Western blot analysis of IFT80 expression in IFT80f/f and IFT80d/d DPSCs. IFT80 protein level was normalized to GAPDH (n = 3). (B) Immunofluorescence analysis of primary cilia in cultured DPSCs. Primary cilia were stained with acetylated α-tubulin (green) antibody. DAPI staining was used as counterstaining. Scale bars represent 20 μm. Cilia length (n = 20 cells) and cilia percentage (n = 3 with at least 200 cells analyzed) were calculated. (C) qPCR analysis of DMP1 and DSPP expression in IFT80f/f and IFT80d/d DPSCs induced with OS medium for 7 days (n = 3, triplicates per group). Target gene expression was normalized to GAPDH (n = 3, triplicates per group). (D) ALP staining of IFT80f/f and IFT80d/d DPSCs at day 7 of OS induction (n = 3, triplicates per group). (E) Alizarin Red staining of IFT80f/f and IFT80d/d DPSCs induced with OS medium for 21 days (n = 3, triplicates per group). Data are expressed as mean ± SEM; ns, not statistically significant; **p < 0.01; ***p < 0.0001.
We first noticed that IFT80d/d DPSCs displayed a defect in cilia formation (Fig. 2B). Quantification results showed that the ciliated population in IFT80f/f was more than 80%, while it was less than 30% in IFT80d/d group. A small portion of DPSCs in the IFT80d/d group showed primary cilia, but the cilia were shorter than that in IFT80f/f group (Fig. 2B).
We next examined the odontogenic differentiation of DPSCs. To eliminate the effect of proliferation on differentiation, we performed all differentiation assays with a high cell density, at which both IFT80f/f and IFT80d/d DPSCs had a minimum proliferation rate (Fig. S2). The result showed that deletion of IFT80 in DPSCs impaired odontogenic differentiation as evidenced by reduced expression of odontoblast differentiation markers DMP1 and DSPP (Fig. 2C), less ALP activity (Fig. 2D) and decreased mineral nodules formation (Fig. 2E).
FGF2-FGFR1 signaling is blocked in IFT80-deficient DPSCs
Our next step was to identify the signaling pathway(s) that IFT80 mediated in DPSCs. FGF signaling regulates the differentiation of many types of cells including dental pulp cells [7, 26]. We tested FGFR1-3, FGF2, FGF8, and FGF9 expression in wild-type DPSCs and found that FGFR1 and FGF2 were highly expressed in DPSCs (Fig. 3A). To further examine the mechanism of IFT80 in the regulation of FGF signaling, we studied whether deletion of IFT80 affects the expression of FGFR and FGF. The results showed that the mRNA levels of FGFR1 and FGF2 were dramatically reduced in IFT80d/d DPSCs compared with these in IFT80f/f DPSCs (Fig. 3B). Interestingly, FGFR2 and FGFR3 expression were not significantly changed (Fig. 3B), suggesting FGF2-FGFR1 is a major FGF signaling affected by IFT80 deletion.
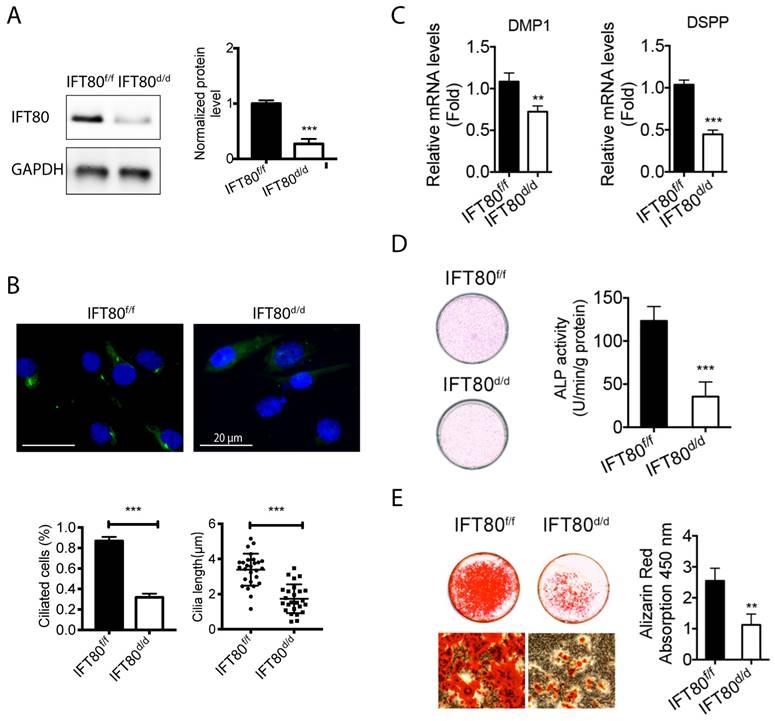
Deletion of IFT80 in DPSCs impairs FGF2-FGFR1 signaling. (A) qPCR analysis of FGFR1-3, FGF2, FGF8, and FGF9 expression in wild-type DPSCs. The target gene expression was normalized to GAPDH expression (n=3, triplicates per group). (B) qPCR analysis of FGF2, and FGFR1-3 expression in IFT80f/f and IFT80d/d DPSCs. The expression levels of FGF2 and FGFR1-3 were normalized to GAPDH expression (n = 3, triplicates per group). (C) ALP activity of IFT80f/f and IFT80d/d DPSCs at day 7 of OS induction with or without FGF2 (10 ng/mL) treatment for the first 3 days (n = 3, triplicates per group). (D) Alizarin Red staining of IFT80f/f and IFT80d/d DPSCs at day 14 of OS induction with or without FGF2 (10 ng/mL) treatment for the first 3 days (n = 3, triplicates per group). Data are expressed as mean ± SEM; ns, not statistically significant; ***p < 0.0001.
When IFT80f/f and IFT80d/d DPSCs were exposed to FGF2 in the early differentiation stage, we found that FGF2 significantly advanced differentiation in IFT80f/f DPSCs as tested by ALP activity and Alizarin Red staining (Figs. 3C and 3D). This promotion effect was completely abolished in cells without IFT80. These data suggested FGF2 signaling is essential for the differentiation of DPSCs, and IFT80 is involved in FGF2 signaling transduction.
