Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. The common types of RIHD
2. Pathogenesis of RIHD
3. Drugs therapy of RIHD
4. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(10):2128-2138. doi:10.7150/ijbs.35460 This issue Cite
Review
Radiation-induced heart disease: a review of classification, mechanism and prevention
Heru Wang1,2, Jinlong Wei1, Qingshuang Zheng2, Lingbin Meng3, Ying Xin4, Xia Yin2 ![]() , Xin Jiang1
, Xin Jiang1 ![]()
1. Department of Radiation Oncology, The First Hospital of Jilin University, Changchun, 130021, China
2. Department of Cardiology, The First Hospital of Jilin University, Changchun, 130021, China
3. Department of Internal Medicine, Florida Hospital, Orlando, FL 32804,USA
4. Key Laboratory of Pathobiology, Ministry of Education, Jilin University, Changchun 130021, China
Received 2019-4-3; Accepted 2019-6-19; Published 2019-8-8
Abstract

With the increasing incidence of thoracic tumors, radiation therapy (RT) has become an important component of comprehensive treatment. RT improves survival in many cancers, but it involves some inevitable complications. Radiation-induced heart disease (RIHD) is one of the most serious complications. RIHD comprises a spectrum of heart disease including cardiomyopathy, pericarditis, coronary artery disease, valvular heart disease and conduction system abnormalities. There are numerous clinical manifestations of RIHD, such as chest pain, palpitation, and dyspnea, even without obvious symptoms. Based on previous studies, the pathogenesis of RIHD is related to the production and effects of various cytokines caused by endothelial injury, inflammatory response, and oxidative stress (OS). Therefore, it is of great importance for clinicians to identify the mechanism and propose interventions for the prevention of RIHD.
Keywords: Radiation-induced heart disease, inflammation, oxidative stress, statins, ACE inhibitors.
Introduction
With an increase in the incidence of tumors, radiation therapy (RT) has become an important treatment method [1]. RT improves survival of patients with tumor, but it also involves some inevitable complications of radiation. Radiation-induced heart disease (RIHD) is one of the most serious complications. Previous studies demonstrated that the heart is well resistant to radiation, and the symptoms of RIHD often require a long incubation period to manifest, so the RIHD has not attracted much attention [2]. With the increasing number and the prolonged survival of patients, researchers have gradually found that the RIHD offset some benefits and many patients succumbed to ischemic heart disease [3-5]. RIHD is now receiving increasing attention from clinicians and patients.
RIHD comprises a spectrum of heart disease including pericarditis, cardiomyopathy, coronary artery disease, vavular heart disease, and cardiac conduction abnormality [6]. Up to now, the total morbidity of RIHD has not been completely censused in the throng of patients with thoracic tumors. However, because of the high 5-year survival rate of breast cancer and Hodgkin's lymphoma patients, the current research on the incidence of RIHD mainly focuses on patients with the above two types of cancer. Because the latency of each disease and the follow-up time in each study are different, the results of studies are quite different. The incidence of each kind of heart disease varies greatly among breast cancer patients, ranging from 0.5% to 37% [7-9]. In lymphoma patients, the incidence of RIHD is higher than that of breast cancer patients, reaching 49.5-54.6%, and the incidence of various heart diseases ranges from 11-31%[10-12]. For other thoracic tumors, true morbidity of RIHD was underestimated because of its short survival rate and short follow-up time. And a number of large clinical studies have confirmed that radiation therapy increases the risk of heart disease-related mortality[12, 13]. Although morbidity of RIHD has been reduced by optimized treatment plan and techniques with radiation, but recent studies have shown that modern technology does not eliminate the risk of RIHD[9, 14].
At present, it is thought that RIHD is a result of various mechanisms interacting with each other through multiple complex pathways. However, endothelial injury, oxidative stress (OS) and inflammation, endoplasmic reticulum and mitochondrial damage are considered to be the main reasons [1]. With the development of research, microRNAs(miRNAs) have gradually become a new focus of research on the pathogenesis of RIHD [15]. Many researchers believe that miRNAs regulate the production of various cytokines, which in turn play an important role in the development of late cardiac injury related with radiation. There is no clear treatment program to effectively eradicate the onset and subsequent development of RIHD. However, reducing heart exposed range and doses have become a recognized primary prevention for RIHD. When the heart has been irradiated secondary prevention is crucial.
In this review, we summarize the common classification and important mechanisms that cause RIHD as well as some treatments.
1. The common types of RIHD
RIHD following chest RT has been recognized fact and demonstrated in numerous clinical trials. The choice of treatment and tumor location has significant influence on RIHD[16, 17]. At the present stage, chemotherapy is a risk factor for RIHD, anthracycline had evident effects on RIHD[18]. Studies have clearly indicated that receiving chemotherapy or radiotherapy alone is more facility to suffer ischaemic heart disease, and chemoradiotherapy increases risk of arrhythmia and heart failure[16]. The dose is linearly associated with the morbidity of RIHD, tumor location makes it extremely variable. The left anterior myocardium, pulmonary valve and atrioventricular are strongly impacted by the left radiation[17]. In addition to the above factors, age, smoking, diabetes, hypertension, dyslipidaemias, obesity etc are also applied to the common types and morbidity of RIHD[14].
1.1 Pericarditis
The clinical process of pericarditis can be divided into 4 stages including acute and chronic pericarditis, fibrinous pericarditis , and the final evolution, constrictive pericarditis[19].The most frequent manifestation of acute stage is exudative pericarditis [2]. Its occurrence is mainly related to damage of capillary endothelial cells and lymphatic stenosis or occlusion[20, 21]. Before the optimization of RT techniquset and scheme, about 80% of patients receiving RT suffered acute pericarditis[22, 23]. Many of patients with pericardial effusion present with hemodynamic abnormalities, but in most cases it is self-limited. The presence of a clear, benign pericardial effusion in acute phase may predispose patients to chronic pericarditis. But only 20% of patients developed symptomatic constrictive pericarditis.[24]. The morbidity is closely related to the radiation dose received by the heart. When the radiation dose is increased by 10 Gy, the morbidity increases five times[25]. Although the incidence of pericarditis has decreased to 6-10% with the optimization of radiotherapy protective techniques and programs, studies have shown that the risk of pericarditis among breast cancer survivors is still increasing[8, 26].
1.2 Cardiomyopathy
The clinical symptoms of myocardial injury caused by RT are quite late, mainly manifested as myocardial fibrosis[27]. Studies demonstrated that the incubation period of myocardial injury can be as long as more than ten years, and by the time of diagnosis most patients have previously experienced irreversible damage[28]. Most radiation-induced myocardial injury has no clinical symptoms, so the clinically diagnostic rate is low, only about 10%[26]. The most common echocardiographic abnormalities are regional wall motion abnormality (usually inferior wall), mild left ventricular hypertrophy and diastolic dysfunction, which can manifest as severe congestive cardiac insufficiency[29]. Myocardial injury is common in patients who have received anthracycline chemotherapy or high dose of radiation (>60 Gy). Patients who have received high dose of radiotherapy are prone to restrictive myocardial injury, and who have received chemoradiotherapy are prone to diastolic myocardial injury[30].
