Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(6):1071-1085. doi:10.7150/ijbs.41230 This issue Cite
Research Paper
The CARM1-p300-c-Myc-Max (CPCM) transcriptional complex regulates the expression of CUL4A/4B and affects the stability of CRL4 E3 ligases in colorectal cancer
Wenzhu Lu1, Chunmei Yang2, Hongbo He1, Hong Liu1 ![]()
1. Department of Integrated Traditional and Western Medicine, West China Hospital of Sichuan University, Chengdu 610041, Sichuan, China
2. Department of Integrated Traditional and Western Medicine, Chengdu Shangjinnanfu Hospital/West China Hospital of Sichuan University, Chengdu 610041, Sichuan, China
Received 2019-10-16; Accepted 2020-1-2; Published 2020-2-4
Abstract

The transcription factor c-Myc and two cullin family members CUL4A/4B function as oncogenes in colorectal cancer. Our recent publication reveals that c-Myc specifically activates the expression of CUL4A/4B through binding to their promoters. However, the underlying mechanism of how c-Myc actions in this process is still unknown. Using mass spectrometry and immunoprecipitation assays, we identified c-Myc formed a transcriptional complex with its partner Max (Myc-associated factor X), a histone acetyltransferase p300 and a coactivator associated arginine methyltransferase 1 (CARM1) in the present study. Knockdown or overexpression of the components of CARM1-p300-c-Myc-Max (CPCM) complex resulted in a decrease or increase of CUL4A/4B levels, respectively. Individual knockdown or inhibition of CPCM components decreased cell proliferation, colony formation, and cell invasion. Biochemically, knockdown or inhibition of CPCM components decreased their occupancies on the promoters of CUL4A/4B and resulted in their downregulation. Importantly, inhibition of CPCM components also caused a decrease of CRL4 E3 ligase activities and eventually led to an accumulation of ST7 (suppression of tumorigenicity 7), the specific substrate of CRL4 E3 ligases in colorectal cancer. Moreover, the in vivo tumor formation results indicated that knockdown or inhibition of CPCM components significantly decreased the tumor volumes. Together, our results suggest that the CPCM complex mediates explicitly the expression of CUL4A/4B, and thus affects the stability of CRL4 E3 ligases and the ubiquitination of ST7. These results provide more options by targeting the CPCM components to inhibit tumor growth in the therapy of colorectal cancer.
Keywords: CARM1, p300, CUL4A, CUL4B, CRL4 E3 ligase, colorectal cancer
Introduction
Colorectal cancer (CRC) is one of the highest incidences and mortality of cancer [1, 2]. According to the clinical data from the North American Association of Central Center Registries (NAACCR), the incidence and mortality of CRC in the United States are 0.04% and 0.016%, respectively [1, 2]. In the past decades, the molecular mechanisms regarding CRC tumorigenesis have been extensively investigated and current evidence recognizes that genomic instability, genetic factors, inflammatory microenvironment, aberrant expression of tumor suppressors and oncogenes, and differentially expressed noncoding RNAs [e.g., microRNAs (miRNAs) and long noncoding RNAs (lncRNAs)] are the major contributors of CRC pathogenesis [3-6]. At least three distinct pathways involved in genomic instability have been reported, and they include chromosomal instability (with an incidence of 65%-70% in sporadic colorectal cancers), microsatellite instability (caused by DNA mismatch repair) and CpG island methylator phenotype (leading to gene silence) [7-9]. Gene mutations inherited from ancestors to descendants are also a significant cause of CRC tumorigenesis and two common inherited CRC syndromes are hereditary nonpolyposis colorectal cancer (HNPCC) and familial adenomatous polyposis (FAP) [10, 11]. Inflammatory microenvironment can affect many tumorigenic phenotypes, such as tumor proliferation and survival, angiogenesis, invasiveness, and metastasis [12]. The activation of oncogenes and suppression of tumor suppressors are the direct causes that lead to tumorigenesis [13]. Bioinformatic analyses in TCGA (The Cancer Genome Atlas) database and gene expression analyses in CRC tumor tissues and cultured tumor cells demonstrate that a variety of tumor suppressors [e.g., p53, APC (Adenomatous Polyposis Coli), LOH (Loss of Heterozygosity), PTEN (Phosphatase and Tensin Homolog) and ST7 (Suppressor of Tumorigenicity protein 7] and oncogenes [e.g., KRAS (Kirsten Rat Sarcoma), c-Myc, Her2 (Human Epidermal Growth factor receptor 2)] are differentially expressed [14, 15]. In recent years, a significant number of miRNAs and lncRNAs are also found to play important roles in the pathogenesis of CRC [16-18]. More than 250 miRNAs (e.g., miR-31, miR-34a, miR-155, and miR-221) and dozens of lncRNAs [e.g., H19, CCAL (Colorectal Cancer-associated lncRNA), CCAT1 (Colon Cancer-associated Transcript 1), CCAT1-L, and CCAT2)] are reported to be differentially expressed in CRC tissues and cells [16-18].