To find out how IFT80 was involved in FGF2 signaling transduction, we first asked whether FGFR1, the receptor for FGF2, was located in primary cilia. We examined the location of both FGFR1 and phosphorylated FGFR1 in DPSCs. DPSCs were isolated from CMV;CiliaGFP mice, in which primary cilia is labeled with GFP. Immunostaining analysis showed that FGFR1 and the phosphorylation form of FGFR1 (p-FGFR1) in CMV;CiliaGFP DPSCs were not located in primary cilia with or without FGF2 stimulation (Fig. 4A), suggesting the reduced FGFR1 expression was not a direct result from cilia loss in IFT80d/d DPSCs. Immunostaining results also showed reduced FGFR1 expression in IFT80d/d DPSCs (Fig. 4A). We next used Western blot to confirm the decreased FGFR1 expression in IFT80-deficient DPSCs (Fig. 4B).
Deletion of IFT80 in DPSCs impairs the expression of FGFR1. (A) CMV;CiliaGFP DPSCs were treated with or without FGF2 (10 ng/mL) for 10 min and stained with FGFR1 antibody or p-FGFR1 antibody. The yellow arrows indicate primary cilia (green). DAPI staining was used as counterstaining. Scale bars represent 10 μm. (B) Western blot analysis of FGFR1 expression in IFT80f/f and IFT80d/d DPSCs. FGFR1 expression was normalized to β-actin (n = 3). (C) Western blot analysis of FGFR1 phosphorylation with FGF2 (10 ng/mL) stimulation in IFT80f/f and IFT80d/d DPSCs. FGFR1 phosphorylation level was normalized to FGFR1 or β-actin (n = 3). PD, PD173074 (1 μM, FGFR1 inhibitor). (D) Western blot analysis of ERK phosphorylation in IFT80f/f and IFT80d/d DPSCs. ERK phosphorylation level was normalized to ERK (n = 3). Data are expressed as mean ± SEM; ns, not statistically significant; *p < 0.05; ***p < 0.0001.
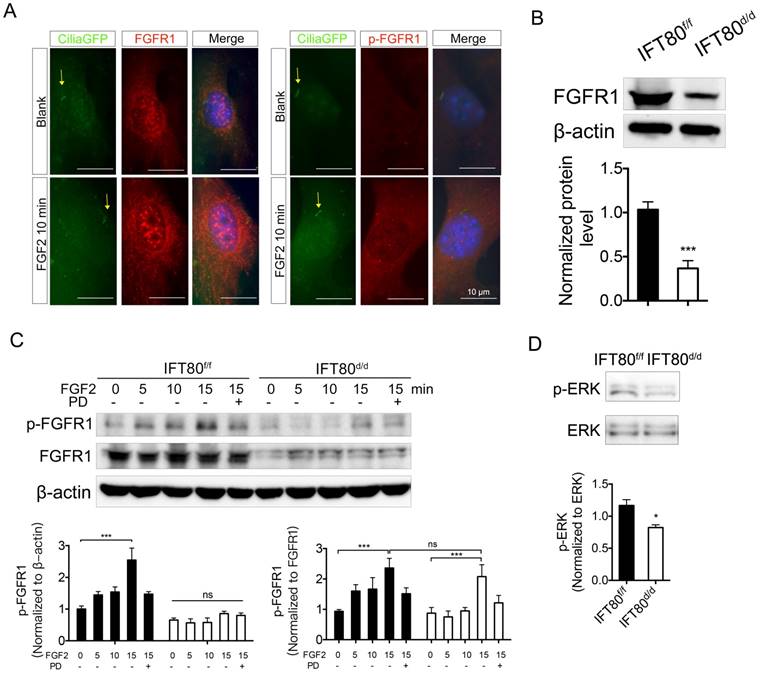
FGFR1 belongs to the transmembrane receptor tyrosine kinases (RTK) family. FGFs binding induces phosphorylation of tyrosine residues and activates downstream signaling pathways. We found phosphorylation of FGFR1 gradually increased in IFT80f/f DPSCs upon FGF2 stimulation (Fig. 4C). Pretreated with PD173074, a selective FGFR1 inhibitor, inhibited FGF2-induced FGFR1 phosphorylation in IFT80f/f DPSCs (Fig. 4C). In contrast, FGF2-induced FGFR1 phosphorylation level was much lower in IFT80d/d DPSCs when normalized to β-actin. Since IFT80d/d DPSCs also showed less FGFR1 expression, we also normalized FGFR1 phosphorylation to total FGFR1 expression. We found that the normalized phosphorylation of FGFR1 in IFT80d/d DPSCs was comparable to that in IFT80f/f DPSCs 15 min after FGF2 stimulation, suggesting FGFR phosphorylation was not blocked in IFT80d/d DPSCs. We also found that ERK phosphorylation level reduced, further confirmed the impaired FGFR1 signaling transduction in IFT80d/d DPSCs (Fig. 4D). Together, these data demonstrated that reduced FGFR1 expression is the primary cause of the FGF2-FGFR1 signaling defect in IFT80d/d DPSCs.
