Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(12):2561-2575. doi:10.7150/ijbs.34985 This issue Cite
Research Paper
EGF suppresses the expression of miR-124a in pancreatic β cell lines via ETS2 activation through the MEK and PI3K signaling pathways
Lin Yang*, Yuansen Zhu*, Delin Kong, Jiawei Gong, Wen Yu, Yang Liang, Yuzhe Nie, Chun-Bo Teng ![]()
College of Life Science, Northeast Forestry University, Harbin, China
*These authors contributed equally to this work
Received 2019-3-18; Accepted 2019-8-11; Published 2019-9-7
Abstract

Diabetes mellitus is characterized by pancreatic β cell dysfunction. Previous studies have indicated that epidermal growth factor (EGF) and microRNA-124a (miR-124a) play opposite roles in insulin biosynthesis and secretion by beta cells. However, the underlying mechanisms remain poorly understood. In the present study, we demonstrated that EGF could inhibit miR-124a expression in beta cell lines through downstream signaling pathways, including mitogen-activated protein kinase kinase (MEK) and phosphatidylinositol 3-kinase (PI3K) cascades. Further, the transcription factor ETS2, a member of the ETS (E26 transformation-specific) family, was identified to be responsible for the EGF-mediated suppression of miR-124a expression, which was dependent on ETS2 phosphorylation at threonine 72. Activation of ETS2 decreased miR-124a promoter transcriptional activity through the putative conserved binding sites AGGAANA/TN in three miR-124a promoters located in different chromosomes. Of note, ETS2 played a positive role in regulating beta cell function-related genes, including miR-124a targets, Forkhead box a2 (FOXA2) and Neurogenic differentiation 1 (NEUROD1), which may have partly been through the inhibition of miR-124 expression. Knockdown and overexpression of ETS2 led to the prevention and promotion of insulin biosynthesis respectively, while barely affecting the secretion ability. These results suggest that EGF may induce the activation of ETS2 to inhibit miR-124a expression to maintain proper beta cell functions and that ETS2, as a novel regulator of insulin production, is a potential therapeutic target for diabetes mellitus treatment.
Keywords: EGFR signaling, MicroRNA-124a, ETS2, Beta cells, Insulin.
Introduction
Diabetes mellitus, including type 1 (T1D) and type 2 diabetes (T2D), is a prevalent disease worldwide. A better understanding of the biological and physiological role of pancreatic β cells could help develop new therapeutic strategies [1-4]. MiRNAs have been indicated to play an important role in human islet β cell biology and to be potential candidates for a new pharmacological strategy [5-8]. Initially described as a brain‐specific miRNA in mammals [9, 10], MiR-124a-3p (miR-124a), is also well represented in the mouse pancreatic MIN6 β‐cell line [11]. Increasing expression levels of miR-124a have been found during embryonic pancreas development [12] and differentiation of human induced pluripotent stem cells (hiPSCs) into insulin-producing cells [13], indicating its function in pancreatic β cell development. Emerging data suggest that miR-124a is overexpressed in pancreatic islets in humans with T2D compared to that in subjects without diabetes [14-16]. MiR-124a has also been shown to play a negative role in insulin biosynthesis and secretion by directly targeting the expression of the glucose-sensing regulator forkhead box a2 (FOXA2) [12, 15], and some key components of the exocytotic machinery, such as Rab27 [17], NOC2 [17], and MTPN [15] in pancreatic β cells. NeuroD1, a transcription factor involved in insulin gene transcription [18] was also predicted to be the targets of miR-124a [19], and was increased in mRNA levels upon miR-124a silencing [15]. These studies indicate that miR-124a is potentially involved in beta cell dysfunction in T2D. Furthermore, the low expression levels of miR-124a in pancreatic islets in subjects without diabetes (in comparison with several other pancreatic islet microRNAs) [14, 20], suggests the need to maintain low miR-124a expression levels in adult insulin-producing cells for proper function.
Epidermal growth factor (EGF) has a complex role in pancreatic development and beta cell maintenance and function. EGF binds to its receptor EGFR for signaling via multiple downstream pathways, including PI3K/AKT, Ras/MEK/ERK1/2, JNK, JAK/STAT3, and others [21, 22]. EGFR signaling is required for normal beta cell development, as EGFR-/- mice display impaired beta cell mass and disrupted islet migration [23], and because constitutively active EGFR (L858R) increases beta cell proliferation and mass in newborn mice [24]. In addition, a recent study has shown that EGF-mediated EGFR signaling could inhibit fetal beta cell differentiation via apical polarity [25]. EGFR signaling is also essential for beta cell proliferation after birth, and even a modest attenuation of this pathway leads to severe defects in the postnatal growth of beta cells [26]. EGFR is also involved in beta cell proliferation and survival in response to high-fat diet, pregnancy [27] and pancreatic duct ligation [28], as well as other extracellular stimuli that induce compensatory expansion of beta cells [24, 29, 30]. Interestingly, in the adult pancreas, EGF plays a role in acinar to beta cell reprogramming in mice with chronic hyperglycemia [31], and it is indispensable for the in vitro generation of insulin-producing beta cells from adult pancreatic acinar cells or human pluripotent stem cells [32-34]. Additionally, there are studies showing that EGF can stimulate insulin secretion by activating phospholipase D2 (PLD2) [35, 36]. Our previous studies have shown that EGFR signaling can inhibit miR-124a expression to regulate pancreatic progenitor cell proliferation [37]. Therefore, we wondered whether there exists a similar mechanism of EGFR signaling modulating miR-124a expression for proper beta cell activity.
In this study, we investigated the regulatory effect of EGFR signaling on miR-124a in pancreatic beta cells. We found that EGF suppressed miR-124a expression in beta cell lines, which was mainly attributed to the ERK and PI3K pathways downstream of EGFR signaling cascades. We further identified that the activation of ETS2, an ETS (E26 transformation-specific) transcription factor, contributed to the inhibition of miR-124a expression by EGF. The putative ETS2-binding sites on the miR-124a promoters were also determined. In addition, we demonstrated the positive effects of ETS2 on insulin biosynthesis.
Materials and Methods
Cell culture
The murine pancreatic β cells MIN6, obtained from ATCC (Manassas, VA, USA), were cultured in High-Glucose DMEM (HyClone™, Logan, UT) containing 15% (v/v) FBS (Biological Industries, Kibbutz Beit Haemek, Israel), 1% (v/v) GlutaMax Supplement (Gibco, Grand Island, NY, USA), 1% (v/v) penicillin-streptomycin (HyClone™, Logan, UT), and 1× B27 (Invitrogen, Carlsbad, CA) at 37°C in a 5% CO2 atmosphere. The rat cell line INS-1 was maintained in RPMI‐1640 medium (HyClone™, Logan, UT) containing 10% FBS, 1% GlutaMax Supplement, 1% penicillin-streptomycin, 50 μmol/L 2‐mercaptoethanol (Sigma, St. Louis, MO) and 1 mM sodium pyruvate (HyClone™, Logan, UT).
Analysis of gene expression
RNA was extracted with Tripure™ reagent (Roche, Basel, Switzerland), and cDNA was synthesized with TransScript II One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China). Mature miR-124a was quantified based on the stem-loop reverse transcript qPCR method using the artificially designed stem-loop-RT primer and qPCR primers. MiR-124a expression was normalized to snRNA U6 (RnU6). Meanwhile, cDNA transcribed through oligo (dT) was used for the analysis of other gene expression, and ACTB (β-actin) was used as an internal control. qPCR was performed using SYBR Green Mix (Roche, Indianapolis, USA) with a Roche LightCycler 480 II Real-Time PCR System. The primer sequences used here are listed Tables S1 and S2 in supplementary materials.
Transient transfection
Small interfering RNAs (siRNAs) and miRNA mimics were designed and synthesized by GenePharma (Shanghai, China, Table S4). Plasmids were extracted and purified using the EasyPure HiPure Plasmid MiniPrep Kit (TransGen Biotech, Beijing, China). Cells were transfected with RNA oligonucleotides or plasmids using Lipofectamine RNAiMAX or 2000 (Thermo Fisher Scientific, Wilmington, USA) according to the manufacturer's instructions. After 24 hours, the cells were treated with or without DMSO (Sigma, St. Louis, MO), 20 μM U0126 (Beyotime, Shanghai, China), 20 μM LY294002 (Beyotime, Shanghai, China), or 5 μM Gefitinib (Sigma, St. Louis, MO). At 36 to 48 hours posttransfection, the cells were harvested for real-time qPCR or Western blotting. For EGF (R&D Systems, Minneapolis, USA) treatment, 24 hours posttransfection, cells were starved for 12 hours and 20 ng/mL EGF was added for another 12 h. Then, cells were harvested for real-time qPCR or Western blotting.