Protein modifications such as phosphorylation and ubiquitination are also involved in many cellular processes during tumorigenesis [19]. In recent years, a class of genes known as Cullins are discovered to contribute to tumorigenesis through affecting the ubiquitination of proteins that function in multiple biological processes such as DNA damage and repair, cell cycle progression and cell death [20, 21]. Protein ubiquitination is mediated by the ubiquitin-proteasome system (UPS), which includes several important components such as ubiquitin (Ub), Ub-activating enzyme (E1), Ub-conjugating enzymes (E2s), Ub-ligases (E3s), substrate proteins and deubiquitinases (DUBs) [20, 21]. Constitutive upregulation of Cullin genes especially CUL4A and CUL4B have been observed in many cancers [22, 23]. Biochemically, CUL4A/4B act as scaffolds to assemble E3 ubiquitin ligases with RING-box proteins (RBX1 and RBX2), adaptor protein DNA damage binding protein 1 (DDB1), and substrate recognition receptors such as DCAFs (DDB1 and CUL4-associated Factors) [20-23]. These E3 ligases are known as Cullin-RING ubiquitin ligases (CRL4s) and they can ubiquitinate a great number of proteins involved in DNA damage and repair [e.g., DDB2 and UNG2 (Uracil-N-glycosylase 2)] [20-23], cell cycle progression (e.g., p21 and p27) [24, 25], and tumor suppression (e.g., PTEN and ST7] [26, 27]. The mammalian CUL4A and CUL4B share over 80% protein sequence identity, however, current findings indicate that they do not show obvious functional redundancy [27]. In the same type of cancer cells, only either CUL4A or CUL4B is overexpressed [27]. One exception is our recent finding in which CUL4A/4B are both overexpressed in colorectal cancer [27]. The mechanical investigation demonstrates that intracellular inflammatory environment induces the expression of a transcription factor c-Myc, which specifically binds to the promoters of CUL4A/4B and activates their expression. Both CUL4A and CUL4B can assemble an E3 ligase with DDB1, RBX1, and DCAF4. These two complexes are termed as CRL4DCAF4 E3 ligases, which can specifically ubiquitinate a tumor suppressor ST7 [27]. However, we did not reveal how c-Myc activated the expression of CUL4A/4B in this process.
c-Myc is a well-known oncogene and it functions as a transcription factor [28]. The amplification of c-Myc has been observed in multiple cancer types such as cervix cancer [29], breast cancer [30], colorectal cancer [31], osteosarcoma and lung cancer [32, 33]. c-Myc contains a basic helix-loop-helix (bHLH) motif and a leucine zipper (LZ)-binding motif. Basically, c-Myc binds to DNA through the bHLH motif, while it dimerizes with its partner Max (Myc-associated Factor X) through the LZ motif [34]. Biochemically, c-Myc recruits the transcriptional coactivators known as histone acetyltransferases (HATs) [e.g., p300 and CBP (CREB binding protein)] to activate the expression of multiple genes such as CCNA2 (Cyclin A2), CCNE1 (Cyclin E1), and NME1 (Nucleoside Diphosphate Kinase 1) [35, 36]. In addition, c-Myc-associated transcriptional complexes can be modulated by many proteins such as BIN1 (Bridging Integrator 1) [37], MIZ1 (Myc-interacting Zn Finger Protein 1) [38], PAM (Peptidylglycine alpha-amidating Monooxygenase) [39], and TRRAP (Transformation/Transcription Domain Associated Protein) [40].
To explore the mechanism of how c-Myc activates the expression of CUL4A/4B in CRC cells, we immunoprecipitated c-Myc-associated complex and applied it to mass spectrometry analysis. After coimmunoprecipitation assay, we discovered that c-Myc dimerized with its partner protein Max, and directly interacted with a histone acetyltransferase p300, which further recruited CARM1 (Coactivator Associated Arginine Methyltransferase 1) to assemble a transcriptional complex known as CARM1-p300-c-Myc-Max (CPCM). We then focused our studies on evaluating the contribution of CPCM components to the expression of CUL4A/4B and CRL4DCAF4 E3 ligase activities.