FGF2 promotes cilia elongation through actin reorganization in IFT80f/f DPSCs but not in IFT80d/d DPSCs
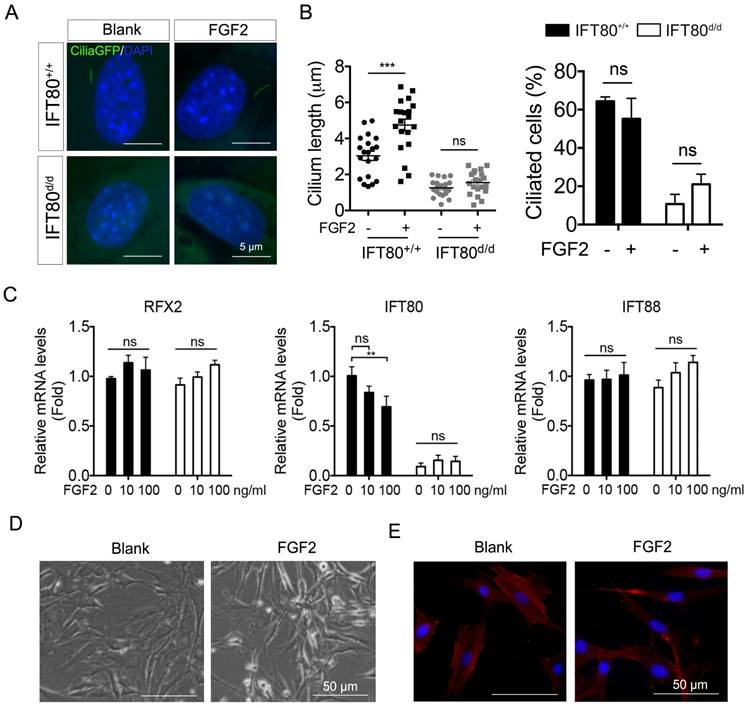
So far, we have showed that during odontogenesis, primary cilia elongated (Fig. 1) and FGF2 promoted odontogenic differentiation (Fig. 3). We next asked whether FGF2 promotes odontogenic differentiation by increasing cilia length. We analyzed cilia formation in FGF2-treated DPSCs. Cilia;IFT80+/+ and Cilia;IFT80f/f DPSCs were treated with Ad-CMV-Cre to turn on GFP expression on primary cilia. The results showed that FGF2 significantly induced cilia elongation in Cilia;IFT80+/+ DPSCs without affecting ciliated rate (Fig. 5A). As we expected, FGF2 failed to increase the cilia length and ciliated population in Cilia;IFT80d/d DPSCs (Fig. 5B), confirming IFT80 was essential for FGF2-induced cilia elongation. We further tested the expression of cilia-related transcription factor RFX2 and ciliary protein IFT80 and IFT88 (Fig. 5C). FGF2 stimulation did not promote the expression of these genes. In contrast, high dose of FGF2 slightly decreased IFT80 expression (Fig. 5C), suggesting that cilia elongation in FGF2 group was not associated with increased ciliary protein expression. Additionally, we noticed that FGF2-treated DPSCs became small and lost the polygonal cell morphology (Fig. 5D). Furthermore, we found the loss of stress fibers in FGF2 group (Fig. 5E), suggesting the role of FGF2 in actin cytoskeleton reorganization. In our previous study, we revealed that actin reorganization promotes cilia formation [25]. Taken these together, FGF promoted cilia elongation through actin reorganization, which consequently promotes differentiation.
FGF2 signaling promotes cilia elongation by changing stress fiber. (A) Analysis of primary cilia in CiliaGFP;IFT80+/+ and CiliaGFP;IFT80d/d DPSCs treated with or without FGF2 (10 ng/mL). DAPI staining was used as counterstaining. Scale bars represent 5 μm. (B) Calculated cilia length (n = 20 cells) and cilia percentage (n = 3 with at least 200 cells analyzed). (C) qPCR analysis of RFX2, IFT80 and IFT88 expression in DPSCs (n = 3, triplicates per group) with FGF2 (10 ng/mL) treatment. (D) Morphology of DPSCs cultured in medium with or without 10 ng/mL FGF2 for 24 hours. (E) DPSCs were stained for actin (red) with or without FGF2 (10 ng/mL) treatment. DAPI staining was used as counterstaining. Scale bars represent 50 μm. Data are expressed as mean ± SEM; ns, not statistically significant; **p < 0.001; ***p < 0.0001.
Deletion of IFT80 inhibits DPSC differentiation through inhibiting FGF signaling, Hh signaling and their crosstalk
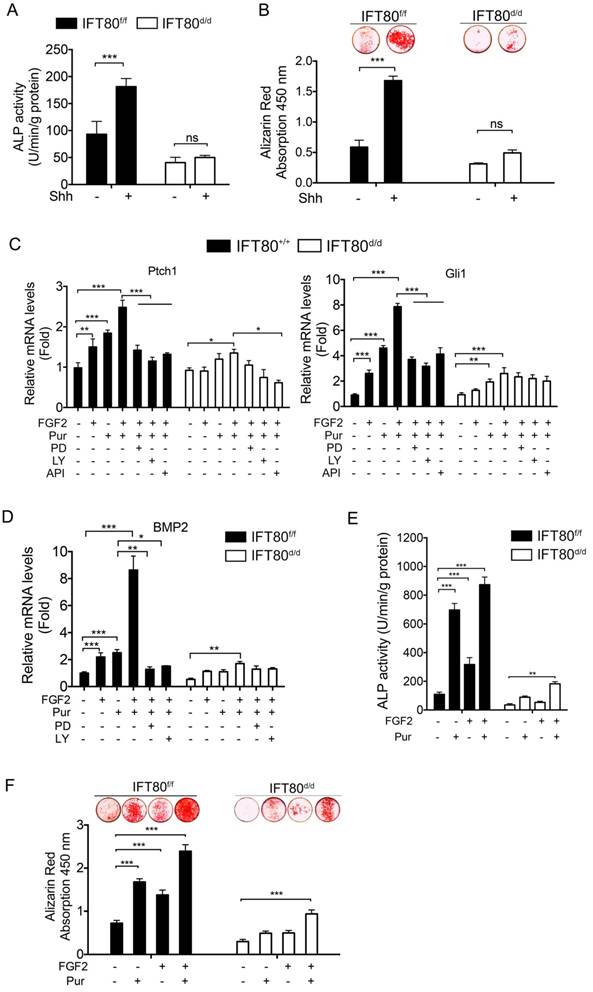
It is widely accepted that Hh signaling transduction highly relays on cilia, and Hh signaling is essential for osteogenic differentiation [27, 28]. Shh promoted differentiation of IFT80f/f DPSCs as examined by ALP activity and Alizarin Red staining, but Shh failed to rescue the differentiation defect in IFT80d/d DPSCs (Fig. 6A and 6B). Since FGF2 promoted cilia formation, we next analyzed whether FGF2 could intensify Hh signaling during differentiation. We found that FGF2 increased Ptch1 and Gli1 expression and this effect was enhanced when FGF2 was combined with Purmorphomine, a Hh signaling agonist, in IFT80f/f DPSCs (Fig. 6C). Inhibition of FGFR1 by PD173074 or inhibition of PI3K-AKT by LY294002 and API-2 significantly blocked FGF2-induced Gli1 and Ptch1 expression (Fig. 6C), suggesting FGF2 aided Hh signaling activation through regulating FGFR1 and PI3K-AKT pathway. We further found that FGF2 promoted BMP2 expression and strongly advanced Purmorphomine-induced BMP2 expression in IFT80f/f DPSCs (Fig. 6D). In consistence with these data, FGF2 plus Purmorphomine treatment great promoted differentiation of IFT80f/f DPSCs (Fig. 6E and 6F). In IFT80-deficient DPSCs, the synergistic effect of FGF signaling and Hh signaling was blocked (Fig. 6C-F), highlighting the critical role of IFT80 in the crosstalk of FGF signaling and Hh signaling.