Plasmids construction
The full-length mouse ETS2 gene (NM_011809.3) was amplified using cDNA from MIN6 cells, and the fragment was cloned into the overexpression vector pcDNA3.1 (+) using One Step Cloning Kit II (Vazyme, Nanjing, China). The ETS2-T72A mutant plasmid was constructed using artificially designed primers and the ClonExpress MultiS One Step Cloning Kit (Vazyme, Nanjing, China). The miR-124 promoter region (-2000 to +1) containing the ETS2-binding motif was amplified by PCR from MIN6 cells. The fragments were cloned into the pGL3-Basic luciferase reporter vector (Promega, Madison, USA), and mutations in the ETS2-binding site were created through multiple pairs of primers and then cloned into pGL3-Basic vector via the ClonExpress MultiS One Step Cloning Kit (Vazyme, Nanjing, China). Primers are listed in Table S3 in supplementary materials.
Western blotting
Cells in 6-well dishes were lysed in RIPA buffer (Beyotime Biotechnology, Shanghai, China) containing protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific, Waltham, MA, USA) and incubated on ice for 15 min. The resulting lysate was centrifuged at 13,000 rpm for 10 min at 4°C. Aliquots of the supernatant were removed for protein quantification using the BCA (bicinchoninic acid assay) kit (Beyotime, Shanghai, China). An equal volume of 5x SDS sample buffer (125 mM Tris-hydrochloric acid, pH 6.8, 4% SDS, 4% 2-mercaptoethanol, and 15% glycerol) was added to each cell lysate sample and incubated for 10 min at 95°C. Equal amounts of protein were loaded on 12% polyacrylamide gels, electrophoresed, and transferred to nitrocellulose filter membranes (Millipore, Billerica, MA, USA). After blocking the membranes with 5% skim milk for 30 min, primary antibodies were reacted with target antigens at 4°C overnight. Secondary antibodies (horseradish peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG) were then incubated with the membranes for 1 h at room temperature. Immunoreactive bands were detected with an ECL Detection Reagent (Tanon, Shanghai, China). Protein levels were normalized to total protein concentrations. The primary antibodies used in this study were against the following: AKT (cat# 4691), p-AKT 473 (cat# 4060), p-AKT 308 (cat# 2965), ERK1/2 (cat# 9102), and p-ERK1/2 (cat# 9101), from Cell Signaling Technology (MA, USA); p-ETS2 (Thr72) (cat# 44-1105G, Thermo Fisher Scientific, Wilmington, USA), ETS2 (cat# ab230519, Abcam, Cambridge, UK), Neurod1 (cat# 12081-1-AP, Proteintech, Chicago, USA), FoxA2 (cat# 22474-1-AP, Proteintech, Chicago, USA), and PDX1 (cat# ab47383, Abcam, Cambridge UK).
Dual-luciferase reporter assay
MIN6 cells were transfected with promoter reporter plasmids and the internal control luciferase vector pRL-TK. Meanwhile, ETS2 overexpression vector or empty vector pCDNA3.1(+) was cotransfected into cells using Lipofectamine 2000 reagent (Invitrogen, California, USA). Forty-eight hours later, cells were lysed, and luciferase activities were measured using a Dual-Luciferase Reporter Assay System (Promega, Madison, USA) on a GloMaxTM 20/20 Luminometer (Promega, Sunnyvale, USA) according to the manufacturer's instructions. For analysis, the firefly luciferase activity was normalized to the Renilla luciferase activity.
Immunofluorescence
After 72 h of transfection, cells were gently washed with 0.01 M PBS (phosphate-buffered saline, pH 7.4), fixed with 4% paraformaldehyde (in PBS) for 20 min at room temperature and then permeabilized with 0.3% Triton X-100 (in PBS) for 13 to 17 min. After blocking with 10% horse serum for 50 min at 37℃, cells were incubated with a primary antibody against PDX1 (cat# ab47383, Abcam, Cambridge UK) or anti-C-peptide (cat# 4593s, Cell Signaling Technology, MA, USA), which had been diluted in 10% horse serum (in PBS), overnight at 4℃. Next, the cells were incubated with secondary antibody diluted in 1% BSA for an hour at 37℃. Cell nuclei were stained with Hoechst 33342 (cat# C1022, Beyotime, Shanghai, China) for 5 min at room temperature. Images were captured using DeltaVision OMX (General Electric Company, Fairfield, Connecticut, USA).
Islets isolation and culture
According to the collagenase digestion method [38], islets were isolated from 8-week-old C57BL/6 female mice with similar weights, which were purchased from the Second Affiliated Hospital of Harbin Medical University (China). Freshly isolated islets were handpicked for purification, and cultured in RPMI 1640 plus 10% FBS and 1× B27 for 3 h before transfected with lentiviral particles. After 24 h of infection, the viral supernatant was replaced by fresh media. 72 h post-infection, they were counted for RNA or protein extraction (200 islets per group) or insulin measurement (10 islets per group). All animal procedures were performed according to protocols approved by the Animal Care and Use Committee of Northeast Forestry University.
Lentivirus preparation
Lentiviral plasmid of shETS2 was constructed through annealing the two primers (Table S3) into pLKO.1-EGFP-puro vector which was cutted with EcoRI and AgeI. Lentivirus was prepared in a polyethylenimine-based method [39]. Briefly, 293T cells were co-transfected with the packaging plasmids pPAX2, pcDNA-MDG, and the transfer plasmids pLKO.1-EGFP-Puro-shETS2-3153 by polyethylenimine for 48 h before harvesting lentiviral particles from the culture medium.
Cellular insulin content measurement and glucose-stimulated insulin secretion assay
One day before transfection, MIN6 cells were seeded in 24-well cell culture plates and then transfected with ETS2 siRNAs or ETS2 expression plasmids or control for 48 h as described above, and the medium was then replaced with DMEM-based complete growth medium (containing 3 mM glucose) 16 h prior to experiments. Next, the cells (or islets infected with shNC or shETS2 lentiviral particles for 72 h) were incubated with glucose-free Krebs-Ringer bicarbonate (KRB) HEPES buffer (115 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl2, 1.2 mM KH2PO4, 20 mM NaHCO3, 16 mM HEPES, 2.56 mM CaCl2, and 0.2% BSA) for 1 h. Then, the cells were collected and homogenized by sonication in phosphate-buffered saline (PBS), while islets were lysed in PBS using a 27-gauge syringe and homogenized by sonication. The cellular insulin content was measured according to a mouse insulin ELISA kit (Njjcbio, Nanjing, China). For the glucose-stimulated insulin secretion assay, after incubation for 1 h in glucose-free KRBH buffer, the cells or islets were treated for 1 hour in KRBH buffer with low (3 mM for MIN6 cells, 2.8 mM for islets) or stimulatory (20 mM for MIN6 cells, 16.7 mmol/l for islets) concentrations of glucose. Supernatants were obtained for the determination of insulin concentration using an ELISA kit. Data were normalized to total protein content, which was measured using a bicinchoninic acid assay.
Statistical analysis
Analysis was performed with GraphPad Prism v.7.0 (GraphPad Software Inc., San Diego, CA, USA). Values were expressed as the mean ± standard error of the mean (SEM) of at least three independent experiments. Significant differences were determined using unpaired two-tailed Student's t-test for two-group comparisons or one-way ANOVA for multiple comparisons. P < 0.05 was considered statistically significant.
Results
EGF inhibits miR-124a expression in both MIN6 and INS-1 cell lines
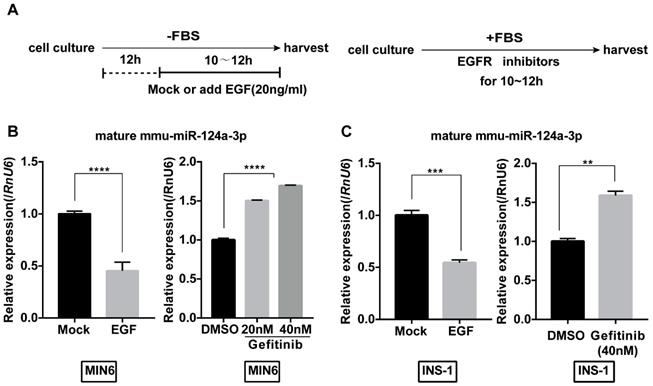
We first examined the effect of EGF on the expression of miR-124a in MIN6 cells. To eliminate the effect of EGF in serum, we starved MIN6 cells for 12 hours in serum-free medium and then treated them with or without EGF (20 ng/mL) under serum-free conditions (Fig. 1A). Levels of mature miR-124a in MIN6 cells started to decrease 6 hours after EGF treatment (Fig. 1B). Moreover, we found that gefitinib, an EGF receptor inhibitor, significantly upregulated the levels of miR-124a to a limited scale in a concentration-dependent manner (Fig. 1B). Interestingly, we also found the same effect of EGF and gefitinib in another β cell line, the rat cell line INS-1 (Fig. 1C). These results indicate that EGF could inhibit miR-124a expression in beta cells and that this pattern of regulation may be conserved among species to some extent.