1.3 Coronary artery disease (CAD)
The injury of coronary artery induced by radiation is consistent with coronary atherosclerosis due to additional factors. The initial trigger was still endothelial cell injury the infiltration of monocytes into the intima, inducing low-density lipoprotein deposition and the formation of fatty streaks [31, 32]. RT as an external influence factor can induce the microvascular injury and accelerate the onset of CAD with high-risk patients [33]. RT induces vascular endothelial dysfunction, which ultimately results in clinical cardiovascular events that occur many years later after completion of RT. However, with the extension of patients' survival and the attention to RIHD, many clinical studies are dedicated to this field. Clinical studies have demonstrated that the incidence of CAD in patients is up to 85%, it closely related to radiation dose, location, time, and other factors [3, 8]. As for high-dose radiotherapy, a study conducted by Netherlands Cancer Institute found that compared with the general population, the cumulative incidence of CAD over 25 years was 34.5% and its risk increased 3-5 times [11].
1.4 Valvular heart disease
Myocardial ischemia and hypoxia caused by myocardial fibrosis and coronary diseases are the basic causes of valve function injury. The incubation period of RT induced valvular heart disease (VHD) is much longer than the aforementioned types of RIHD. Therefore, VHD lesions are very rare clinically. A previous study of Hodgkin's lymphoma survivors showed a cumulative VHD incidence of 10% in 13 years increasing to 20% in 30 years. This suggested that previous history of RT increased the likelihood of VHD in these patients to a large extent [11]. The incidence of VHD is closely related to the doses and anthracycline. Dose decrease resulted the gradual decline in the accumulated VHD incidence over 30 years from 12.4% at >40 Gy to 3% at <30 Gy [34]. The earliest changes in the general pathology of VHD seem to include valvular contraction and associated reflux within the first 10 years after RT, with preferential involvement of the mitral and aortic valves. The progression of valvular fibrosis thickening and calcification occurs much later, with stenosis often occurring 20 years after RT [35]. Mitral regurgitation and aortic regurgitation are the most common defects. When stenosis occurs, aortic regurgitation is frequently involved [36].
1.5 Conduction system abnormality
Conduction system abnormality caused by RT usually manifests as atrioventricular block, pathological sinus node syndrome, QTc prolongation, supraventricular arrhythmia and ventricular tachycardia[37]. The incidence of conduction system abnormality is about 5%, often occurs within 2 months after the end of RT, and 70% of ECG abnormalities can return to normal after half year of RT, but the incidence rate was still increased compared with that before the treatment[33, 38]. This shows that the effect of RT on the heart is partly reversible, but it will still cause some damage to heart.
2. Pathogenesis of RIHD
Although the effect of radiation on the heart has been clear in pre-clinical trials, the underlying mechanism of RIHD gradual progression from no clinical manifestations in the early stage to chronic heart disease in the later stage is not fully understood. There are many cytokines involved in the process and the regulation and control mechanisms are affected by various factors. These factors interact with each other so that the mechanism of RIHD is very complex. At present, it is believed that RIHD is associated with endothelial cell injury, inflammatory reaction, OS, mitochondria and endoplasmic reticulum injury, various cytokines, calcium overload, and micro-RNAs [21, 27, 39]. It is accepted that the early damage of RT is mostly caused by acute and chronic inflammatory changes, and the late toxicity is partly caused by OS and inflammation together. These changes can lead to heart disease[40]. Understanding the biological mechanism of RIHD is very important to clarify the pathogenesis of related diseases, and it will also be an important step to evaluate the feasible therapeutic targets. (Figure 1)
The Pathogenesis of RIHD
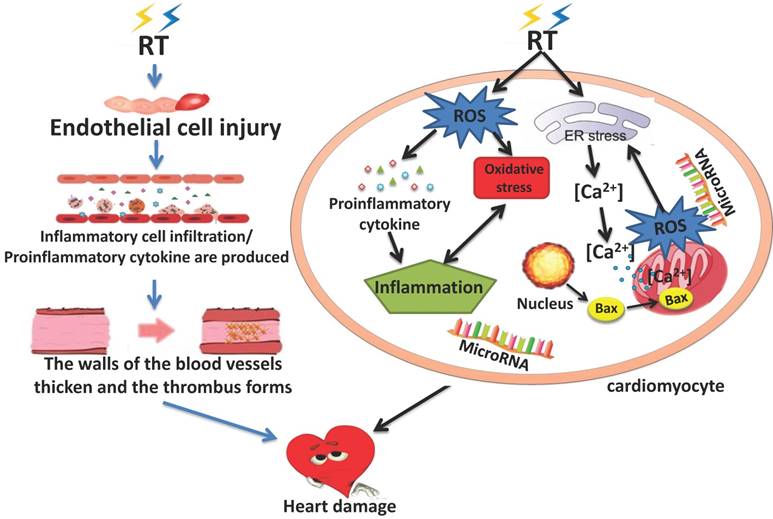
2.1 The endothelial cell injury and inflammation
Radiation-induced endothelial cell injury is deemed to be the primary and fundamental cause of myocardial injury[41-44]. RT can influence cardiac capillary endothelial cells, leading to their proliferation, injury, swelling and degeneration, and significantly reduce the number of capillaries. Although endothelial cells can regenerate, capillary network damage is irreversible [45], this may reduce the blood supply of myocardium. Radiation exposure of heart not only induces endothelial cell damage and the decrease of capillaries, but also changes coagulation function and platelet activity. The deposition and release of von Willebrand factor (vWF) in endothelial cells increased after radiation exposure of heart. The changes of vWF expression eventually lead to increasing platelet adhesion and thrombosis in capillaries[46, 47]. Animal studies have also shown that the inflammatory thrombotic plaque emerged in the blood vessels after the rat heart exposured to high doses of radiation[48].Thrombosis and decreased cardiac blood supply together lead to myocardial ischemia[49].
Both in vivo and vitro experiments showed that, in addition to the increased expression of vWF, the expression of e-selectin, p-selectin, intercellular cell adhesion molecule (ICAM), plaet-endothelial cell adhesion molecule-1(PECAM1) and other pro-inflammatory adhesion factors also increased a few hours after the endothelial cells exposure to radiation, mediating the infiltration of inflammatory cells into tissues and promoting the acute inflammation[50-54]. The regulation of irradiation-induced pro-inflammatory adhesion factors may be the key to early endothelial response. In addition to the increased expression of these adhesion factors, inflammatory mediators such as tumor necrosis factor-α, interleukin (IL-6, IL-8 and IL-10) also appeared in the myocardium to participate in the formation of acute inflammation.
Experiments have shown that IL-8 can not only mediate inflammatory responses, but also induce apoptosis. Then platelet-derived growth factor (PDGF), transforming growth factor Beta(TGF-β), nuclear factor kappa B (NF-κB), and connective tissue growth factor (CTGF) are released, leading to the chronic inflammation. [55-58].
The above inflammatory factors can not only mediate the production of inflammation, but also promote the proliferation of endothelial cells and fibroblasts, the increase of collagen deposition can cause the thickening of vessel walls and the stenosis of lumen[41, 42]. This may exacerbate the lack of blood flow to the myocardium due to the reduced capillary network. The heart is the main oxygen consuming organ in the human body, decreased blood supply can give rise to myocardial hypoxia which will aggravated the myocardial injury. Myocardial ischemia and hypoxia, inflammatory responses, collagen deposition, and proliferation of endothelial cells and fibroblasts lead to tissue remodeling, cardiac fibrosis, and atherosclerosis, and these changes are the primary endpoints of RIHD[49, 59].