Crosstalk between FGF2 and Hh/BMP2 signaling during DPSC differentiation is blocked in IFT80-deficient DPSCs. (A) ALP activity of IFT80f/f and IFT80d/d DPSCs at day 7 of OS induction with or without Shh (1 μg/mL) treatment (n = 3, triplicates per group). (B) Alizarin Red staining of IFT80f/f and IFT80d/d DPSCs at day 14 of OS induction with or without Shh (1 μg/mL) treatment (n = 3, triplicates per group). (C) qPCR results showing Ptch1 and Gli1 expression in IFT80f/f and IFT80d/d DPSCs. Cells were treated with FGF2 (10 ng/mL), Purmorphomine (Pur, 2 μM), PD173074 (PD, 1 μM), LY294002 (LY, 15 μM) or API-2 (API, 1 µM) as indicated (n=3, triplicates per group). (D) qPCR results showing BMP2 expression in IFT80f/f and IFT80d/d DPSCs. Cells were treated with FGF2 (10 ng/mL), Purmorphomine (Pur, 2 μM), PD173074 (PD, 1 μM), or LY294002 (LY, 15 μM) as indicated (n=3, triplicates per group). (E) ALP activity of IFT80f/f and IFT80d/d DPSCs at day 7 of OS induction treated with FGF2 (10 ng/mL) and Purmorphomine (Pur, 2 μM) as indicated (n = 3, triplicates per group). (F) Alizarin Red staining of IFT80f/f and IFT80d/d DPSCs at day 21 of OS medium treated with FGF2 (10 ng/mL) and Purmorphomine (Pur, 2 μM) as indicated (n = 3, triplicates per group). Data are expressed as mean ± SEM; ns, not statistically significant; *p<0.05; **p < 0.01; ***p < 0.0001.
FGF2 promotes BMP2 signaling through PI3K-AKT signaling
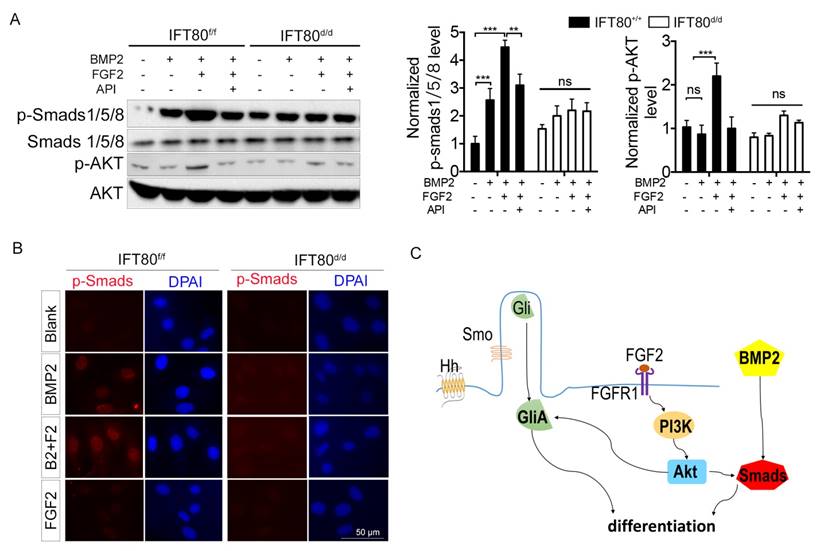
To further uncover the crosstalk among FGF2, Hh and BMP2, we examined Smads1/5/8, the critical downstream mediator of BMP2 signaling, using both Western blot and immunostaining method. We found that FGF2 aided phosphorylation of Smads1/5/8 (Fig. 7A) and nuclear translocation of p-Smads1/5/8 (Fig. 7B). PI3K-AKT signaling is essential for the activity of BMP2 signaling [29], and our results showed that FGF2 could activate PI3K-AKT cascade (Fig. 7A), demonstrating FGF2 promoted BMP2 signaling through PI3K-AKT signaling. Impaired FGF2-PI3K-AKT transduction in IFT80d/d DPSCs disrupted synergistic effect with BMP2 signaling. Indeed, BMP2 activated phosphorylation of Smads1/5/8 and BMP2 induced p-Smads1/5/8 nuclear translocation also impaired in IFT80d/d DPSCs (Fig. 7A and 7B). Overexpression of BMP2 by transfection partially rescued the differentiation of IFT80d/d DPSCs (Fig. S4A and S4B). However, BMP2 did not change the cilia length and ciliated cell population (Fig. S4C). Smad1 linker phosphorylation is an essential downstream of FGF signaling to antagonist BMP2 signaling [30, 31], as we expected, impaired FGF signaling in IFT80d/d DPSCs caused impaired Smad linker phosphorylation compared with that in IFT80f/f DPSCs (Fig. S5).
Crosstalk between FGF2 and Hh/BMP2 signaling during DPSC differentiation is blocked in IFT80-deficient DPSCs. (A) Western blot analysis of Smads1/5/8 and AKT phosphorylation in IFT80f/f and IFT80d/d DPSCs. Cells were treated with BMP2 (100 ng/mL), FGF2 (10 ng/mL), and API-2 (API, 1 μM) as indicated. Phosphorylation of Smads1/5/8 and AKT was normalized to Smads1/5/8 and AKT, respectively (n = 3). (B) Immunofluorescence analysis of p-Smads1/5/8 in IFT80f/f and IFT80d/d DPSCs (red). DAPI staining was used as counterstaining. Scale bars represent 50 μm. (C) Schematic representation of the proposed function of IFT80/cilia in FGF2 signaling, Hh signaling, and their crosstalk. Data are expressed as mean ± SEM; ns, not statistically significant; **p < 0.01; ***p < 0.0001.
Discussion
DPSCs give rise to odontoblasts that produce dentin. Primary cilia have been observed in both DPSCs and odontoblasts more than a decade ago [11]; however, the roles of primary cilia and ciliary proteins in DPSC differentiation and function were not well-addressed. Our study reveals for the first time that IFT80 is required for DPSC differentiation. Deletion of IFT80 in DPSCs reduces FGFR1 expression and consequently impairs FGF2-FGFR1-PI3K-AKT signaling. Moreover, IFT80 deletion impairs cilia formation, Hh signaling, as well as the coupling of FGF2 and Hh/BMP2 signaling, which eventually blocks DPSC differentiation.