Materials and methods
Cells and cell culture
Human CRC cell lines including HT29, HT55, HCT-15, HCT-116, HCA-24, SW620 and T84 were acquired from American Type Culture Collection (ATCC) (Manassas, VA, USA). Cells were grown in DMEM containing 10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO, USA, #F2442) and 50 U/mL penicillin-streptomycin (PS) (Sigma-Aldrich, #P4333). The source and growth condition of the human colon epithelial cell line (HCEC-1CT) were the same as described previously [27]. Cells were cultured in a 37°C humidified atmosphere supplemented with 5% CO2 and cells were split twice one week.
Cell transfection
Cells were seeded into 6-well plates and incubated overnight to reach a density of 50% confluence. Specific siRNAs including sip300 (Thermo Fisher Scientific, Waltham, MA, USA, assay ID:106444), sic-Myc (assay ID: 103828), siCARM1 (assay ID: 112501), and siMax (assay ID: 143519) for gene knockdown or plasmids for gene overexpression were mixed with Lipofectamine 2000 (Thermo Fisher Scientific, #11668019) to form siRNA (or plasmid) duplex-Lipofectamine, which were then added into 6-well plates containing cells. Three replicates were performed for each siRNA or plasmid. After mixing gently, cells were further incubated 24 h at 37°C in a CO2 incubator. The resulting cells were subjected to RNA and protein extraction.
Immunoprecipitation and mass spectrometry
Cells (1×108) expressing pCDNA3-2×Flag-3×HA-c-Myc or pCDNA3-2×Flag-3×HA (Empty vector, control) were lysed in RIPA lysis and extraction buffer (Thermo Fisher Scientific, #89900) containing 1 × protease inhibitor cocktail (Sigma-Aldrich, #P8340). Cell extracts were centrifuged at 14,000 rpm for 30 min and the supernatant was incubated with anti-Flag agarose (Sigma-Aldrich, #A2220) to pull down Flag-associated proteins at 4°C overnight. The resulting Flag-associated proteins were washed five times with RIPA lysis and extraction buffer and then eluted with 100 μg/mL Flag peptide (Sigma-Aldrich, #F4799). The obtained protein complex was further incubated with anti-HA agarose (Thermo Fisher Scientific, #26181) to pull down HA-associated proteins at 4°C overnight. The resulting HA-associated proteins were washed five times with RIPA lysis and extraction buffer, followed by loading onto a 10% SDS-PAGE gel for separation. The gel was subsequently performed sliver staining with a kit (Thermo Fisher Scientific, #24612). Protein bands were cut into small slices and then digested with a Trypsin Kit (Thermo Fisher Scientific, #60109101). Mass spectrometry analysis was performed to determine c-Myc-associated proteins following a protocol as described previously [27].
Cell proliferation assay
Cell viability was determined using a CellTiter 96 non-radioactive cell proliferation kit (Promega, Madison, WI, USA, #G4000). Briefly, HT29 and HCT-116 cells were transfected with sip300, sic-Myc, siCARM1, and siMax to generate their corresponding knockdown cells. These cells were plated onto 96-well plates and cell viability was determined every day for five days according to the manufacturer's method. In addition, HT29 and HCT-116 cells were grown in DMEM and the same medium containing 5 μM sAJM, 20 μM CARM-IN-1 or 50 nM C646. Cell viability was also determined every day for five days.
Colony formation assay
The c-Myc-knockdown (KD), p300-KD, CARM1-KD and Max-KD cells in HT-29 and HCT-116 backgrounds were seeded onto 6-well plates with a density of 103 cells per well. Cells were incubated at 37°C for two weeks with a medium change every three days. For colony formation assay in cells treated with CPCM component inhibitors, the HT-29 and HCT-116 were seed to 6-well plates with a density of 103 cells per well. Cells were cultured in a 37°C incubator to adhere for 16 h, followed by treatment with 5 μM sAJM, 20 μM CARM-IN-1 or 50 nM C646 for two weeks with a medium change every three days. Colonies were fixed with 70% ethanol for 10 min and stained with 0.2% crystal violet and the 6-well plates were photographed.
Cell invasion assay
The knockdown cells (5×104) of CPCM components and cells treated with individual CPCM component inhibitor were suspended into 100 μL serum-free DMEM medium and plated on the top filter membrane in a Boyden chambers insert (Millipore, Burlington, MA, USA, #P18P01250). The lower chamber was filled with DMEM medium containing 10% FBS. After incubation at 37°C for 24 h, cells on the lower chamber were fixed with 70% ethanol for 10 min and then were stained with 0.1% crystal violet, followed by a photograph.