2.2 Oxidative stress
In normal cell, reactive oxygen species (ROS) are important mediators of cellular processes such as immune response, cell signal transduction, microbial defense, differentiation, cell adhesion and apoptosis[60], therefore, the production of ROS is beneficial to cells under physiological conditions[61]. When ROS are produced in large quantities, their activity can be eliminated by reduction of intracellular antioxidants, including glutathione, to remove excess free radicals [62, 63]. However, glutathione and other antioxidants are also consumed during their activity, and the cell's ability to maintain redox balance is ultimately impaired.When the amount of endogenous and/or exogenous ROS exceeds the scavenging capacity of antioxidants, ROS begins to dominate and cause damage to cardiac myocytes. Some scholars believe that ROS - mediated OS is an important cause of atherosclerosis, hypertension and congestive heart failure[64, 65]. Like other heart diseases, OS also plays an important role in RIHD[36]. 80% of tissues and cells are composed of water, and most of the radiation damage (X-ray, gamma rays, rapid electrons) after exposure to radiation is caused by the generation of ROS and reactive nitrogen species (RNS) caused by the radiation decomposition of water, which is an important source of normal tissue damage after ionizing radiation [66]. DNA damage is likely to occur when intracellular antioxidants cannot adequately remove ROS. It has been reported that DNA damage has many forms, which can significantly change the structure of DNA and eventually lead to cell cycle arrest, apoptosis, mutation and other effects[67, 68]. The DNA damage repair (DDR) pathway, mediated by multiple functional proteins in cells, is an important mechanism to repair DNA damage and ensure the integrity of the genome[69]. P53 gene is one of the main effectors of DDR signaling pathway[70]. Same as P53, Bcl-2 gene family also changes after radiation, leading to increased apoptosis[71]. In addition to DNA damage, ROS can also lead to peroxidation of lipids and proteins and activate multiple signaling pathways[72].
ROS can not only directly damage the intracellular macromolecular structure, but also altered the expression of multiple proteomes in the cytoplasm, activation of pro-inflammatory factors in connection with ROS[73]. As a second messenger signal in cells, ROS participates in and regulates signaling pathways, including mitogen-activated protein kinases(MAPK) and NF-κB, and promote the occurrence of inflammation[74, 75]. NF-κB regulates DNA transcription and protein complexes engage in various cellular stress responses, and may be a key regulator of the link between OS and inflammatory[76]. ROS acts as a second messenger to activate NF-κB and induce the production of inflammatory cytokines. Therefore, proinflammatory cytokines and chemokines are believed to be closely related to the occurrence of OS, while OS enhanced inflammation in turn drives the progression of disease, leading to a vicious cycle [77].However, how OS and inflammation interact to promote RIHD remains unclear.
2.3 Apoptosis(endoplasmic reticulum and mitochondria signaling pathway)
Cell apoptosis and necrosis occur in various types of cells in the heart after exposure to radiation, among which mitochondrial dysfunction and irreversible damage are the key links of cell apoptosis and necrosis, and the occurrence of mitochondrial dysfunction is closely related to endoplasmic reticulum(ER)stress. Mitochondria are organelles that account for an important proportion of the total volume of cardiac myocytes, and mitochondria carry extranuclear DNA, so they are important targets for radiation-induced cell damage[78]. Mitochondrial permeability transition (MPT) and loss of mitochondrial membrane potential are important mechanisms of mitochondrial dysfunction and are involved in the pathogenesis of a variety of cardiovascular diseases[79]. Multiple stimuli, such as calcium ions flowing into mitochondria, inorganic phosphates, reactive oxygen species and other oxidants, can also induce MPT[80]. After cardiac myocytes are irradiated, the stimulated ER releases calcium ions from the calcium pool of the ER into the cytoplasm. This process will cause mitochondrial calcium overload and lead to its membrane swelling and release of apoptotic factors from it. Moreover, severe MPT can lead to mitochondrial membrane depolarization and the decoupling of oxidative phosphorylation, which is closely related to the opening of mitochondrial permeability transition pore(mPTP)[39, 81]. Bax is one of the important pro-apoptotic proteins in the Bcl-2 family [82]. It has been reported that exposure to RT leads to increased expression and activation of Bax, leading to its translocation and insertion into the mitochondrial outer membrane [83, 84]. This accelerated the opening of mitochondrial voltage - dependent anion channels. MPT and the insertion and ectopia increase of Bax improve the permeability of mitochondrial membrane and reduce the mitochondrial membrane potential together. This prolongs and enhances calcium-induced mitochondrial membrane swelling, leading to apoptosis[71].
Excessive ROS production by mitochondria in human cells was observed immediately after irradiation[64]. A large amount of ROS can cause lipid peroxidation and protein damage of ER. Then the ER produces a small amount of ROS and releases it into the cytoplasm. These reactions can reduce mitochondrial membrane potential, inhibit respiratory chain, and accelerate the generation of peroxidation[39, 85, 86]. The increased permeability of mitochondrial membrane leads to a cascade reaction, which produces a large amount of ROS. ROS further promotes the release of calcium from the calcium pool of the ER, leading to the overload of calcium in mitochondria, thus increasing the generation of ROS. This is consistent with the results of cell experiments. Oqura et al. found that once the acute increase of ROS subside, the subsequent generation of ROS will be observed[87]. Kobashigawa et al. also pointed out that ROS levels produced by mitochondria continued to increase one week after radiation exposure[88]. A vicious cycle of ROS produced by mitochondria and Ca2+ release caused by ER may lead to long-term toxicity induced by radiation, which eventually leads to cell cycle arrest[68]. This leads to apoptosis and premature aging. In addition, mitochondrial damage can also cause bystander effect in neighboring mitochondria, which amplifies radiation effect and leads to further cell damage [39, 89].
2.4 Micro-RNA
With the development of research, several studies have indicated that micro-RNAs (miRNAs) play an important role in the occurrence and progression of RIHD [90-92]. Alterations in miRNA expression may occur following exposure to several OS-inducing factors including ionizing radiation [90]. Many studies confirmed that miRNAs are implicated in the pathological processes connected with cardiac radiation damage, OS, inflammation, endothelial dysfunction, hypertrophy, and fibrosis resulting in heart failure [91, 92]. Recently, miRNAs have been found to be involved in the regulation of radiation-induced DNA damage and the induction of premature aging[93]. MiRNA-21 has the function of promoting cell proliferation and anti-apoptosis [94]. Csilla et al. found that the expression of miRNA-21 in the myocardium was significantly increased after radiation and this change was more obvious in the left ventricle than in the right ventricle [95]. MiRNA-1 expression was altered in many cardiovascular diseases and down-regulated in irradiated animal models, consistent with changes in cardiac hypertrophy and heart failure [91]. Furthermore, changes in miRNA-34a expression are also related to heart injury. A study showed that miRNA-34a expression was up-regulated after the heart was exposed to radiation [96].
In addition to the above mi-RNAs, miRNA-29, miRNA-199b, miRNA-221, miRNA-222, and miRNA-15 are also believed to be associated with the occurrence of heart disease [97-99]. However, it is not clear whether they are related to RIHD. The role of miRNAs in RIHD is a relatively new research topic, which has significant therapeutic potential in clinic. Much work has been done on miRNAs as important regulators of the cardiovascular system, and the understanding of their role in RIHD is currently limited. Further studies are needed to clarify the mechanisms underlying the regulation of RIHD by miRNAs.