The primary cilium is a signaling hub in a cell [17, 32, 33]. Excepted for the well-known Hh signaling and Wnt signaling, emerging studies have suggested that IFT proteins and primary cilia mediate platelet-derived growth factor (PDGF) signaling [34], Notch [35], transforming growth factor beta (TGF-β), insulin-like growth factor (IGF) [36, 37], mammalian target of rapamycin (mTOR) [33], and epidermal growth factor (EGF) [32] signaling pathways. The role of primary cilia in FGF signaling was not well-defined. It has been reported that mutation of FGFR1, a receptor of FGF signaling, in zebrafish shortens cilia, implying signaling through FGFR1 is required for cilia formation [38]. Since FGF signaling is also a critical signaling pathway for tooth development, we hypothesized that primary cilia/IFT proteins could regulate FGF signaling in DPSC differentiation. We found that loss of IFT80 in DPSCs dramatically reduced FGFR1 expression (Fig. 3). FGFR1 or p-FGFR1 was not accumulated in cilia even with FGF2 stimulation (Fig. 4), suggesting cilia loss was not a direct cause for the reduced FGFR1 expression in IFT80-deficient DPSCs. One possible explanation is that the FGFR1 reduction results from the disruption of Hh-Gli signaling in IFT80-deficient DPSCs. This hypothesis was supported by the study from Qin et al. [39], showing inhibition of Gli with Gli-antagonist 61 (GANT61), a Gli specific inhibitor, inhibits FGFR1 expression. Another possibility is FGFR1 expression is regulated by ciliary proteins that are impaired in IFT80-deficient DPSCs. Centrin-2, a small calcium-binding protein, might be one of these cilia-associated proteins that regulated by IFT80. Centrin-2 is required for FGF8 and FGFR1 expression in Xenopus laevis because it is associated with their RNA polymerase II binding site [40]. Since Centrin-2 localizes in the basal body of cilia [40], its function or expression might be affected by IFT80 and cilia loss. It is also possible that proteins involved in FGFR1 degradation are associated with primary cilia. The exact mechanism of how IFT80 regulates FGFR1 expression still needs further study.
The role of FGF2 in osteogenic differentiation is quite controversial [41] and seems stage-specific as suggested by recent studies [7, 42]. Early and limited FGF2 treatment promotes differentiation through FGFR-MEK-ERK1/2 and BMP signaling pathway, whereas continuous FGF2 treatment inhibits mineralization [7]. In our study, we confirmed that FGF2 has stimulatory effects on DPSC differentiation at an early stage beside its proliferation effect. More importantly, we found that FGF2 treatment significantly promotes cilia elongation and Hh signaling activation in DPSCs. It is well established that Hh signaling transduction requires functional cilia. Thus, pretreating DPSCs with FGF2 modulates cilia formation (Fig. 5), which aids Hh signaling activation (Fig. 6C). The exact mechanism of FGF2 in cilia formation is not entirely clear. Several recent studies demonstrated the regulatory role of actin cytoskeleton in ciliogenesis in different types of cells [43-46]. Cytoskeleton remodeling shows both direct and indirect effects in ciliogenesis. For example, inhibiting branched actin network or actin destabilization directly facilitates ciliogenesis [45, 46] and cytoskeleton remodeling could also modulate transcriptional coactivator such as YAP/TAZ to regulate ciliogenesis [44]. Our data demonstrated FGF2 causes cytoskeleton reorganization in DPSCs, and most likely the cytoskeleton remodeling promotes cilia formation in DPSCs.
We also found FGF2 activated PI3K-AKT (Fig. 6C) to promote Hh signaling, which is supported by the findings from Riobo et al. that Akt promotes Shh signaling by controlling PKA-mediated Gli inactivation [47]. In addition to Hh signaling, we found FGF2 also promoted BMP2 signaling, an important differentiation signaling pathway. FGF2 signaling, as well as Hh signaling, stimulated BMP2 expression, and the combination of FGF2 and Hh signaling showed a synergistic effect. Similar to Hh activation, BMP2 signaling is greatly advanced by AKT activation [29]. BMP2 itself could not activate AKT (Fig. 7A); therefore, FGF2-induced AKT activation serves as an enhancer for BMP2 expression, and then BMP2-induced Smads1/5/8 activation (Fig. 7A and 7B) and DPSC differentiation. Besides the activation effect, FGF2 signaling also promotes the phosphorylation of Smad1 linker to antagonist BMP2 signaling [30, 31]. Although this negative feedback was blocked in IFT80d/d DPSC, the overall effect still favors BMP2 signaling inhibition, eventually leading to the impaired DPSC differentiation.
Collectively, this study demonstrated that IFT80 is a critical regulator for DPSC differentiation. IFT80 maintains cilia and is required for FGFR1 expression. At the same time, FGF2/FGFR1-induced stress fiber rearrangement and AKT activation aid Hh signaling and BMP2 signaling activation to drive DPSC differentiation. In IFT80-deficient DPSC, FGF2/FGFR1 signaling, Hh/BMP2 signaling, and their coupling are impaired, which eventually leads to a differentiation defect. Thus, we revealed a novel role and mechanism of IFT80 in the regulation of DPSC differentiation and provided new insights for bone and tooth regenerative therapeutic design and therapy.
Materials and Methods
Mice
All experiments performed on mice were approved by the University at Buffalo Institutional Animal Care and Use Committee. The generation of IFT80f/f mice model (two LoxP sites flanking exon 6 of IFT80) was previously described [25].
CiliaGFP mice [48] were mated with CMV-Cre mice [49] to generate CMV;CiliaGFP mice, in which all the cilia are labeled with GFP. CiliaGFP mice were also mated with IFT80f/f mice to generate CiliaGFP;IFT80f/f mice.
Regents
Recombinant mouse Shh N-terminus (1 μg/mL, R&D systems, Minneapolis, MN) or Purmorphomine (2 μM, Tocris Bioscience, 4551) were used to activate Hh signaling. FGF2 (10 ng/mL, R&D systems, Minneapolis, MN) was used to activate FGF signaling. BMP2 (100 ng/mL) was used to activate BMP2 signaling. PD173074 (1 μM, Tocris 3044) was used to inhibit FGFR. LY294002 (15 μM, Sigma, L9908) and API-2 (1 μM, Tocris 2151), were employed to inhibit PI3K and AKT respectively.