MiR124a expression is regulated by EGFR signaling in insulinoma cells. (A) Schematic diagram for EGF or inhibitor treatment is displayed. For growth factor treatment (left), cells were first serum-starved for 12 hours (h) and then incubated with serum free of EGF (20 ng/mL) or not for the indicated times, while inhibitors were added without the serum starvation step (right). (B) qRT-PCR was used to determine the expression of mature miR-124a after treatment with EGF (left) or the EGFR inhibitor gefitinib at the indicated concentration (right) in MIN6 cells. (C) The same as in (B), but in INS1 cells. RnU6, U6 small nuclear RNA, internal control for miRNA detection. Data represent the mean ± SE of three independent experiments (each n = 3). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
EGF inhibits miR-124 expression by activating ERK and AKT signaling cascades
EGF-EGFR is well known to stimulate the PI3K/AKT and ERK1/2 signaling pathways [21, 22]. To determine whether these pathways are required for EGF-induced suppression of miR-124a expression, we first treated MIN6 cells with specific inhibitors of these pathways, including U0126 (MEK inhibitor) and LY294002 (PI3K inhibitor). U0126 suppressed the phosphorylation of ERK1/2 in MIN6 cells (Fig. 2C) and was found to stimulate miR-124a expression (Fig. 2A). Additionally, treatment with LY294002 resulted in a similar increase in the level of miR-124a (Fig. 2B) and a decrease in the phosphorylation of AKT at Ser473 (Fig. 2C). Furthermore, both U0126 and LY294002 nearly abrogated the EGF-stimulated decrease in miR-124a expression (Fig. 2D). These observations suggest that the ERK members of the MAPK family and PI3K pathway played an essential role in miR-124a expression in β cells in response to EGF.
Ras/ERK and PI3K/AKT signaling pathways control miR-124a expression. (A) Expression of miR-124a was determined by qRT-PCR in MIN6 cells treated with the inhibitor to MEK (U0126) at the indicated concentration or DMSO for 12 h. (B) The same as in (A), but in cells treated with the inhibitor to PI3K (LY294002) or DMSO. (C) Top: Western blotting was used to monitor the expression of phosphorylated ERK, total ERK, phosphorylated ETS2 (Thr72) and total ETS2 in the presence or absence of U0126; bottom: Western blotting for the levels of phosphorylated AKT (Ser308 and Thr473), total AKT, phosphorylated ETS2 (Thr72) and total ETS2 in the presence or absence of LY294002. (D) qRT-PCR (top) for the expression of miR-124a in MIN6 cells, which were serum-starved overnight and then treated with U0126, LY294002 or DMSO for 30 min prior to the addition of EGF; Western blot for protein levels of phosphorylated ETS2 (Thr72) and total ETS2 in cells under the same conditions as above. (E) Western blotting was used to monitor the expression of phosphorylated AKT, total AKT, phosphorylated ERK, total ERK, phosphorylated ETS2 (Thr72) and total ETS2 in MIN6 cells treated with EGF (left), gefitinib (right) or control medium. RnU6, U6 small nuclear RNA, internal control for microRNA detection. ACTB, actin gene, internal control for cDNA detection. ACTB, actin, internal control for protein detection. Data represent the mean ± SE of three independent experiments (each n = 3). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. ns, not significant.
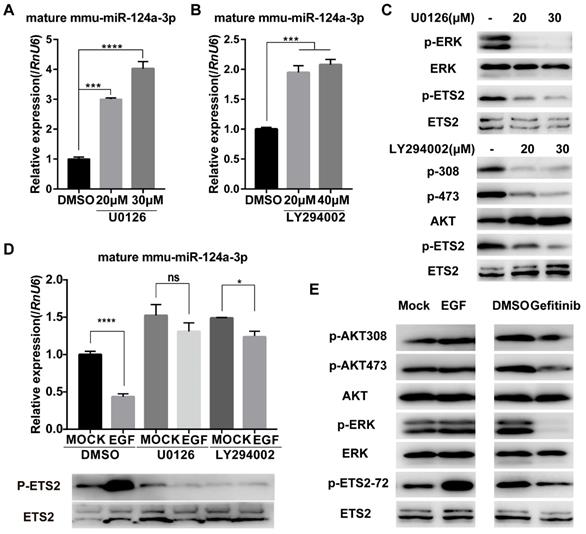
We proposed that EGF may inhibit miR-124a transcriptionally by activating some transcription factors that are downstream effectors of the EGFR signaling pathway and can associate with the miR-124a promoter. The well-defined nuclear targets of the Ras/MAPK pathway include members of the ETS family of transcription factors, including ELK1 and ETS2 [40]. Studies have shown that Ras can stimulate ETS2 phosphorylation at threonine 72 through the Raf/MAPK pathway [41, 42] and some studies have shown that the PI3K/AKT pathway can also lead to the phosphorylation of ETS2 [43], resulting in the ability of ETS2 to activate or repress specific target genes. A decrease in ETS2 phosphorylation at threonine 72 was observed after the treatment of MIN6 cells with the MEK or PI3K inhibitor (Fig. 2C). We also confirmed that EGF could stimulate ETS2 phosphorylation at threonine 72, accompanied by a marked increase in MAPK phosphorylation and a slight increase in AKT phosphorylation in MIN6 cells. The EGFR inhibitor affected ETS2 phosphorylation by abrogating ERK1/2 phosphorylation with slight dephosphorylation of AKT (Fig. 2E). In addition, miR-124a levels were negatively correlated with ETS2 phosphorylation after U0126 and LY294002 treatment prior to EGF incubation (Fig. 2D). Therefore, we speculate that ETS2 was one of the targets downstream of the EGF-MAPK/PI3K axis, leading to the downregulation of miR-124a expression.
ETS2 represses the expression of miR-124a dependent on ERK- or/and AKT- mediated phosphorylation
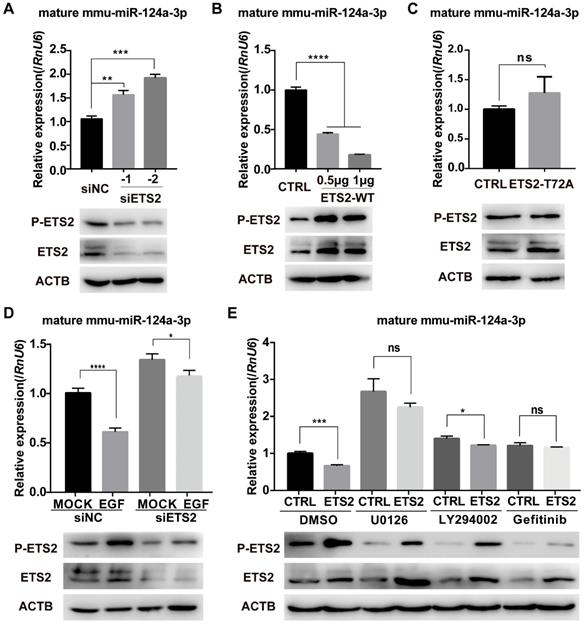
To further determine the role of ETS2 in EGF-induced inhibition of miR-124a expression, we transfected MIN6 cells with ETS2-targeting siRNAs or NC (negative control, scrambled sequences), and an increase in miR-124a expression was observed 48 h post-transfection (Fig. 3A), while ETS2 overexpression indeed blocked the expression of miR-124a as expected (Fig. 3B). In addition, when the mutant type of ETS2 (with the phosphorylation site at threonine 72 replaced by alanine) was overexpressed, no change in miR-124a expression was observed (Fig. 3C). We also examined miR-124a expression levels in cells in which ETS2 was knocked down by siRNAs before EGF treatment. Pretreatment of MIN6 cells with siETS2 was found to eliminate the EGF-stimulated inhibition of miR-124a expression (Fig. 3D), indicating that ETS2 was essential for this inhibition. Moreover, the ETS2 overexpression-induced decrease in miR-124a expression was reversed by inhibitors of MEK, PI3K and EGFR (Fig. 3E), which are important for EGF-induced ETS2 activation, as shown in Fig. 2. Additionally, a negative correlation between ETS2 phosphorylation and miR-124a expression was observed (Fig. 3D, 3E). These results indicated that ETS2 participated in the regulation of miR-124a levels, and its negative role in this process was dependent on its phosphorylation at threonine 72, which was activated by EGF-induced MAPK or/and PI3K signaling pathways. Since other growth factors and related stimuli could stimulate the MAPK pathway-mediated phosphorylation of ETS2 [44], it was not surprising that treatment with U0126 resulted in a greater increase in miR-124a expression levels than treatment with gefitinib, even with ETS2 overexpression (Fig. 3E).