3. Drugs therapy of RIHD
There is currently no effective treatment for RIHD, in particular for the prevention of over-exposure. Improvements in radiotherapy regimens to decrease exposure to normal healthy tissue near tumor cells are considered to be primary prevention [18]. As early as in the 1980s, technologies for reducing cardiac radiation dose have been applied in clinical practice, such as deep inspiratory breath hold (DIBH), three-dimensional conformal radiotherapy, intensity modulated radiation therapy, and volumetric arc therapy etc., which greatly reduce the radiation dose and volume received by the heart during radiotherapy[100-102]. However, the heart inevitably receives radiation doses during RT. It may be impossible for cardiovascular tissues to be fully protected, therefore secondary prevention (follow-up visits for patients and drug treatment) is crucial [103]. Studies suggest that RIHD can be prevented by using some drugs including statins, ACE inhibitors, and antioxidants [104-106]. (Table 1)
Summary of studies investigating the effect of different drug classes on RHID
| Drug class | Drug | Target | Observations | References |
|---|---|---|---|---|
| Statins | Atorvastatin | Atorvastatin reduced the expression of TGF-β1, Smad3/P-Smad3, ROCK I and p-Akt | Atorvastatin Ameliorate radiation-induced cardiac fibrosis in Sprague-Dawley rats. | Zhang et al.[70] |
| ACEIs | Captopril | Captopril inhibits the the renin-angiotensin system and scavenges free radical | Captopril improves breathing rate and cardiopulmonary density/structure | van der Veen et al.[71] |
| Anti-inflammation and Anti-OS compounds | Tocotrienol-rich mix | Tocotrienol-rich mix protects mitochondrial dysfunction | Tocotrienol-rich mix relieves mitochondrial changes and achieves anti-OS and anti-apoptosis in hearts. | Sridharan et al.[89] |
| Pentoxifylline + α-tocopherol | Pentoxifylline and α-tocopherol inhibits expression of intracellular TGF-β and CTGF and defends endothelial function | Pentoxifylline and α-tocopherol inhibit myocardial fibrosis | Boerma et al.[90] | |
| caffeic acid phenethyl ester (CAPE) | No description | CAPE suppresses acute immune system, inflammatory response and induces antioxidant properties | Mansour et al.[44] | |
| L-carnitine | L-carnitine administration activated p38MAPK/Nrf2 signaling | L-carnitine attenuates cardiac function loss | Fan et al.[91] | |
| Melatonin | Melatonin scavenges free radical | Melatonin prevents vasculitis and decreases fibrosis and necrosis | Gurses et al.[92] | |
| Amifostine | No description | Amifostine prevents vascular damage and vasculitis | Kruse et al.[93] | |
| TGF-β1 inhibitors | IPW-5371 | IPW-5371 antagonizes TGF-βR1 | IPW-5371 reduces collagen deposition in the heart and lungs and significantly improve the cardiopulmonary function of mice after irradiation | Rabender et al.[95] |
| Recombinant human neuregulin-1 | rhNRG-1β | rhNRG-1β activats ErbB2-ERK-SIRT1 signaling transduction | rhNRG-1β reduces myocardial damage and protects heart function | Gu et al.[96] |
3.1 Statins
Statins are 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors that are used in clinical practice to reduce cholesterol and lipoprotein density. Recent studies have shown that statins, in addition to lowering cholesterol, can reduce OS and activate adenosine 5'-monophosphate-activated protein kinase (AMPK) to achieve anti-inflammatory effects. Consequently, stains can protect the heart by inhibiting inflammatory reactions and OS [107, 108]. Researchers of previous studies found that pravastatin not only inhibited the early inflammation of the lungs caused by bleomycin, but also reduced the expression of transforming growth factor (TGF)-β1, CTGF, RhoA, and cyclin D1[109]. This means that in addition to lowering blood lipids, statins not only inhibit inflammation and oxidative stress, but also reduce the production of tissue fibrosis. The protective effect of statins on the heart against radiation damage was consistent with the above results.
In the acute response period after radiation exposure to normal tissues, lovastatin can inhibit the activation of transcription factor NF-κB and the expression of inflammatory cell adhesion molecules, thereby inhibiting the acute inflammatory response. For the latter chronic toxicity phase, lovastatin can inhibit the mRNA expression of fibrotropic factor CTGF which induced by RT, and the formation of tissue fibrosis may be alleviated by this change[110, 111]. Pravastatin can also inhibit tissue fibrosis caused by radiation, but its inhibitory ability is weaker than that of lovastatin[112]. Zang et al. further confirmed through experiments that atorvastatin can reduce radiation-induced myocardial fibrosis by inhibiting multiple inflammatory responses and OS pathway activation [104]. Doi et al. showed that pravastatin could also protect tissue damage caused by radiation by reducing DNA double-strand breakage in normal tissue cells [113]. However, it has been reported that atorvastatin can improve the repair of oxidative DNA damage in vascular smooth muscle cells (VSMCs) without affecting the initial level of DNA damage[114]. Therefore, the protective effect of statins on RT-induced myocardial injury is probably related to the repair of DNA damage. A few studies have been conducted on their application in radiation-induced myocardial injury [95, 115], but statins have the potential to be effective protectors of myocardial radiation. Therefore, it is important to clarify the mechanism of action for the discovery of RIHD protection targets.
3.2. Angiotensin-converting enzyme inhibitors (ACEIs)
The renin-angiotensin-aldosterone system (RAAS) is known to play an important role in cardiac remodeling. ACEIs in the RAAS system not only inhibits the production of ROS to reduce myocardial injury caused by OS and inflammation, but also increases the production of NO to protect vascular cells by reducing the negative effects on the bradykinin system [116]. ACEIs are usually used to treat hypertension or congestive heart failure. Studies have indicated that ACEIs may ameliorate radiation-induced toxicity in different organs, including the heart [117], central nervous system [118], and lungs [119].
Interestingly, ACEI drugs can reduce myocardial perivascular fibrosis and myocardial cell apoptosis through anti-inflammation and reducing oxygen free radicals after simultaneous exposure of the heart and lungs, thereby inhibiting myocardial fibrosis and decreased cardiac diastolic function [105]. Rats treated with captopril shortly after the lung was exposed to radiation demonstrated dramatically increased survival and improved vasoreactivity, as well as decreased perivascular fibrosis and inflammatory cell infiltration [119]. Furthermore, rats treated with captopril exhibited reduced diastolic dysfunction and perivascular necrosis in the left ventricle following radiation [105]. There are many differences between research and clinical radiation therapy, but clinical trials have also indicated that ACEI drugs can reduce the incidence of pneumonia induced by radiotherapy [120]. Although these data are interesting, prospective studies evaluating the efficacy of ACEIs in patients undergoing radiation have not been reported. In order to further clarify whether ACEI drugs can be a protective agent to reduce RIHD, a large number of clinical research patients would be required.
3.3. Anti-inflammation and Anti-OS compounds
Inflammation and OS play an important role in the development of RIHD and they interact with each other in various ways. Colchicine inhibits the inflammatory response by inhibiting microtubule polymerization and can reduce platelet aggregation, protecting the heart through its anti-inflammatory and anti-coagulant properties [106]. Some Chinese herbal extracts have been shown to inhibit the inflammatory response induced by radiation and the formation of myocardial fibrosis [121, 122].