Histology
Mouse mandibles were excised, fixed with 10% natural buffered formalin (VWR International, West Chester, PA, USA), and decalcified in 10% EDTA (ethylenediaminetetraacetic acid, Thermo Fisher Scientific) for two weeks at 4 °C. Paraffin-embedded samples were sectioned.
DPSC isolation, culture, and differentiation
The incisors were isolated from the mandibles that dissected from 6-week old IFT80f/f mice. Whole dental pulp was gently collected from the interior of the incisor and exposed to enzymatic digestion with collagenase type I (3 mg/mL) and dispase (4 mg/mL) for one hour at 37°C with shaking. The digested tissues were homogenized by repetitive pipetting, and the released cells were centrifuged at 200 g for 10 minutes. The cells were cultured in alpha-modified Eagle's medium (α-MEM, Life Technologies) containing 10% fetal bovine serum (FBS, Life Technologies), 2 mM L-glutamine (Life Technologies), 100 U/mL penicillin and 100 μg/mL streptomycin (Life Technologies). DPSCs were allowed to adhere to the plastic dish for 24 hours, and then the medium was changed to remove floating debris. Culture medium was replaced every three days until the cells reach 80% confluence. Then the cells were detached by 0.25% Trypsin-EDTA (Life Technologies) and sub-cultivated at a ratio of 1:2 [6, 50].
g for 10 minutes. The cells were cultured in alpha-modified Eagle's medium (α-MEM, Life Technologies) containing 10% fetal bovine serum (FBS, Life Technologies), 2 mM L-glutamine (Life Technologies), 100 U/mL penicillin and 100 μg/mL streptomycin (Life Technologies). DPSCs were allowed to adhere to the plastic dish for 24 hours, and then the medium was changed to remove floating debris. Culture medium was replaced every three days until the cells reach 80% confluence. Then the cells were detached by 0.25% Trypsin-EDTA (Life Technologies) and sub-cultivated at a ratio of 1:2 [6, 50].
Odontogenic differentiation was induced with Odontogenic medium (OS medium) consisting of α-MEM (Gibico), 10% fetal bovine serum (FBS, Gibico), 10 mM β-glycerophosphate (Sigma), 50 μg/mL ascorbic acid (Sigma), 10-8 M dexamethasone (Sigma). ALP activity assay was performed 7 days after odontogenic induction. The cells were lysed with harvest buffer containing 10 mM Tris-Cl (pH 7.4), 0.2% NP40, and 2 mM PMSF. The lysates were homogenized and cleared by centrifuging. The supernatants were mixed with assay buffer (100 mM glycine and 1 mM MgCl2, pH 10.5) and p-Nitrophenyl Phosphate (PNPP) solution (50 mM in 0.1 M glycine buffer), and incubated at 37  for 5-15 min. The reaction was stopped by the NaOH solution (0.1 N). The optical density was measured at 405 nm using AD 340 microplate reader (Beckman Coulter, Fullerton, CA). ALP activity was normalized to total cellular protein determined by BCA protein assay kit (Thermo Fisher Scientific) and expressed as units per minute per gram of total protein. To measure mineralization, Alizarin Red S staining was performed either 14 days or 21 days after odontogenic induction. The cells were washed with PBS, fixed with 2% formaldehyde, and then stained with 40 mM of Alizarin Red S solution (pH 4.2) at room temperature for 30 min. Cells were rinsed five times with dH2O to reduce nonspecific staining. Quantification was performed with a destaining solution containing 10% cetylpyridinium chloride and 10 mM sodium phosphate (pH 7.0) and measured at the wavelength of 562 nm. The experiment was done in triplicate.
for 5-15 min. The reaction was stopped by the NaOH solution (0.1 N). The optical density was measured at 405 nm using AD 340 microplate reader (Beckman Coulter, Fullerton, CA). ALP activity was normalized to total cellular protein determined by BCA protein assay kit (Thermo Fisher Scientific) and expressed as units per minute per gram of total protein. To measure mineralization, Alizarin Red S staining was performed either 14 days or 21 days after odontogenic induction. The cells were washed with PBS, fixed with 2% formaldehyde, and then stained with 40 mM of Alizarin Red S solution (pH 4.2) at room temperature for 30 min. Cells were rinsed five times with dH2O to reduce nonspecific staining. Quantification was performed with a destaining solution containing 10% cetylpyridinium chloride and 10 mM sodium phosphate (pH 7.0) and measured at the wavelength of 562 nm. The experiment was done in triplicate.
Adipogenesis was induced with adipogenic medium containing α-MEM supplemented with 10% FBS, 10-7 M dexamethasone (Sigma), and 5 10-6 M insulin (Sigma). To examine adipogenesis, Oil Red O staining was performed. The cells were fixed with 10% formaldehyde for 10 min and stained with Oil Red O solution (Sigma). Hematoxylin was used as the counterstain.
10-6 M insulin (Sigma). To examine adipogenesis, Oil Red O staining was performed. The cells were fixed with 10% formaldehyde for 10 min and stained with Oil Red O solution (Sigma). Hematoxylin was used as the counterstain.
Chondrogenesis was induced with chondrogenic medium containing DMEM supplemented with 10% FBS and ITS supplement (insulin-10 μg/mL, transferrin-5.5 μg/mL, and sodium selenite-5 ng/mL, Sigma-13146) for three weeks. To examine chondrogenesis, Alician Blue staining was performed. The cells were fixed with 4% glutaraldehyde for 15 min, stained with an acidic solution containing 1% Alician Blue (Sigma) for 30 min at room temperature and washed three times with hydrochloric acid 0.1 N.
DPSCs from IFT80f/f mice were infected with the adenovirus that overexpresses either Cre (Ad-CMV-Cre, #1405, Vector Biolabs) or GFP (Ad-GFP, #1060, Vector Biolabs). The Ad-CMV-Cre infection causes IFT80 deletion in IFT80f/f DPSCs, which were then marked as IFT80d/d. Ad-GFP was used as an infection control and Ad-GFP treated IFT80f/f DPSCs were still marked as IFT80f/f. DPSCs from CiliaGFP mice were infected with Ad-CMV-Cre to turn on GFP express to mark cilia. DPSCs were infected with Ad-BMP2 to overexpress BMP2.
Western blot
Cells were harvested and homogenized with RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, and protease inhibitor cocktail). Protein was denatured in sodium dodecyl sulfate (SDS) buffer and separated with SDS-PAGE gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes and then blocked with 5% skim milk (OXOID). Membranes were incubated with primary antibody overnight at 4 , and then incubated with horseradish peroxidase (HRP) conjugated goat anti-rabbit IgG antibody (1:10000, Novex, Carlsbad, CA) at room temperature for 1 hour. Visualization was performed with WesternBright ECL HRP (Bio-Rad). β-actin (1:500, Santa Cruz) was used as the internal control.