Phosphorylated ETS2 affects miR-124a expression. (A) Expression of ETS2 (phosphorylated and total) and mature miR-124a was determined in MIN6 cells by Western blotting and qRT-PCR, respectively, after transfection with siRNAs targeting ETS2 (siETS2, 50nM) or negative control (siNC) for 48 h. (B) The same as in (A), but in cells transfected with the indicated amount of wild-type ETS2 overexpression vector (ETS2-WT) or empty vector for 48 h. (C) The same as in (B), but in cells transfected with the overexpression vector of mutant ETS2, whose phosphorylation site at threonine 72 was replaced to alanine (ETS2-T72A), or empty vector. (D) Expression of mature miR-124a was determined by qRT-PCR (top) in MIN6 cells transfected first with siETS2 or siNC for 36 h and subsequently serum-starved for 12 h before EGF treatment. Expression of phosphorylated and total ETS2 was monitored by Western blotting (bottom) under the same conditions. (E) Expression of mature miR-124a and ETS2 (phosphorylated and total) was determined by qRT-PCR (top) and immunoblotting (bottom), respectively, in MIN6 cells transfected first with vector containing the wide-type ETS2 coding sequence or control vector for 48 h and subsequently treated with DMSO, U0126, LY294002, or gefitinib. Data represent the mean ± SE of three independent experiments (each n = 3). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. ns, not significant.
ETS2 targets the promoters of miR-124a for inhibition
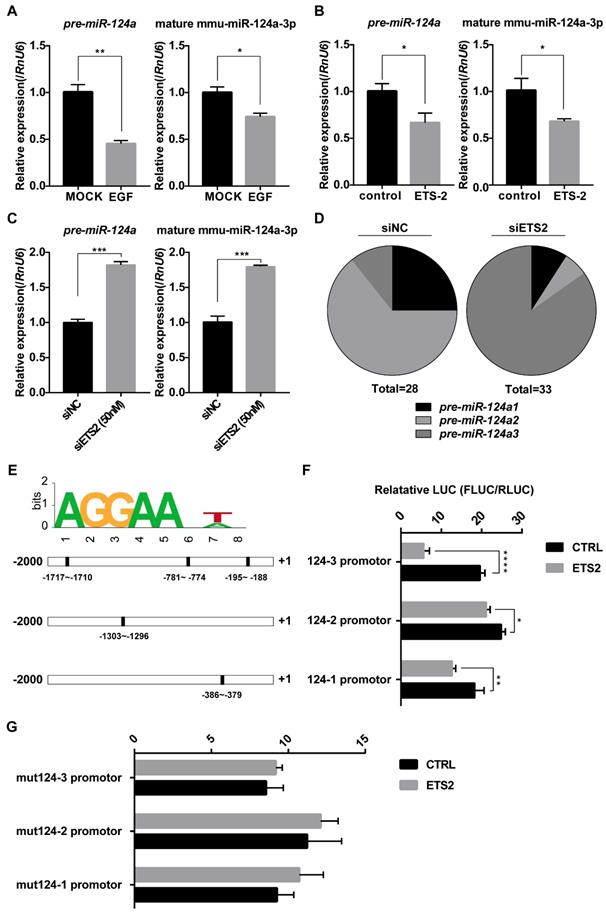
There are three miR-124 precursor genes located on chromosomes 14, 3, and 2 in the mouse genome, encoding mature miR-124a sequences, miR-124a, miR-124a2, and miR-124a3, respectively. To evaluate whether the negative regulation of miR-124a by ETS2 was a direct transcriptional modulation, we first investigated pre-miR-124a expression either under EGF-treated or ETS2-overexpressing conditions or under ETS2-silenced conditions using one pair of primers simultaneously amplifying isoforms 1, 2, and 3 (as shown in Fig. S1A). Corresponding changes in pre-miR-124a expression were observed, as expected (Fig. 4A-4C), indicating that promoter control by ETS2 transcription factor may be responsible for the suppression of miR-124 expression. In addition, after ETS2 knockdown, a significant variation in the proportion of three miR-124a precursors existing in MIN6 cells was observed, and the proportion of pre-miR-124a3 was markedly increased (Fig. 4D). The miR-124a promoter was next investigated using the online analysis tool ALEEGN-PROMO [45] and the JASPAR database for the identification of putative transcription factor-binding sites. Regions 2500 bases upstream and 100 downstream from the transcription start site, which is highly conserved among human, mouse and rat genomes [9, 46], were analyzed for the miR-124a genes. Three of the putative ETS2-binding sites (AGGAANA/TN) were identified on the miR-124a3 promoter, while only one putative ETS2-binding motif was found to be located on the miR-124a1 and miR-124a2 promoters. As shown in the schematic in Fig. 4E, the positions are indicated relative to the transcription initiation site, and most of these binding sites are evolutionarily conserved between mice and rats (Fig. S1B). Subsequently, miR-124a promoter constructs fused to a luciferase reporter were cotransfected with plasmids encoding ETS2, and the results showed that the overexpression of ETS2 could repress the activity of all three miR-124a promoters, especially the miR-124a3 promoter (Fig. 4F). These data were consistent with the results showing that pre-miR-124a3 expression levels were markedly increased after ETS2 knockdown. Furthermore, when mutations were introduced into the ETS2-binding sites by deleting the AGGAA sequence and the seventh nucleotide A/T, the transrepression activity of ETS2 nearly disappeared (Fig. 4F). Together, these findings indicate that ETS2 could bind to highly conserved sites on the miR-124a promoter to induce transcriptional repression.
ETS2 targets the promoters of miR-124a for inhibition. (A) Expression of mature miR-124a and pre-miR-124a was determined by qRT-PCR in MIN6 cells under EGF treatment. (B) The same as in (A), but in MIN6 cells transfected with siETS2 or siNC. (C) The same as in (A), but in MIN6 cell transfected with ETS2 overexpression vector or control vector. (D) Proportion of pre-miR-124a encoded from three different genomic loci (pre-miR-124a1, 2, and 3) was analyzed by clone sequencing (as describe in material and methods) in MIN6 cells treated with siNC (left) or siETS2 (right). Total = 28 or 33 indicates the number of positive clones used in this analysis. (E) A putative ETS2-binding sequence is illustrated (top). Bottom: putative ETS2-binding elements (filled rectangle) are represented in the promoters (-2000 to +1) of miR-124a1, 2 and 3 (outlined rectangle). (F) Luciferase assays were performed to test the effect of ETS2 on miR-124a promoters 1, 2, and 3 in MIN6 cells as described in methods. FLUC: firefly luciferase activity. RLUC: Renilla luciferase activity. (G) Luciferase assays were performed to test critical binding sites for ETS2 to targeting using mutated miR-124a promoters. Mutations were introduced by deleting the first five conserved AGGAA and the seventh nucleotide A/T of the putative ETS2-binding motif. Data represent the mean ± SE of three independent experiments (each n = 3). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. ns, not significant.
ETS2 regulates Foxa2 and NeuroD1 expression partly by inhibiting miR-124a expression
Several reports have indicated a complicated role of miR-124a in pancreatic beta cell development and insulin production and secretion. One of the miR-124a target genes validated in pancreatic β cells is the transcription factor FOXA2 [12], which regulates the expression of important β cell-specific genes such as PDX1 [47, 48]. It has also been reported that miR-124a inhibition increases the expression of NEUROD1 [15], a transcription factor involved in insulin gene transcription [18, 49], and it has been proven that miR-124a can directly target neurod1 mRNA in the 3'-UTR in human cells [19]. Using a miR-124a mimic transfection assay, we validated that miR-124a inhibited the expression of FOXA2 and NEUROD1 in MIN6 cells (Fig. 5A). However, although the EGFR inhibitor markedly blocked the expression of NEUROD1, EGF treatment did not result in a corresponding upregulation of this protein. Moreover, the level of FOXA2 was downregulated in the presence of EGF, while it was slightly increased under conditions of EGFR inhibitor or MAPK inhibitor treatment (Fig. 5A). Thus, our results show that EGF could not simply enhance the expression of miR-124a target genes via the downregulation of miR-124a, and clearly, EGF had a complex influence on beta cell function.