As described above, ROS and RNS are released in large quantities after irradiation, which promotes an acute inflammatory response and subsequent OS. A pre-clinical trial indicated that rats exposed to 7 Gy gamma radiation and injected with caffeic acid phenethyl ester (CAPE), had an suppressed acute immune system and inflammatory response, as well as induced antioxidant properties, thereby alleviating the myocardial injury caused by radiation[123]. Tocomin SupraBio (TSB) enriched with tocotrienols can retain stability of the membrane potential and confront radiation-induced alterations in succinate driven mitochondrial respiration in the rat model of local heart exposed to irradiation. TSB also significantly improved the redox state and maintain the ratio of pro-apoptotic protein Bax and Bcl-2 through regulating the ratio of GSH and GSSH in left ventricular tissues. Therefore TSB can relieve radiation-induced mitochondrial changes and achieve anti-OS and anti-apoptosis in hearts. However, this drug cannot effectively inhibit the generation of radiation-induced myocardial fibrosis in the later stages, so whether it can be applied in clinical practice remains unknown[124]. A combination of antioxidant pentoxifylline and α-tocopherol inhibited myocardial fibrosis in irradiated rats by inhibiting expression of intracellular TGF-β and CTGF. In addition, pentoxifylline also changes endothelial function and prevents downregulation of endothelial cell surface thrombomodulin to defends endothelial function[125]. It's worth noting that nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a transcription factor which encodes many antioxidants and anti-electrophile enzymes. The activation of p38MAPK/Nrf2 signaling expression and the activation of downstream pathways may significantly suppress the degree of OS, reduce myocardial injury, and protect cardiac function[126]. In addition, there are many drugs such as melatonin and amifostine that are thought to reduce radiation-induced heart toxicity through anti-inflammatory and anti-OS, but the underlying mechanisms require further study [127, 128].
3.4. Others
It is well known that TGF-β is not only involved in inflammation and OS, but also induce collagen deposition and play an important role in the formation of myocardial fibrosis. Irradiated rats were given xaliproden (an orally active non-peptide agonist) to increase circulating TGF-β1 levels by 300% which significantly induced the expression of TGF-β1 and TGF-β1 target genes in the heart tissue. Similarly, in the same RIHD model, induction of TGF-β1 augmented radiation-induced changes in cardiac function and myocardial fibrosis [129]. IPW-5371, as a TGF-βR1 inhibitor, was reported to reduce collagen deposition in the heart and lungs and significantly improve the cardiopulmonary function of mice after irradiation [130]. These results further demonstrate the direct involvement of TGF-β1 in radiation-induced adverse remodeling and damage in the heart.
In addition, a recent animal trial has also shown that rhNRG-1β can reduce the damage to myocardial nuclei caused by radiation, maintain mitochondrial homeostasis, improve energy metabolism of myocardial cells, and alleviate the reduction of cardiac function and cardiac structural changes [131]. Furthermore, GSTA4-4 can eliminate Nrf2 activator 4-HNE and reduce the activation of antioxidant stress pathway. The cardiac function and capillary density of GSTA4-4 KO mice were improved compared with WT mice and the expression of Nrf2 transcription factor was up-regulated after receive local radiation. Therefore, GSTA4-4 inhibitors and recombinant Nrf2 activators have also become research hotspots for anti-OS drugs which can reduce cardiac radiation toxicity [117]. However, the mechanisms of these drugs are complex and indistinct. Further clinical trials and studies must be conducted before these drugs are actually used in clinical settings.
4. Conclusion
Survivors receiving chest radiotherapy are at an increased risk of RIHD. RIHD represents a spectrum of cardiac pathology including CAD, Cardiomyopathy, pericardial disease, arrhythmias, and valvular abnormalities. Although pre-clinical animal and cell models have been used to study the potential pathophysiological mechanisms of RIHD, the exact mechanisms of the various RIHD pathogenesis are not entirely understood. We have reviewed several common pathways involved in the development of RIHD including endothelial injury, inflammation and OS, and endoplasmic reticulum and mitochondrial dysfunction. The development of therapeutic targets to prevent microvascular damage, inflammation, and late fibrosis will hinge on our increased understanding of RIHD. The use of certain drugs can be quite helpful in reducing radiation-induced heart damage. However, these drugs may be not the most accurate treatment for RIHD and need to be developed for specific disease progression.
Acknowledgements
This work was supported in part by grants from the National Natural Science Foundation of China (81570344, to Ying Xin; 81670353 to Xia Yin), the Norman Bethne Program of Jilin University (2015225, to Ying Xin and 2015203, to Xin Jiang), the Jilin Provincial Science and Technology Foundations (20180414039GH to Ying Xin and 20190201200JC to Xin Jiang).
Author Contributions
JX and YX conceived and designed the study. WHR, WJL, and ZQS wrote the paper. MLB and XY reviewed and edited the manuscript. All authors read and approved the manuscript.
Competing Interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Slezak J, Kura B, Babal P, Barancik M, Ferko M, Frimmel K. et al. Potential markers and metabolic processes involved in the mechanism of radiation-induced heart injury. Canadian journal of physiology and pharmacology. 2017;95:1190-203
2. Lee PJ, Mallik R. Cardiovascular effects of radiation therapy: practical approach to radiation therapy-induced heart disease. Cardiol Rev. 2005;13:80-6
3. Darby SC, Ewertz M, McGale P, Bennet AM, Blom-Goldman U, Bronnum D. et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med. 2013;368:987-98
4. Davis M, Witteles RM. Radiation-induced heart disease: an under-recognized entity? Curr Treat Options Cardiovasc Med. 2014;16:317
5. Andratschke N, Maurer J, Molls M, Trott KR. Late radiation-induced heart disease after radiotherapy. Clinical importance, radiobiological mechanisms and strategies of prevention. Radiother Oncol. 2011;100:160-6
6. Donnellan E, Phelan D, McCarthy CP, Collier P, Desai M, Griffin B. Radiation-induced heart disease: A practical guide to diagnosis and management. Cleve Clin J Med. 2016;83:914-22
7. Hooning MJ, Botma A, Aleman BM, Baaijens MH, Bartelink H, Klijn JG. et al. Long-term risk of cardiovascular disease in 10-year survivors of breast cancer. J Natl Cancer Inst. 2007;99:365-75
8. McGale P, Darby SC, Hall P, Adolfsson J, Bengtsson NO, Bennet AM. et al. Incidence of heart disease in 35,000 women treated with radiotherapy for breast cancer in Denmark and Sweden. Radiother Oncol. 2011;100:167-75
9. Boero IJ, Paravati AJ, Triplett DP, Hwang L, Matsuno RK, Gillespie EF. et al. Modern Radiation Therapy and Cardiac Outcomes in Breast Cancer. Int J Radiat Oncol Biol Phys. 2016;94:700-8