, and then incubated with horseradish peroxidase (HRP) conjugated goat anti-rabbit IgG antibody (1:10000, Novex, Carlsbad, CA) at room temperature for 1 hour. Visualization was performed with WesternBright ECL HRP (Bio-Rad). β-actin (1:500, Santa Cruz) was used as the internal control.
The same procedure was used to determine the IFT80 (1:400, PAB15842, Abnova), FGFR1 (1:1000, ab10646, Abcam), p-FGFR1 (1:1000, ab59194, Abcam), Smad1/5/8 (1:300, sc-6031-R, Santa Cruz), p-Smad1/5/8 (1:300, sc-12353-R, Santa Cruz), AKT (1:300, sc-8312, Santa Cruz), p-AKT (1:300, sc-7985-r, Santa Cruz).
qPCR
Total RNA was extracted from cultured DPSCs with Trizol reagent (Invitrogen, Carlsbad, CA) and then synthesized to cDNA with total RNA by RNA to cDNA EcoDry Premix kit (Clontech, Palo Alto, CA). qPCR was performed with ABI PRISM 7500 real-time PCR machine (Invitrogen, Carlsbad, CA) and SYBR Green PCR Master Mix (Invitrogen). Sequence and product length for each primer pair were listed in Supplementary Table S1. Gene expression was normalized to the housekeeping gene GAPDH and calculated according to the 2-ddCT method [51]. All reactions were run in triplicate.
Immunocytochemistry and Immunofluorescence
Deparaffinized sections or fixed cells were permeabilized, blocked and incubated with acetylated α-tubulin (1:200, T6793, Sigma) overnight at 4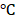 . The slides were washed and stained with Alexa Fluor 568 conjugated anti-rabbit IgG (1:1000, Invitrogen) antibody for 1 hour at room temperature. DAPI (6-diamidino-2-phenylindole, Sigma) staining was used as the counterstain for nuclei.
. The slides were washed and stained with Alexa Fluor 568 conjugated anti-rabbit IgG (1:1000, Invitrogen) antibody for 1 hour at room temperature. DAPI (6-diamidino-2-phenylindole, Sigma) staining was used as the counterstain for nuclei.
The same staining procedure was used for acetylated α-tubulin (1:500, T6793, Sigma), FGFR1 (1:100, ab10646, Abcam), p-FGFR1 (1:100, ab59194, Abcam), Phospho-SMAD1 (Ser206) (1:100, PA517092, Invitrogen), and p-Smad1/5/8 (1:50, sc-12353-R, Santa Cruz) staining.
Statistical analysis
All data are presented as mean ± SEM (n = 3 or more as indicated in figure legends). Comparisons between two groups were performed by Student's t-test and comparisons among grouped samples were analyzed by two-way ANOVA followed by Tukey's multiple comparisons. P<0.05 was considered to be of statistical significance. The program GraphPad Prism (GraphPad Software, Inc., San Diego, USA) was used for these analyses.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Dr. Aimee B. Stablewski and Gene Targeting and Transgenic Shared Resource at Roswell Park Cancer Institute [supported by the National Cancer Institute (NCI) grant P30CA016056], for technical assistance with the ES cell injections for generating IFT80 floxed mice. Research reported in this publication was supported by the National Institute of Dental and Craniofacial Research and the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under Award Numbers DE023105, AR066101 and AG048388 to S.Y. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
X.Y. and M.L. designed, performed experiments and analyzed the data. S.Y. and X.C. designed the research and analyzed the data.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sharpe PT. Dental mesenchymal stem cells. Development. 2016;143:2273-80
2. Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13625-30
3. Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A. et al. Stem cell properties of human dental pulp stem cells. Journal of dental research. 2002;81:531-5
4. Alge DL, Zhou D, Adams LL, Wyss BK, Shadday MD, Woods EJ. et al. Donor-matched comparison of dental pulp stem cells and bone marrow-derived mesenchymal stem cells in a rat model. J Tissue Eng Regen Med. 2010;4:73-81
5. Grottkau BE, Purudappa PP, Lin YF. Multilineage differentiation of dental pulp stem cells from green fluorescent protein transgenic mice. International journal of oral science. 2010;2:21-7
6. Akmal M, Zarina Z, Rohaya M, Sahidan S, Zaidah Z, Hisham Z. Isolation and Characterization of Dental Pulp Stem Cells from Murine Incisors. Journal of Biological Sciences. 2014:14
7. Sagomonyants K, Kalajzic I, Maye P, Mina M. Enhanced Dentinogenesis of Pulp Progenitors by Early Exposure to FGF2. Journal of dental research. 2015;94:1582-90
8. Kim SG, Zhou J, Solomon C, Zheng Y, Suzuki T, Chen M. et al. Effects of growth factors on dental stem/progenitor cells. Dent Clin North Am. 2012;56:563-75
9. Kim J, Park JC, Kim SH, Im GI, Kim BS, Lee JB. et al. Treatment of FGF-2 on stem cells from inflamed dental pulp tissue from human deciduous teeth. Oral Dis. 2014;20:191-204
10. Osathanon T, Nowwarote N, Pavasant P. Basic fibroblast growth factor inhibits mineralization but induces neuronal differentiation by human dental pulp stem cells through a FGFR and PLCγ signaling pathway. Journal of cellular biochemistry. 2011;112:1807-16
11. Magloire H, Couble ML, Romeas A, Bleicher F. Odontoblast primary cilia: facts and hypotheses. Cell Biol Int. 2004;28:93-9
12. Lyu R, Zhou J. The Multifaceted Roles of Primary Cilia in the Regulation of Stem Cell Properties and Functions. Journal of cellular physiology. 2016
13. Yuan X, Cao X, Yang SY. IFT80 is required for stem cell proliferation, differentiation, and odontoblast polarization during tooth development. Cell Death & Disease. 2019:10
14. Bodle JC, Loboa EG. Concise Review: Primary Cilia: Control Centers for Stem Cell Lineage Specification and Potential Targets for Cell-Based Therapies. Stem cells. 2016;34:1445-54
15. Jaafar Marican NH, Cruz-Migoni SB, Borycki AG. Asymmetric Distribution of Primary Cilia Allocates Satellite Cells for Self-Renewal. Stem Cell Reports. 2016;6:798-805