ETS2 regulates Foxa2 and NeuroD1 expression partly by inhibiting miR-124a expression. (A) Expression of FOXA2 and NEUROD1 was determined by immunoblotting in MIN6 cells transfected with negative control (NC) or miR-124a mimics, or incubated with EGF or not, or incubated with DMSO, LY294002, U0126 or gefitinib. (B) Expression of FOXA2 and NEUROD1 were determined by qRT-PCR in MIN6 cells following siETS2 or siNC transfection for 72 h. Subsequently, Western blotting was used to monitor the protein expression of FOXA2, NEUROD, phosphorylated ETS2 (Thr72) and total ETS2. ACTB (β-actin) was used as a loading control. (C) The same as in (B), but in MIN6 cells transfected with plasmids overexpressing wide-type (ETS2-WT) or mutant ETS2 (ETS2-T72A) or empty vector for control. (D) Expression of miR-124a, FOXA2 and NEUROD1 were determined by qRT-PCR and western blot in ETS2-overexpression or control MIN6 cells following miR-124a mimics or NC transfection. To restore miR-124a expression, MIN6 cells were co-transfected with the indicated combinations of mimics and vectors for 72 h. ACTB, the actin gene, internal control for cDNA detection. ACTB, actin, internal control for protein detection. Data represent the mean ± SE of three independent experiments (each n = 3). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.

Since the effective distance of ETS2 to miR-124a was closer than that of EGF, we wondered whether ETS2 could regulate miR-124a target genes and then play a role in beta cell functions. We measured FOXA2 and NEUROD1 levels in ETS2 knockdown or ETS2-overexpressing MIN6 cells. As illustrated in Fig. 5B and 5C, FOXA2 and NEUROD1 expression was suppressed in ETS2-siRNA-transfected cells, while ETS2-overexpressing cells demonstrated a significant increase both at RNA and protein levels. Similar results were obtained in rat beta cells, INS-1 (Fig. S1C). Surprisingly, overexpression of the mutant type of ETS2 (when the phosphorylation site indispensable for the suppression of miR-124a expression was mutated) led to a similar upregulation in FOXA2 and NEUROD1 (Fig. 5C, S1C). To further determine if activation of Foxa2 and NeuroD1 by ETS2 was independent of miR-124a, we enforced miR-124a expression by using miR-124a mimics in ETS2-transfected cells. We found that excess miR-124a prevented the ETS2-mediated upregulation in NEUROD1, while it had little effect on the enhancement of FOXA2 expression induced by ETS2 (Fig. 5D). Therefore, ETS2 could regulate miR-124a target genes FOXA2 and NEUROD1, which may be partly through the inhibition of miR-124a expression.
ETS2 has a positive role in insulin biosynthesis
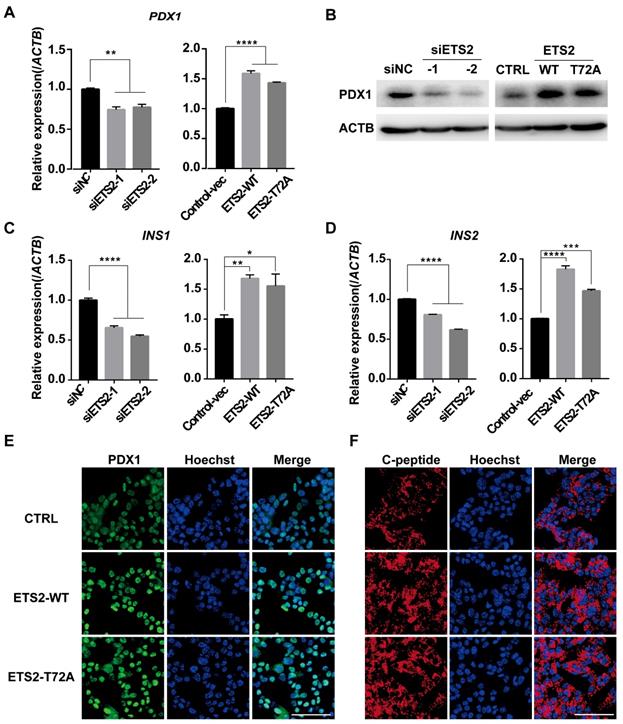
As a downstream gene activated by FOXA2, PDX1 was regulated by ETS2 (Fig. 6A, 6B, 6E and Fig. S1C). Importantly, PDX1 and NEUROD1 are known to play key roles in regulating insulin gene transcription [18, 48]. As expected, similar effects on insulin mRNA level and protein content (represented by C-peptide) were also observed under conditions of ETS2 knockdown or overexpression (Fig. 6C, 6D, 6F, and Fig. S1C). ELISA for total cellular insulin content further illustrated the positive role of ETS2 in insulin biosynthesis (Fig. 7A and 7C). However, we did not find significant variation in the ability of insulin secretion (determined by glucose-stimulated index and the insulin secretion normalized to insulin content) after ETS2 silencing or overexpression (Fig. 7B and 7D). Although the level of insulin secreted from ETS2-silenced or overexpressed MIN6 cells was significantly varied in the presence of 20 mM glucose (Fig. S1D), the percentage of secreted insulin was similar to controls after standardizing with total insulin content (Fig. 7B and 7D). This indicated that, during glucose stimulated insulin secretion, the change of total insulin content in siETS2 or ETS2 transfected MIN6 cells was the major cause of varied insulin release. We also verified the function of ETS2 in isolated mouse islets. As showed in Fig. 7E, knockdown of ETS2 by lentivirus infection led to elevated miR-124 expression, following decreased expression of insulin biosynthesis related genes such as FOXA2, NEUROD1, and PDX1. Impaired insulin biosynthesis and unaffected insulin secretion were also observed in shETS2-islets (Fig. 7F, S1E). These were consistent to the results in beta cell lines.
ETS2 has a positive role in regulation of insulin synthesis key genes. (A) Expression of PDX1 was determined by qRT-PCR and in MIN6 cells transfected with siNC or siETS2, or control vector, ETS2-WT or ETS2-T72A overexpression plasmids. (B) Expression of PDX1 was determined by western blot in ETS2-knockdown or overexpression cells. (C) Expression of INS1 was determined by qRT-PCR in ETS2-Knockdown or overexpression cells. (D) The same as with (C), but for INS2 expression detection. (E) Expression and location of PDX1 (green) were determined by immunofluorescence in MIN6 cells following control, ETS2-WT or ETS2-T72A overexpression plasmids transfection for 72h. Hoechst (blue), dye for nucleus staining. Scale bars, 50 μM. (F), the same with (E), but for C-peptide (red) staining.
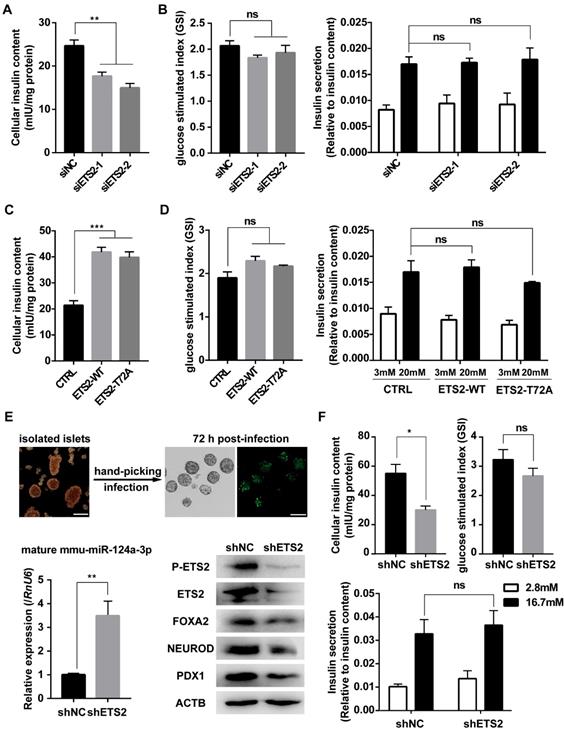
ETS2 promotes insulin biosynthesis in insulinoma cells and isolated murine islets. (A) ELISA was used to monitor the cellular insulin content in MIN6 cells following siNC or siETS2 transfection for 48 h. IU: International Unit. One unit of mouse insulin is contained in 0.0345 mg of the first International Standard. (B) ELISA was used to measure the glucose stimulated index (the ratio of the insulin value after high glucose stimulation divided by insulin value after low glucose stimulation) in MIN6 cells following indicated siRNA transfection for 48 h. And insulin secretion normalized to insulin content (3 mmol/l glucose, white bars; 20 mmol/l glucose, black bars) was measured. (C), the same in (A), but for cell transfected with control, ETS2-WT or ETS2-T72A overexpression plasmids. (D), the same in (B), but for cell transfected with control, ETS2-WT or ETS2-T72A overexpression plasmids. (E) Murine islets were isolated to confirm ETS2 function on miR-124a, FOXA2, NEUROD1 and PDX1 expression, after infection with pLKO.1-EGFP-shNC or shETS2 lentiviral particles for 72 h. Infection efficiency was visualized by EGFP fluorescence. Scale bars, 200 μM. (F) ELISA was used to monitor the cellular insulin content, glucose stimulated index, and insulin secretion normalized to insulin content (2.8 mmol/l glucose, white bars; 16,7 mmol/l glucose, black bars) in ETS2-knockdown or control islets. Values are means ± SE of three independent experiments (each n = 3). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. ns, not significant.