10. van Nimwegen FA, Schaapveld M, Janus CP, Krol AD, Petersen EJ, Raemaekers JM. et al. Cardiovascular disease after Hodgkin lymphoma treatment: 40-year disease risk. JAMA Intern Med. 2015;175:1007-17
11. Aleman BM, van den Belt-Dusebout AW, De Bruin ML, van 't Veer MB, Baaijens MH, de Boer JP. et al. Late cardiotoxicity after treatment for Hodgkin lymphoma. Blood. 2007;109:1878-86
12. van Leeuwen FE, Ng AK. Long-term risk of second malignancy and cardiovascular disease after Hodgkin lymphoma treatment. Hematology Am Soc Hematol Educ Program. 2016;2016:323-30
13. Hancock SL, Tucker MA, Hoppe RT. Factors affecting late mortality from heart disease after treatment of Hodgkin's disease. JAMA. 1993;270:1949-55
14. Boekel NB, Schaapveld M, Gietema JA, Russell NS, Poortmans P, Theuws JC. et al. Cardiovascular Disease Risk in a Large, Population-Based Cohort of Breast Cancer Survivors. Int J Radiat Oncol Biol Phys. 2016;94:1061-72
15. Kura B, Babal P, Slezak J. Implication of microRNAs in the development and potential treatment of radiation-induced heart disease. Canadian journal of physiology and pharmacology. 2017;95:1236-44
16. Hardy D, Liu CC, Cormier JN, Xia R, Du XL. Cardiac toxicity in association with chemotherapy and radiation therapy in a large cohort of older patients with non-small-cell lung cancer. Ann Oncol. 2010;21:1825-33
17. Wollschlager D, Karle H, Stockinger M, Bartkowiak D, Buhrdel S, Merzenich H. et al. Radiation dose distribution in functional heart regions from tangential breast cancer radiotherapy. Radiother Oncol. 2016;119:65-70
18. Curigliano G, Cardinale D, Suter T, Plataniotis G, de Azambuja E, Sandri MT. et al. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO Clinical Practice Guidelines. Ann Oncol. 2012;23(Suppl 7):vii155-66
19. Yusuf SW, Sami S, Daher IN. Radiation-induced heart disease: a clinical update. Cardiol Res Pract. 2011;2011:317659
20. Carver JR, Shapiro CL, Ng A, Jacobs L, Schwartz C, Virgo KS. et al. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol. 2007;25:3991-4008
21. Taunk NK, Haffty BG, Kostis JB, Goyal S. Radiation-induced heart disease: pathologic abnormalities and putative mechanisms. Front Oncol. 2015;5:39
22. Brosius FC 3rd, Waller BF, Roberts WC. Radiation heart disease. Analysis of 16 young (aged 15 to 33 years) necropsy patients who received over 3,500 rads to the heart. Am J Med. 1981;70:519-30
23. Veinot JP, Edwards WD. Pathology of radiation-induced heart disease: a surgical and autopsy study of 27 cases. Hum Pathol. 1996;27:766-73
24. Walker CM, Saldana DA, Gladish GW, Dicks DL, Kicska G, Mitsumori LM. et al. Cardiac complications of oncologic therapy. Radiographics. 2013;33:1801-15
25. Schultz-Hector S, Trott KR. Radiation-induced cardiovascular diseases: is the epidemiologic evidence compatible with the radiobiologic data? Int J Radiat Oncol Biol Phys. 2007;67:10-8
26. Chang HM, Okwuosa TM, Scarabelli T, Moudgil R, Yeh ETH. Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 2. J Am Coll Cardiol. 2017;70:2552-65
27. Tapio S. Pathology and biology of radiation-induced cardiac disease. J Radiat Res. 2016;57:439-48
28. Heidenreich PA, Hancock SL, Lee BK, Mariscal CS, Schnittger I. Asymptomatic cardiac disease following mediastinal irradiation. J Am Coll Cardiol. 2003;42:743-9
29. Filopei J, Frishman W. Radiation-induced heart disease. Cardiol Rev. 2012;20:184-8
30. Madan R, Benson R, Sharma DN, Julka PK, Rath GK. Radiation induced heart disease: Pathogenesis, management and review literature. J Egypt Natl Canc Inst. 2015;27:187-93
31. Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D. et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293:293-7
32. Hendry JH, Akahoshi M, Wang LS, Lipshultz SE, Stewart FA, Trott KR. Radiation-induced cardiovascular injury. Radiat Environ Biophys. 2008;47:189-93
33. Finch W, Shamsa K, Lee MS. Cardiovascular complications of radiation exposure. Rev Cardiovasc Med. 2014;15:232-44
34. Cutter DJ, Schaapveld M, Darby SC, Hauptmann M, van Nimwegen FA, Krol AD. et al. Risk of valvular heart disease after treatment for Hodgkin lymphoma. J Natl Cancer Inst. 2015:107
35. Wethal T, Lund MB, Edvardsen T, Fossa SD, Pripp AH, Holte H. et al. Valvular dysfunction and left ventricular changes in Hodgkin's lymphoma survivors. A longitudinal study. Br J Cancer. 2009;101:575-81
36. Cuomo JR, Sharma GK, Conger PD, Weintraub NL. Novel concepts in radiation-induced cardiovascular disease. World J Cardiol. 2016;8:504-19
37. Jaworski C, Mariani JA, Wheeler G, Kaye DM. Cardiac complications of thoracic irradiation. J Am Coll Cardiol. 2013;61:2319-28
38. Giraud P, Cosset JM. [Radiation toxicity to the heart: physiopathology and clinical data]. Bulletin du cancer. 2004;91(Suppl 3):147-53
39. Liu LK, Ouyang W, Zhao X, Su Sh F, Yang Y, Ding WJ. et al. Pathogenesis and Prevention of Radiation-induced Myocardial Fibrosis. Asian Pac J Cancer Prev. 2017;18:583-7
40. Zhao W, Robbins ME. Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: therapeutic implications. Curr Med Chem. 2009;16:130-43
41. Boerma M, Hauer-Jensen M. Preclinical research into basic mechanisms of radiation-induced heart disease. Cardiol Res Pract. 2010. 2011
42. Boerma M, Sridharan V, Mao XW, Nelson GA, Cheema AK, Koturbash I. et al. Effects of ionizing radiation on the heart. Mutat Res. 2016;770:319-27
43. Lauk S, Trott KR. Endothelial cell proliferation in the rat heart following local heart irradiation. Int J Radiat Biol. 1990;57:1017-30
44. Fajardo LF, Stewart JR. Capillary injury preceding radiation-induced myocardial fibrosis. Radiology. 1971;101:429-33
45. Carr ZA, Land CE, Kleinerman RA, Weinstock RW, Stovall M, Griem ML. et al. Coronary heart disease after radiotherapy for peptic ulcer disease. Int J Radiat Oncol Biol Phys. 2005;61:842-50
46. Verheij M, Dewit LG, Boomgaard MN, Brinkman HJ, van Mourik JA. Ionizing radiation enhances platelet adhesion to the extracellular matrix of human endothelial cells by an increase in the release of von Willebrand factor. Radiat Res. 1994;137:202-7
47. Boerma M, Kruse JJ, van Loenen M, Klein HR, Bart CI, Zurcher C. et al. Increased deposition of von Willebrand factor in the rat heart after local ionizing irradiation. Strahlenther Onkol. 2004;180:109-16
48. Hoving S, Heeneman S, Gijbels MJ, te Poele JA, Russell NS, Daemen MJ. et al. Single-dose and fractionated irradiation promote initiation and progression of atherosclerosis and induce an inflammatory plaque phenotype in ApoE(-/-) mice. Int J Radiat Oncol Biol Phys. 2008;71:848-57
49. Seemann I, Te Poele JA, Hoving S, Stewart FA. Mouse bone marrow-derived endothelial progenitor cells do not restore radiation-induced microvascular damage. ISRN Cardiol. 2014;2014:506348
50. Hallahan DE, Virudachalam S. Intercellular adhesion molecule 1 knockout abrogates radiation induced pulmonary inflammation. Proc Natl Acad Sci U S A. 1997;94:6432-7
51. Hallahan DE, Virudachalam S. Ionizing radiation mediates expression of cell adhesion molecules in distinct histological patterns within the lung. Cancer Res. 1997;57:2096-9
52. Heckmann M, Douwes K, Peter R, Degitz K. Vascular activation of adhesion molecule mRNA and cell surface expression by ionizing radiation. Exp Cell Res. 1998;238:148-54