16. Li G, Liu M, Zhang S, Wan H, Zhang Q, Yue R. et al. Essential Role of IFT140 in Promoting Dentinogenesis. Journal of dental research. 2017:22034517741283
17. Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nature reviews Genetics. 2010;11:331-44
18. Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance. Journal of cell science. 2010;123:499-503
19. Taschner M, Weber K, Mourao A, Vetter M, Awasthi M, Stiegler M. et al. Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin-binding IFT-B2 complex. The EMBO journal. 2016 e201593164
20. Katoh Y, Terada M, Nishijima Y, Takei R, Nozaki S, Hamada H. et al. Overall architecture of the intraflagellar transport (IFT)-B complex containing Cluap1/IFT38 as an essential component of the IFT-B peripheral subcomplex. Journal of Biological Chemistry. 2016 jbc. M116. 713883
21. Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. American Journal of Medical Genetics Part C: Seminars in Medical Genetics: Wiley Online Library. 2012;160C(3):165-74
22. Haycraft CJ, Serra R. Cilia involvement in patterning and maintenance of the skeleton. Current topics in developmental biology. 2008;85:303-32
23. Nakatomi M, Hovorakova M, Gritli-Linde A, Blair HJ, MacArthur K, Peterka M. et al. Evc regulates a symmetrical response to Shh signaling in molar development. Journal of dental research. 2013;92:222-8
24. Yang S, Wang C. The intraflagellar transport protein IFT80 is required for cilia formation and osteogenesis. Bone. 2012;51:407-17
25. Yuan X, Cao J, He X, Serra R, Qu J, Cao X. et al. Ciliary IFT80 balances canonical versus non-canonical hedgehog signalling for osteoblast differentiation. Nat Commun. 2016;7:11024
26. Su N, Jin M, Chen L. Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Res. 2014;2:14003
27. Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191:415-28
28. Yang J, Andre P, Ye L, Yang YZ. The Hedgehog signalling pathway in bone formation. Int J Oral Sci. 2015;7:73-9
29. Mukherjee A, Rotwein P. Akt promotes BMP2-mediated osteoblast differentiation and bone development. Journal of cell science. 2009;122:716-26
30. Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. 2009;136:1093-104
31. Sapkota G, Alarcon C, Spagnoli FM, Brivanlou AH, Massague J. Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol Cell. 2007;25:441-54
32. Christensen ST, Clement CA, Satir P, Pedersen LB. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. The Journal of pathology. 2012;226:172-84
33. Pala R, Alomari N, Nauli SM. Primary Cilium-Dependent Signaling Mechanisms. Int J Mol Sci. 2017:18
34. Noda K, Kitami M, Kitami K, Kaku M, Komatsu Y. Canonical and noncanonical intraflagellar transport regulates craniofacial skeletal development. Proc Natl Acad Sci U S A. 2016;113:E2589-97
35. Grisanti L, Revenkova E, Gordon RE, Iomini C. Primary cilia maintain corneal epithelial homeostasis by regulation of the Notch signaling pathway. Development. 2016
36. Yeh C, Li A, Chuang JZ, Saito M, Caceres A, Sung CH. IGF-1 activates a cilium-localized noncanonical Gbetagamma signaling pathway that regulates cell-cycle progression. Dev Cell. 2013;26:358-68
37. Dalbay MT, Thorpe SD, Connelly JT, Chapple JP, Knight MM. Adipogenic Differentiation of hMSCs is Mediated by Recruitment of IGF-1r Onto the Primary Cilium Associated With Cilia Elongation. Stem Cells. 2015;33:1952-61
38. Neugebauer JM, Amack JD, Peterson AG, Bisgrove BW, Yost HJ. FGF signalling during embryo development regulates cilia length in diverse epithelia. Nature. 2009;458:651-4
39. Qin S, Sun D, Li H, Li X, Pan W, Yan C. et al. The Effect of SHH-Gli Signaling Pathway on the Synovial Fibroblast Proliferation in Rheumatoid Arthritis. Inflammation. 2016;39:503-12
40. Shi J, Zhao Y, Vonderfecht T, Winey M, Klymkowsky MW. Centrin-2 (Cetn2) mediated regulation of FGF/FGFR gene expression in Xenopus. Scientific reports. 2015;5:10283
41. Takei Y, Minamizaki T, Yoshiko Y. Functional diversity of fibroblast growth factors in bone formation. Int J Endocrinol. 2015;2015:729352
42. Li P, Bai Y, Yin G, Pu X, Huang Z, Liao X. et al. Synergistic and sequential effects of BMP-2, bFGF and VEGF on osteogenic differentiation of rat osteoblasts. Journal of bone and mineral metabolism. 2014;32:627-35
43. Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K. et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464:1048-51
44. Kim J, Jo H, Hong H, Kim MH, Kim JM, Lee JK. et al. Actin remodelling factors control ciliogenesis by regulating YAP/TAZ activity and vesicle trafficking. Nat Commun. 2015;6:6781
45. Cao J, Shen Y, Zhu L, Xu Y, Zhou Y, Wu Z. et al. miR-129-3p controls cilia assembly by regulating CP110 and actin dynamics. Nat Cell Biol. 2012;14:697-706
46. Sanchez de Diego A, Alonso Guerrero A, Martinez AC, van Wely KH. Dido3-dependent HDAC6 targeting controls cilium size. Nat Commun. 2014;5:3500
47. Riobo NA, Lu K, Ai X, Haines GM, Emerson CP Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4505-10
48. O'Connor AK, Malarkey EB, Berbari NF, Croyle MJ, Haycraft CJ, Bell PD. et al. An inducible CiliaGFP mouse model for in vivo visualization and analysis of cilia in live tissue. Cilia. 2013;2:8
49. Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080-1
50. Ellis KM, O'Carroll DC, Lewis MD, Rychkov GY, Koblar SA. Neurogenic potential of dental pulp stem cells isolated from murine incisors. Stem Cell Res Ther. 2014;5:30
51. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
Author contact
![]() Corresponding author: Dr. Shuying Yang, Department of Anatomy & Cell Biology, School of Dental Medicine, University of Pennsylvania, PA, United States. Email: shuyingyedu
Corresponding author: Dr. Shuying Yang, Department of Anatomy & Cell Biology, School of Dental Medicine, University of Pennsylvania, PA, United States. Email: shuyingyedu