Discussion
Studies have shown that miR-124a plays an important role in pancreatic endocrine development [13, 37], insulin biosynthesis and secretion, and insulin signal transduction [12, 15]. Despite these important biological roles, little is known about the regulation of miR-124 biogenesis in the pancreas. Our previous study showed a mutual inhibition between EGF and miR-124a to determine the proliferation and differentiation of pancreatic progenitor cells [37], while in differentiated beta cells, it was unknown whether EGFR contributes to control miR-124 expression to regulate beta cell function. We described here that EGF activated EGFR signaling cascades, including the MAPK and PI3K pathways, to down-regulate the expression of miR-124a in MIN6 and INS-1 cells (Fig. 1 and 2). However, we did not detect a consistent positive effect of EGFR signaling on miR-124a targets (FOXA2 and NEUROD1) according to EGF and EGFR inhibitor treatments (Fig. 5A). Considering the multiplicity of interactions between microRNA and its targets and the complexity of effects of EGFR signaling on cell survival and proliferation, EGFR signaling-mediated inhibition of miR-124a expression may comprehensively function to regulate the proper activity of beta cells.
ETS2 is a member of the ETS (E26 transformation-specific sequence) family of transcription factors that participates in cellular proliferation, differentiation and apoptosis, thus playing important roles in embryo development [50-53], senescence [54], vasculogenesis [55], immunity [56-59], osteogenesis [60], adipogenesis [61], and tumorigenesis [62, 63]. Some members of the ETS family, such as Etv4 and Etv5, have been indicated in pancreatic development mediating mesenchymal-to-epithelial signaling [64]. ELK1 [65] and ETS1 [66-68] have been reported to inhibit glucose-stimulated insulin secretion by pancreatic β cells and to be related to beta cell dysfunction. Compared with other ETS family members, ETS2 was the most abundant factor expressed during pancreatic morphogenesis [64], as well as in normal adult pancreatic islets (data not shown), which indicated the vital role of ETS2 in pancreatic development and function [64]; however, the relevant details have not yet been addressed. In this study, we showed that ETS2 inhibited miR-124a expression at the transcriptional level and in an ERK1/2- or/and PI3K-dependent manner. We also demonstrated that ETS2 has a positive role in insulin biosynthesis in pancreatic β cells.
ETS2 is an important substrate of the MAPKs ERK1/2 or PI3K, which can be activated by growth factors or cellular stress [43, 44, 69-71]. In our study, we determined that both the ERK1/2 and PI3K signaling pathways are involved in the stimulation of ETS2 phosphorylation in MIN6 cells, since the ERK1/2 or PI3K inhibitor alone could decrease the EGF-induced phosphorylation of ETS2 at threonine 72 (Fig. 2). Here, we found that the suppression of ETS2 activity attenuated EGF-mediated regulation of miR-124a expression by siETS2 or inhibitors of ERK1/2 or PI3K (Fig. 2D and Fig. 3D, 3E), supporting the notion that ETS2 acts as a downstream effector of the EGF/Ras/ERK or EGF/PI3K/AKT pathway to repress miR-124a expression in pancreatic β cells. Phosphorylation at threonine 72 is important for ETS2 activation and has been well studied [71], and it was also found to be essential for ETS2 function in miR-124a regulation (Fig. 3C), and this phenomenon was also found to be essential for ETS2 functions in miR-124a regulation (Fig. 3C). However, ETS2 is phosphorylated on other residues through the ERK or AKT pathway, and the role(s) that these other sites play in the regulation of miR-124a expression remains to be determined. ETS2 has been reported to control the transcription of miRNAs. ETS2 regulates microRNA-126 in endothelial cells [72], stimulates miR-155 during the inflammatory response [73], and represses the expression of miR-196b in gastric cancer cells [74] by binding to motifs containing the conserved sequence GGAA on the promoter. A similar mechanism was identified for the regulatory effect of ETS2 on the miR-124 promoter, and the core conserved sequence AAGGAANA/TN was found to be indispensable in this process (Fig. 4E-4G). Luciferase assay results showed that ETS2 displayed a strong suppressive ability on the miR-124a3 promoter, which may be the result of multiple binding sites located in this region (Fig. 4E, 4F). We identified 100% identical evolutionarily conserved binding motifs between mice and rats (Fig. S1B), in accordance with the similar inhibition of miR-124a expression by EGFR signaling (Fig. 1), further indicating a transrepression role of ETS2 in miR-124a regulation. Nevertheless, it remains to be investigated whether the repressor role of ETS2 in the regulation of miR-124a is dependent on other posttranslational modifications or specific binding partners, as described in [75] that ETS2 can switch between being an activator and being a suppressor by specifically interacting with the co-repressor ZMYND11.
We next found that ETS2 has a positive effect on the transcription factors FOXA2, NEUROD1 and PDX1 (Fig. 5B-5D and Fig. 6), which are known to not only control beta cell development but also maintain mature beta cell function [18, 47-49, 76-79]. NEUROD1, which has been reported to modulate insulin expression, has been confirmed to be regulated by miR-124a (Fig. 5A). Studies have shown that miR-124a targets FOXA2 expression, consequently regulating PDX1 and pre-proinsulin genes (INS1, INS2) transcription [12]. We demonstrated that ETS2-silencing induced a decrease in FOXA2, NEUROD1, PDX1, INS1 and INS2 expression. A reverse effect on these genes was observed in response to ETS2 overexpression (Figs. 5 and 6), potentially reflecting a decrease in miR-124a level. However, mutant ETS2 without the phosphorylation site (T was mutated to A), which has been shown disappeared inhibition on miR-124a expression (Fig. 2C), still participated in the positive regulation of these genes. Thus, ETS2 might regulate NEUROD1 and FOXA2 partly or completely independent of miR-124a. Paradoxically, restored miR-124a eliminated ETS2-overexpression-induced promotion of NEUROD1 expression (Fig. 5D). We suggested that it might result from faster speed of mRNA degradation by excess exogenous miR-124a than that of mRNA production activated by EST2. For FOXA2, results showed that ETS2 could promote FOXA2 expression at mRNA levels (Fig. 5B, 5C, 5D), while miR-124a mimics slightly reduced FOXA2 mRNA levels, but significantly decreased its protein levels in cells without ETS2 overexpression, which was consistent with the previous report [12], indicating translational regulation role of miR-124a on FOXA2 (Fig. 5A, 5D). Therefore, even excess exogenous miR-124a could not block the transcriptional activation of FOXA2 by ETS2. And these results indicate that there exist other pathways or mechanisms by which ETS2 regulates FOXA2 and NEUROD genes independent of miR-124a. Changes in PDX1 and INS1 and INS2 expression may result from variations in FOXA2 and NEUROD and/or direct control of ETS2, following corresponding variation in insulin production both in beta cell lines and isolated mouse islets (Fig. 6 and 7), which was consistent with PDX1 and insulin expression under the same conditions. No change was apparent in ability of glucose-induced insulin secretion after ETS2 treatment (Fig. 6, 7B, 7D and 7F). Thus, ETS2 may not influence the final step of the secretory pathways such as molecular machinery that drives insulin exocytosis, although miR-124a has been reported to impair insulin secretion [15, 17]. There may be homeostatic mechanisms for ETS2 to avoid excess insulin secretion by antagonizing its inhibition on miR-124a, thus maintaining a proper beta cell function.
Conclusions
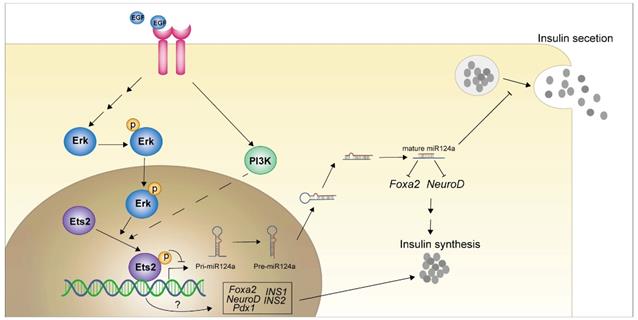
In present study, we showed that EGF activated MEK/ERK and PI3K/AKT signaling cascades to down-regulate miR-124a expression, and a member of ETS family of transcription factors, ETS2, acted as the downstream effector of EGFR signaling to suppression the promoter activity of miR-124a (a kind of miRNA that related to beta cell dysfunction in diabetes). Moreover ETS2 was discovered to positively regulate insulin biogenesis related genes FOXA2, NEUROD1, PDX1, INS1 and INS2 in a T72 phosphorylation independent manner (Fig. 8). Our results expand our understanding of mechanisms of inappropriate regulation of miR-124a involved in processes leading to human disease such as diabetes mellitus, and imply a series of new ETS2 functions, and support future work in exploring this pathway in vivo.