53. Gallo RL, Dorschner RA, Takashima S, Klagsbrun M, Eriksson E, Bernfield M. Endothelial cell surface alkaline phosphatase activity is induced by IL-6 released during wound repair. J Invest Dermatol. 1997;109:597-603
54. Van Der Meeren A, Squiban C, Gourmelon P, Lafont H, Gaugler MH. Differential regulation by IL-4 and IL-10 of radiation-induced IL-6 and IL-8 production and ICAM-1 expression by human endothelial cells. Cytokine. 1999;11:831-8
55. Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675-80
56. Meeren AV, Bertho JM, Vandamme M, Gaugler MH. Ionizing radiation enhances IL-6 and IL-8 production by human endothelial cells. Mediators Inflamm. 1997;6:185-93
57. Kruse JJ, Floot BG, te Poele JA, Russell NS, Stewart FA. Radiation-induced activation of TGF-beta signaling pathways in relation to vascular damage in mouse kidneys. Radiat Res. 2009;171:188-97
58. Schultz-Hector S, Balz K. Radiation-induced loss of endothelial alkaline phosphatase activity and development of myocardial degeneration. An ultrastructural study. Lab Invest. 1994;71:252-60
59. Mathias D, Mitchel RE, Barclay M, Wyatt H, Bugden M, Priest ND. et al. Low-dose irradiation affects expression of inflammatory markers in the heart of ApoE -/- mice. PLoS One. 2015;10:e0119661
60. Bae YS, Oh H, Rhee SG, Yoo YD. Regulation of reactive oxygen species generation in cell signaling. Mol Cells. 2011;32:491-509
61. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524-37
62. Matthews PM, Taivassalo T. Applications of magnetic resonance spectroscopy to diagnosis and monitoring of mitochondrial disease. Ital J Neurol Sci. 1997;18:341-51
63. Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93:903-7
64. Chen K, Keaney JF Jr. Evolving concepts of oxidative stress and reactive oxygen species in cardiovascular disease. Curr Atheroscler Rep. 2012;14:476-83
65. Sugamura K, Keaney JF Jr. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med. 2011;51:978-92
66. Vorotnikova E, Rosenthal RA, Tries M, Doctrow SR, Braunhut SJ. Novel synthetic SOD/catalase mimetics can mitigate capillary endothelial cell apoptosis caused by ionizing radiation. Radiat Res. 2010;173:748-59
67. Marnett LJ, Riggins JN, West JD. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J Clin Invest. 2003;111:583-93
68. Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H. et al. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic Biol Med. 2012;53:260-70
69. Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002;106:927-32
70. Bhattacharya S, Asaithamby A. Ionizing radiation and heart risks. Semin Cell Dev Biol. 2016;58:14-25
71. Sridharan V, Aykin-Burns N, Tripathi P, Krager KJ, Sharma SK, Moros EG. et al. Radiation-induced alterations in mitochondria of the rat heart. Radiat Res. 2014;181:324-34
72. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885-96
73. Bakshi MV, Barjaktarovic Z, Azimzadeh O, Kempf SJ, Merl J, Hauck SM. et al. Long-term effects of acute low-dose ionizing radiation on the neonatal mouse heart: a proteomic study. Radiat Environ Biophys. 2013;52:451-61
74. Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780:1325-36
75. Yarnold J, Brotons MC. Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol. 2010;97:149-61
76. Weintraub NL, Jones WK, Manka D. Understanding radiation-induced vascular disease. J Am Coll Cardiol. 2010;55:1237-9
77. Moro C, Jouan MG, Rakotovao A, Toufektsian MC, Ormezzano O, Nagy N. et al. Delayed expression of cytokines after reperfused myocardial infarction: possible trigger for cardiac dysfunction and ventricular remodeling. Am J Physiol Heart Circ Physiol. 2007;293:H3014-9
78. Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A. et al. Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol. 2013;4:102
79. Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA. et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680-7
80. Di Lisa F, Carpi A, Giorgio V, Bernardi P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim Biophys Acta. 2011;1813:1316-22
81. Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora's box opens. Nat Rev Mol Cell Biol. 2001;2:67-71
82. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99-163
83. An J, Li P, Li J, Dietz R, Donath S. ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity. J Mol Med (Berl). 2009;87:401-10
84. Salata C, Ferreira-Machado SC, De Andrade CB, Mencalha AL, Mandarim-De-Lacerda CA, de Almeida CE. Apoptosis induction of cardiomyocytes and subsequent fibrosis after irradiation and neoadjuvant chemotherapy. Int J Radiat Biol. 2014;90:284-90
85. Indo HP, Inanami O, Koumura T, Suenaga S, Yen HC, Kakinuma S. et al. Roles of mitochondria-generated reactive oxygen species on X-ray-induced apoptosis in a human hepatocellular carcinoma cell line, HLE. Free Radic Res. 2012;46:1029-43
86. Motoori S, Majima HJ, Ebara M, Kato H, Hirai F, Kakinuma S. et al. Overexpression of mitochondrial manganese superoxide dismutase protects against radiation-induced cell death in the human hepatocellular carcinoma cell line HLE. Cancer Res. 2001;61:5382-8
87. Ogura A, Oowada S, Kon Y, Hirayama A, Yasui H, Meike S. et al. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett. 2009;277:64-71
88. Kobashigawa S, Suzuki K, Yamashita S. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem Biophys Res Commun. 2011;414:795-800
89. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001;61:3894-901
90. Simone NL, Soule BP, Ly D, Saleh AD, Savage JE, Degraff W. et al. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS One. 2009;4:e6377
91. Kura B, Yin C, Frimmel K, Krizak J, Okruhlicova L, Kukreja RC. et al. Changes of microRNA-1, -15b and -21 levels in irradiated rat hearts after treatment with potentially radioprotective drugs. Physiol Res. 2016;65(Suppl 1):S129-37
92. Slezak J, Kura B, Ravingerova T, Tribulova N, Okruhlicova L, Barancik M. Mechanisms of cardiac radiation injury and potential preventive approaches. Can J Physiol Pharmacol. 2015;93:737-53
93. Wang Y, Scheiber MN, Neumann C, Calin GA, Zhou D. MicroRNA regulation of ionizing radiation-induced premature senescence. Int J Radiat Oncol Biol Phys. 2011;81:839-48
94. Zhu H, Fan GC. Role of microRNAs in the reperfused myocardium towards post-infarct remodelling. Cardiovasc Res. 2012;94:284-92
95. Viczenczova C, Kura B, Egan Benova T, Yin C, Kukreja RC, Slezak J. et al. Irradiation-Induced Cardiac Connexin-43 and miR-21 Responses Are Hampered by Treatment with Atorvastatin and Aspirin. International journal of molecular sciences. 2018:19
96. Hu Y, Xia W, Hou M. Macrophage migration inhibitory factor serves a pivotal role in the regulation of radiation-induced cardiac senescencethrough rebalancing the microRNA-34a/sirtuin 1 signaling pathway. Int J Mol Med. 2018;42:2849-58
97. Park SY, Lee JH, Ha M, Nam JW, Kim VN. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol. 2009;16:23-9
98. da Costa Martins PA, Salic K, Gladka MM, Armand AS, Leptidis S, el Azzouzi H. et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat Cell Biol. 2010;12:1220-7
99. Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM. et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. 2012;110:71-81