Graphical abstract of how EGFR signaling pathway and downstream effector inhibit the miR-124a expression and the role of ETS2 in insulin production in pancreatic beta cells.
Supplementary Material
Supplementary figure and tables.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (No. 31472159), Natural Science Foundation of Heilongjiang Province (ZD2017001) and the Fundamental Research Funds for the Central Universities of China (No. 2572016EAJ3).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brody H. Diabetes. Nature. 2012;485:S1
2. Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148:1160-1171
3. Dolgin E. Diabetes: Encapsulating the problem. Nature. 2016;540:S60-S62
4. Cheng CW, Villani V, Buono R, Wei M, Kumar S, Yilmaz OH. et al. Fasting-Mimicking Diet Promotes Ngn3-Driven beta-Cell Regeneration to Reverse Diabetes. Cell. 2017;168:775-788 e712
5. Yamada Y, Tabata M, Yasuzaki Y, Nomura M, Shibata A, Ibayashi Y. et al. A nanocarrier system for the delivery of nucleic acids targeted to a pancreatic beta cell line. Biomaterials. 2014;35:6430-6438
6. Filios SR, Shalev A. beta-Cell MicroRNAs: Small but Powerful. Diabetes. 2015;64:3631-3644
7. Poy MN. MicroRNAs: An adaptive mechanism in the pancreatic beta-cell…and beyond? Best Pract Res Clin Endocrinol Metab. 2016;30:621-628
8. Zheng Y, Wang Z, Zhou Z. miRNAs: novel regulators of autoimmunity-mediated pancreatic beta-cell destruction in type 1 diabetes. Cell Mol Immunol. 2017;14:488-496
9. Baroukh NN, Van Obberghen E. Function of microRNA-375 and microRNA-124a in pancreas and brain. FEBS J. 2009;276:6509-6521
10. Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13
11. Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE. et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226-230
12. Baroukh N, Ravier MA, Loder MK, Hill EV, Bounacer A, Scharfmann R. et al. MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J Biol Chem. 2007;282:19575-19588
13. Sebastiani G, Valentini M, Grieco GE, Ventriglia G, Nigi L, Mancarella F. et al. MicroRNA expression profiles of human iPSCs differentiation into insulin-producing cells. Acta Diabetol. 2017;54:265-281
14. Bolmeson C, Esguerra JL, Salehi A, Speidel D, Eliasson L, Cilio CM. Differences in islet-enriched miRNAs in healthy and glucose intolerant human subjects. Biochem Biophys Res Commun. 2011;404:16-22
15. Sebastiani G, Po A, Miele E, Ventriglia G, Ceccarelli E, Bugliani M. et al. MicroRNA-124a is hyperexpressed in type 2 diabetic human pancreatic islets and negatively regulates insulin secretion. Acta Diabetol. 2015;52:523-530
16. Kong L, Zhu J, Han W, Jiang X, Xu M, Zhao Y. et al. Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: a clinical study. Acta Diabetol. 2011;48:61-69
17. Lovis P, Gattesco S, Regazzi R. Regulation of the expression of components of the exocytotic machinery of insulin-secreting cells by microRNAs. Biol Chem. 2008;389:305-312
18. Wong WP, Tiano JP, Liu S, Hewitt SC, Le May C, Dalle S. et al. Extranuclear estrogen receptor-alpha stimulates NeuroD1 binding to the insulin promoter and favors insulin synthesis. Proc Natl Acad Sci U S A. 2010;107:13057-13062
19. Liu K, Liu Y, Mo W, Qiu R, Wang X, Wu JY. et al. MiR-124 regulates early neurogenesis in the optic vesicle and forebrain, targeting NeuroD1. Nucleic Acids Res. 2011;39:2869-2879
20. Kameswaran V, Bramswig NC, McKenna LB, Penn M, Schug J, Hand NJ. et al. Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab. 2014;19:135-145
21. Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44:195-217
22. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12:553-563
23. Miettinen PJ, Huotari M, Koivisto T, Ustinov J, Palgi J, Rasilainen S. et al. Impaired migration and delayed differentiation of pancreatic islet cells in mice lacking EGF-receptors. Development. 2000;127:2617-2627
24. Hakonen E, Ustinov J, Eizirik DL, Sariola H, Miettinen PJ, Otonkoski T. In vivo activation of the PI3K-Akt pathway in mouse beta cells by the EGFR mutation L858R protects against diabetes. Diabetologia. 2014;57:970-979
25. Lof-Ohlin ZM, Nyeng P, Bechard ME, Hess K, Bankaitis E, Greiner TU. et al. EGFR signalling controls cellular fate and pancreatic organogenesis by regulating apicobasal polarity. Nat Cell Biol. 2017;19:1313-1325
26. Miettinen PJ, Ustinov J, Ormio P, Gao R, Palgi J, Hakonen E. et al. Downregulation of EGF receptor signaling in pancreatic islets causes diabetes due to impaired postnatal beta-cell growth. Diabetes. 2006;55:3299-3308
27. Hakonen E, Ustinov J, Mathijs I, Palgi J, Bouwens L, Miettinen PJ. et al. Epidermal growth factor (EGF)-receptor signalling is needed for murine beta cell mass expansion in response to high-fat diet and pregnancy but not after pancreatic duct ligation. Diabetologia. 2011;54:1735-1743
28. Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L. et al. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci U S A. 2014;111:E1211-1220
29. Suarez-Pinzon WL, Yan Y, Power R, Brand SJ, Rabinovitch A. Combination therapy with epidermal growth factor and gastrin increases beta-cell mass and reverses hyperglycemia in diabetic NOD mice. Diabetes. 2005;54:2596-2601
30. Lemper M, De Groef S, Stange G, Baeyens L, Heimberg H. A combination of cytokines EGF and CNTF protects the functional beta cell mass in mice with short-term hyperglycaemia. Diabetologia. 2016;59:1948-1958
31. Baeyens L, Lemper M, Leuckx G, De Groef S, Bonfanti P, Stange G. et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol. 2014;32:76-83
32. Baeyens L, De Breuck S, Lardon J, Mfopou JK, Rooman I, Bouwens L. In vitro generation of insulin-producing beta cells from adult exocrine pancreatic cells. Diabetologia. 2005;48:49-57
33. Minami K, Okuno M, Miyawaki K, Okumachi A, Ishizaki K, Oyama K. et al. Lineage tracing and characterization of insulin-secreting cells generated from adult pancreatic acinar cells. Proc Natl Acad Sci U S A. 2005;102:15116-15121
34. Kumar SS, Alarfaj AA, Munusamy MA, Singh AJ, Peng IC, Priya SP. et al. Recent developments in beta-cell differentiation of pluripotent stem cells induced by small and large molecules. Int J Mol Sci. 2014;15:23418-23447
35. Lee HY, Jung H, Jang IH, Suh PG, Ryu SH. Cdk5 phosphorylates PLD2 to mediate EGF-dependent insulin secretion. Cell Signal. 2008;20:1787-1794
36. Lee HY, Yea K, Kim J, Lee BD, Chae YC, Kim HS. et al. Epidermal growth factor increases insulin secretion and lowers blood glucose in diabetic mice. J Cell Mol Med. 2008;12:1593-1604
37. Zhang Z, Zhai W, Liang J, Chen Z, Ma M, Zhao Y. et al. Mutual inhibitions between epidermal growth factor receptor signaling and miR-124a control pancreatic progenitor proliferation. J Cell Physiol. 2018;234:12978-12988
38. Zmuda EJ, Powell CA, Hai T. A method for murine islet isolation and subcapsular kidney transplantation. J Vis Exp. 2011;50:2096
39. Toledo JR, Prieto Y, Oramas N, Sanchez O. Polyethylenimine-based transfection method as a simple and effective way to produce recombinant lentiviral vectors. Appl Biochem Biotechnol. 2009;157:538-544
40. Wasylyk B, Hagman J, Gutierrez-Hartmann A. Ets transcription factors: nuclear effectors of the Ras-MAP-kinase signaling pathway. Trends Biochem Sci. 1998;23:213-216
41. McCarthy SA, Chen D, Yang BS, Garcia Ramirez JJ, Cherwinski H, Chen XR. et al. Rapid phosphorylation of Ets-2 accompanies mitogen-activated protein kinase activation and the induction of heparin-binding epidermal growth factor gene expression by oncogenic Raf-1. Mol Cell Biol. 1997;17:2401-2412
42. Fowles LF, Martin ML, Nelsen L, Stacey KJ, Redd D, Clark YM. et al. Persistent activation of mitogen-activated protein kinases p42 and p44 and ets-2 phosphorylation in response to colony-stimulating factor 1/c-fms signaling. Mol Cell Biol. 1998;18:5148-5156
43. Smith JL, Schaffner AE, Hofmeister JK, Hartman M, Wei G, Forsthoefel D. et al. ets-2 is a target for an akt (Protein kinase B)/jun N-terminal kinase signaling pathway in macrophages of motheaten-viable mutant mice. Mol Cell Biol. 2000;20:8026-8034
44. Weng LP, Brown JL, Baker KM, Ostrowski MC, Eng C. PTEN blocks insulin-mediated ETS-2 phosphorylation through MAP kinase, independently of the phosphoinositide 3-kinase pathway. Hum Mol Genet. 2002;11:1687-1696
45. Farre D, Roset R, Huerta M, Adsuara JE, Rosello L, Alba MM. et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003;31:3651-3653
46. Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F. et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424-1429
47. Lee CS, Sund NJ, Vatamaniuk MZ, Matschinsky FM, Stoffers DA, Kaestner KH. Foxa2 controls Pdx1 gene expression in pancreatic beta-cells in vivo. Diabetes. 2002;51:2546-2551
48. Liu XD, Ruan JX, Xia JH, Yang SL, Fan JH, Li K. The study of regulatory effects of Pdx-1, MafA and NeuroD1 on the activity of porcine insulin promoter and the expression of human islet amyloid polypeptide. Mol Cell Biochem. 2014;394:59-66
49. Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T. et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23:323-328
50. Georgiades P, Rossant J. Ets2 is necessary in trophoblast for normal embryonic anteroposterior axis development. Development. 2006;133:1059-1068
51. Staun-Ram E, Goldman S, Shalev E. Ets-2 and p53 mediate cAMP-induced MMP-2 expression, activity and trophoblast invasion. Reprod Biol Endocrinol. 2009;7:135
52. Odiatis C, Georgiades P. New insights for Ets2 function in trophoblast using lentivirus-mediated gene knockdown in trophoblast stem cells. Placenta. 2010;31:630-640
53. Polydorou C, Georgiades P. Ets2-dependent trophoblast signalling is required for gastrulation progression after primitive streak initiation. Nat Commun. 2013;4:1658
54. Dwyer J, Li H, Xu D, Liu JP. Transcriptional regulation of telomerase activity: roles of the the Ets transcription factor family. Ann N Y Acad Sci. 2007;1114:36-47
55. Wei G, Srinivasan R, Cantemir-Stone CZ, Sharma SM, Santhanam R, Weinstein M. et al. Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood. 2009;114:1123-1130
56. Zaldumbide A, Carlotti F, Pognonec P, Boulukos KE. The role of the Ets2 transcription factor in the proliferation, maturation, and survival of mouse thymocytes. J Immunol. 2002;169:4873-4881
57. Stankiewicz MJ, Crispino JD. ETS2 and ERG promote megakaryopoiesis and synergize with alterations in GATA-1 to immortalize hematopoietic progenitor cells. Blood. 2009;113:3337-3347
58. Cheng C, Tempel D, Den Dekker WK, Haasdijk R, Chrifi I, Bos FL. et al. Ets2 determines the inflammatory state of endothelial cells in advanced atherosclerotic lesions. Circ Res. 2011;109:382-395
59. Yoshimatsu Y, Yamazaki T, Mihira H, Itoh T, Suehiro J, Yuki K. et al. Ets family members induce lymphangiogenesis through physical and functional interaction with Prox1. J Cell Sci. 2011;124:2753-2762
60. Raouf A, Seth A. Ets transcription factors and targets in osteogenesis. Oncogene. 2000;19:6455-6463
61. Birsoy K, Berry R, Wang T, Ceyhan O, Tavazoie S, Friedman JM. et al. Analysis of gene networks in white adipose tissue development reveals a role for ETS2 in adipogenesis. Development. 2011;138:4709-4719
62. Tynan JA, Wen F, Muller WJ, Oshima RG. Ets2-dependent microenvironmental support of mouse mammary tumors. Oncogene. 2005;24:6870-6876
63. Munera J, Cecena G, Jedlicka P, Wankell M, Oshima RG. Ets2 regulates colonic stem cells and sensitivity to tumorigenesis. Stem Cells. 2011;29:430-439
64. Kobberup S, Nyeng P, Juhl K, Hutton J, Jensen J. ETS-family genes in pancreatic development. Dev Dyn. 2007;236:3100-3110
65. Zhang XF, Zhu Y, Liang WB, Zhang JJ. The ETS-Domain Transcription Factor Elk-1 Regulates COX-2 Gene Expression and Inhibits Glucose-Stimulated Insulin Secretion in the Pancreatic beta -Cell Line INS-1. Int J Endocrinol. 2013;2013:843462
66. Luo Y, He F, Hu L, Hai L, Huang M, Xu Z. et al. Transcription factor Ets1 regulates expression of thioredoxin-interacting protein and inhibits insulin secretion in pancreatic beta-cells. PLoS One. 2014;9:e99049
67. Zhang XF, Zhu Y, Liang WB, Zhang JJ. Transcription factor Ets-1 inhibits glucose-stimulated insulin secretion of pancreatic beta-cells partly through up-regulation of COX-2 gene expression. Endocrine. 2014;46:470-476
68. Chen F, Sha M, Wang Y, Wu T, Shan W, Liu J. et al. Transcription factor Ets-1 links glucotoxicity to pancreatic beta cell dysfunction through inhibiting PDX-1 expression in rodent models. Diabetologia. 2016;59:316-324
69. Foulds CE, Nelson ML, Blaszczak AG, Graves BJ. Ras/mitogen-activated protein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Mol Cell Biol. 2004;24:10954-10964
70. Liu J, Puscheck EE, Wang F, Trostinskaia A, Barisic D, Maniere G. et al. Serine-threonine kinases and transcription factors active in signal transduction are detected at high levels of phosphorylation during mitosis in preimplantation embryos and trophoblast stem cells. Reproduction. 2004;128:643-654
71. Yordy JS, Muise-Helmericks RC. Signal transduction and the Ets family of transcription factors. Oncogene. 2000;19:6503-6513
72. Harris TA, Yamakuchi M, Kondo M, Oettgen P, Lowenstein CJ. Ets-1 and Ets-2 regulate the expression of microRNA-126 in endothelial cells. Arterioscler Thromb Vasc Biol. 2010;30:1990-1997
73. Quinn SR, Mangan NE, Caffrey BE, Gantier MP, Williams BR, Hertzog PJ. et al. The role of Ets2 transcription factor in the induction of microRNA-155 (miR-155) by lipopolysaccharide and its targeting by interleukin-10. J Biol Chem. 2014;289:4316-4325
74. Liao YL, Hu LY, Tsai KW, Wu CW, Chan WC, Li SC. et al. Transcriptional regulation of miR-196b by ETS2 in gastric cancer cells. Carcinogenesis. 2012;33:760-769
75. Plotnik JP, Hollenhorst PC. Interaction with ZMYND11 mediates opposing roles of Ras-responsive transcription factors ETS1 and ETS2. Nucleic Acids Res. 2017;45:4452-4462
76. Gao N, White P, Doliba N, Golson ML, Matschinsky FM, Kaestner KH. Foxa2 controls vesicle docking and insulin secretion in mature Beta cells. Cell Metab. 2007;6:267-279
77. Jia S, Ivanov A, Blasevic D, Muller T, Purfurst B, Sun W. et al. Insm1 cooperates with Neurod1 and Foxa2 to maintain mature pancreatic beta-cell function. EMBO J. 2015;34:1417-1433
78. Bastidas-Ponce A, Roscioni SS, Burtscher I, Bader E, Sterr M, Bakhti M. et al. Foxa2 and Pdx1 cooperatively regulate postnatal maturation of pancreatic beta-cells. Mol Metab. 2017;6:524-534
79. Li H, Wang Z, Li Y, Fang R, Wang H, Shi H. et al. Hepatitis B X-interacting protein promotes the formation of the insulin gene-transcribing protein complex Pdx-1/Neurod1 in animal pancreatic beta-cells. J Biol Chem. 2018;293:2053-2065
Author contact
![]() Corresponding author: Hexing Road 26, Harbin, 150040, People's Republic of China. E-mail: chunbotengedu.cn (CB. Teng)
Corresponding author: Hexing Road 26, Harbin, 150040, People's Republic of China. E-mail: chunbotengedu.cn (CB. Teng)