100. Yeboa DN, Evans SB. Contemporary Breast Radiotherapy and Cardiac Toxicity. Semin Radiat Oncol. 2016;26:71-8
101. Maraldo MV, Brodin NP, Aznar MC, Vogelius IR, Munck af Rosenschold P, Petersen PM. et al. Estimated risk of cardiovascular disease and secondary cancers with modern highly conformal radiotherapy for early-stage mediastinal Hodgkin lymphoma. Ann Oncol. 2013;24:2113-8
102. Reardon KA, Read PW, Morris MM, Reardon MA, Geesey C, Wijesooriya K. A comparative analysis of 3D conformal deep inspiratory-breath hold and free-breathing intensity-modulated radiation therapy for left-sided breast cancer. Med Dosim. 2013;38:190-5
103. Taylor CW, Povall JM, McGale P, Nisbet A, Dodwell D, Smith JT. et al. Cardiac dose from tangential breast cancer radiotherapy in the year 2006. Int J Radiat Oncol Biol Phys. 2008;72:501-7
104. Zhang K, He X, Zhou Y, Gao L, Qi Z, Chen J. et al. Atorvastatin Ameliorates Radiation-Induced Cardiac Fibrosis in Rats. Radiat Res. 2015;184:611-20
105. van der Veen SJ, Ghobadi G, de Boer RA, Faber H, Cannon MV, Nagle PW. et al. ACE inhibition attenuates radiation-induced cardiopulmonary damage. Radiother Oncol. 2015;114:96-103
106. O'Herron T, Lafferty J. Prophylactic use of colchicine in preventing radiation induced coronary artery disease. Med Hypotheses. 2018;111:58-60
107. Sun W, Lee TS, Zhu M, Gu C, Wang Y, Zhu Y. et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006;114:2655-62
108. Ichihara S, Noda A, Nagata K, Obata K, Xu J, Ichihara G. et al. Pravastatin increases survival and suppresses an increase in myocardial matrix metalloproteinase activity in a rat model of heart failure. Cardiovasc Res. 2006;69:726-35
109. Kim JW, Rhee CK, Kim TJ, Kim YH, Lee SH, Yoon HK. et al. Effect of pravastatin on bleomycin-induced acute lung injury and pulmonary fibrosis. Clin Exp Pharmacol Physiol. 2010;37:1055-63
110. Ostrau C, Hulsenbeck J, Herzog M, Schad A, Torzewski M, Lackner KJ. et al. Lovastatin attenuates ionizing radiation-induced normal tissue damage in vivo. Radiother Oncol. 2009;92:492-9
111. Haydont V, Mathe D, Bourgier C, Abdelali J, Aigueperse J, Bourhis J. et al. Induction of CTGF by TGF-beta1 in normal and radiation enteritis human smooth muscle cells: Smad/Rho balance and therapeutic perspectives. Radiother Oncol. 2005;76:219-25
112. Eberlein M, Heusinger-Ribeiro J, Goppelt-Struebe M. Rho-dependent inhibition of the induction of connective tissue growth factor (CTGF) by HMG CoA reductase inhibitors (statins). Br J Pharmacol. 2001;133:1172-80
113. Doi H, Matsumoto S, Odawara S, Shikata T, Kitajima K, Tanooka M. et al. Pravastatin reduces radiation-induced damage in normal tissues. Exp Ther Med. 2017;13:1765-72
114. Mahmoudi M, Gorenne I, Mercer J, Figg N, Littlewood T, Bennett M. Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ Res. 2008;103:717-25
115. Monceau V, Pasinetti N, Schupp C, Pouzoulet F, Opolon P, Vozenin MC. Modulation of the Rho/ROCK pathway in heart and lung after thorax irradiation reveals targets to improve normal tissue toxicity. Current drug targets. 2010;11:1395-404
116. Bertrand ME. Provision of cardiovascular protection by ACE inhibitors: a review of recent trials. Curr Med Res Opin. 2004;20:1559-69
117. Boerma M, Singh P, Sridharan V, Tripathi P, Sharma S, Singh SP. Effects of Local Heart Irradiation in a Glutathione S-Transferase Alpha 4-Null Mouse Model. Radiat Res. 2015;183:610-9
118. Lee TC, Greene-Schloesser D, Payne V, Diz DI, Hsu FC, Kooshki M. et al. Chronic administration of the angiotensin-converting enzyme inhibitor, ramipril, prevents fractionated whole-brain irradiation-induced perirhinal cortex-dependent cognitive impairment. Radiat Res. 2012;178:46-56
119. Ghosh SN, Zhang R, Fish BL, Semenenko VA, Li XA, Moulder JE. et al. Renin-Angiotensin system suppression mitigates experimental radiation pneumonitis. International journal of radiation oncology, biology, physics. 2009;75:1528-36
120. Alite F, Balasubramanian N, Adams W, Surucu M, Mescioglu I, Harkenrider MM. Decreased Risk of Radiation Pneumonitis With Coincident Concurrent Use of Angiotensin-converting Enzyme Inhibitors in Patients Receiving Lung Stereotactic Body Radiation Therapy. Am J Clin Oncol. 2018;41:576-80
121. Yang WS, Yang E, Kim MJ, Jeong D, Yoon DH, Sung GH. et al. Momordica charantia Inhibits Inflammatory Responses in Murine Macrophages via Suppression of TAK1. Am J Chin Med. 2018;46:435-52
122. Gu J, Liu K, Li H, Wang X, Yang K. Astragalus saponin attenuates the expression of fibrosis-related molecules in irradiated cardiac fibroblasts. Acta Biochim Biophys Sin (Shanghai). 2014;46:492-501
123. Mansour HH, Tawfik SS. Early treatment of radiation-induced heart damage in rats by caffeic acid phenethyl ester. Eur J Pharmacol. 2012;692:46-51
124. Sridharan V, Tripathi P, Aykin-Burns N, Krager KJ, Sharma SK, Moros EG. et al. A tocotrienol-enriched formulation protects against radiation-induced changes in cardiac mitochondria without modifying late cardiac function or structure. Radiat Res. 2015;183:357-66
125. Boerma M, Roberto KA, Hauer-Jensen M. Prevention and treatment of functional and structural radiation injury in the rat heart by pentoxifylline and alpha-tocopherol. Int J Radiat Oncol Biol Phys. 2008;72:170-7
126. Fan Z, Han Y, Ye Y, Liu C, Cai H. l-carnitine preserves cardiac function by activating p38 MAPK/Nrf2 signalling in hearts exposed to irradiation. Eur J Pharmacol. 2017;804:7-12
127. Gurses I, Ozeren M, Serin M, Yucel N, Erkal HS. Histopathological evaluation of melatonin as a protective agent in heart injury induced by radiation in a rat model. Pathol Res Pract. 2014;210:863-71
128. Kruse JJ, Strootman EG, Wondergem J. Effects of amifostine on radiation-induced cardiac damage. Acta Oncol. 2003;42:4-9
129. Boerma M, Wang J, Sridharan V, Herbert JM, Hauer-Jensen M. Pharmacological induction of transforming growth factor-beta1 in rat models enhances radiation injury in the intestine and the heart. PLoS One. 2013;8:e70479
130. Rabender C, Mezzaroma E, Mauro AG, Mullangi R, Abbate A, Anscher M. et al. IPW-5371 Proves Effective as a Radiation Countermeasure by Mitigating Radiation-Induced Late Effects. Radiation Research. 2016;186:478-88
131. Gu AX, Jie YM, Sun L, Zhao SP, Mingyan E, You QS. RhNRG-1 beta Protects the Myocardium against Irradiation-Induced Damage via the ErbB2-ERK-SIRT1 Signaling Pathway. Plos One. 2015:10
Author contact
![]() Corresponding author: Prof. Xia Yin, Ph.D, Department of Cardiology, The First Hospital of Jilin University, 71 Xinmin Street, Changchun, 130021 China, Phone: +86-15804303063, email: xiayintongcom Prof. Xin Jiang, Ph.D, Department of Radiation Oncology, The First Hospital of Jilin University, 71 Xinmin Street, Changchun, 130021 China, Phone: +86-15804302750, email: jiangxedu.cn
Corresponding author: Prof. Xia Yin, Ph.D, Department of Cardiology, The First Hospital of Jilin University, 71 Xinmin Street, Changchun, 130021 China, Phone: +86-15804303063, email: xiayintongcom Prof. Xin Jiang, Ph.D, Department of Radiation Oncology, The First Hospital of Jilin University, 71 Xinmin Street, Changchun, 130021 China, Phone: +86-15804302750, email: jiangxedu.cn