Total RNA isolation and quantitative real-time PCR (qRT-PCR) analysis
Total RNA was isolated from cultured cells using a TRIZOL reagent (Thermo Fisher Scientific, #15596026) according to the method provided by the manufacturer. The purified RNA was reverse-transcribed into cDNA using an M-MuLV reverse transcriptase kit (New England Biolabs, Beijing, China, #M0253S). After dilution 10-fold, cDNAs were applied to qRT-PCR using an SYBR Green Kit (Bio-Rad, Shanghai, China, #1725150) to quantify the expression of genes with primers listed in Supplementary Table 1. The PCR procedures included: 95°C for 2 min, followed by 40 cycles of 30 seconds at 95°C and 20 seconds at 68°C. The individual gene expression level was normalized to β-actin.
Western blotting assay
Cells and tissues were lysed in RIPA lysis and extraction buffer containing 1 × protease inhibitor cocktail. Cell extracts were centrifuged at 14,000 rpm for 15 min and equal amounts of supernatant were loaded onto 10% SDS-PAGE gel for separation. After transferring to PVDF membrane (Millipore, #IPVH00010) and incubating in 5% milk for 1 h, proteins were probed with primary antibodies including anti-Flag (#ab49763), anti-Myc (#ab32), anti-c-Myc (#ab39688), anti-CARM1 (#ab131520), anti-p300 (#ab10485), anti-Max (#ab53570), anti-HA (#ab18181), anti-CUL4A (#ab92554), anti-CUL4B (#ab67035), anti-ST7 (#ab122460) and anti-GAPDH (#ab8245). The membranes were washed with PBST buffer for five times and then probed with HRP-labeled 2nd antibodies (mouse-#ab6728; rabbit-#ab205718). All antibodies were purchased from Abcam (Shanghai, China). The signals were determined by an enhanced chemiluminescence (ECL) kit (GE Healthcare, Piscataway, NJ, USA).
Co-immunoprecipitation (Co-IP)
The HT29 cells under 80% confluence were transfected with the following different combinations of plasmids: pCDNA3-2×Flag + pCDNA3-6×Myc; pCDNA3-2×Flag + pCDNA3-6×Myc-p300; pCDNA3-2×Flag + pCDNA3-6×Myc-CARM1; pCDNA3-2×Flag-c-Myc + pCDNA3-6×Myc; pCDNA3-2×Flag-c-Myc + pCDNA3-6×Myc-p300; pCDNA3-2×Flag-c-Myc + pCDNA3-6×Myc-CARM1; pCDNA3-2×Flag-CARM1 + pCDNA3-6×Myc; and pCDNA3-2×Flag-CARM1 + pCDNA3-6×Myc-p300, respectively. The resulting cells were incubated at 37°C for 48 h, followed by lysing in RIPA lysis and extraction buffer containing 1 × protease inhibitor cocktail. Cell extracts were centrifuged at 14,000 rpm for 30 min and the supernatant was equally divided into two parts to incubate with anti-Flag agarose and anti-Myc agarose (Sigma-Aldrich, #A7470) at 4°C overnight, respectively. The Flag- or Myc-associated proteins were washed five times with RIPA lysis and extraction buffer and then were subjected to western blotting assays to determine protein-protein interactions.
In vivo ubiquitination assay
The in vivo ubiquitination of ST7 was performed following a previous method [27]. Briefly, HCEC-1CT cells (5×107) expressing pCDNA3-2×Flag-ST7 and HA-ubiquitin were treated with 5 μM sAJM, 20 μM CARM-IN-1 or 50 nM C646 for 6 h, followed by lysing RIPA lysis and extraction buffer containing 1 × protease inhibitor cocktail. After centrifuging at 14,000 rpm for 30 min, the supernatant was incubated with anti-Flag-agarose to pull down Flag-ST7-associated proteins. The ubiquitination of ST7 was detected with an anti-HA antibody.
Chromatin immunoprecipitation (ChIP) assay
Cells (1×108) under 80% confluence were washed twice with cold PBS buffer (Thermo Fisher Scientific, #20012027), and then were crosslinked with 1% formaldehyde (Polysciences, Warrington, PA, USA, #18814) at 23°C for 15 min. The crosslinked cells were applied to a ChIP assay using a high-sensitivity ChIP kit (Abcam, #ab185913) according to a protocol provided by the manufacturer. The antibodies used in this assay included anti-c-Myc, anti-p300, anti-CARM1 and anti-Max. The information of these antibodies was the same as described in western blotting assay. The purified DNA samples were applied to qRT-PCR analyses using an SYBR green kit (same as described in mRNA detection) with the primers listed in Supplementary Table 2. The relative enrichment of individual CPCM components on the promoters of CUL4A and CUL4B were normalized to the input.
In vivo tumor formation and growth inhibition
The Athymic nu/nu mice were sourced from Shanghai SLAC Laboratory Animal Co. Ltd (Shanghai, China) and were maintained in accordance with a guideline approved by the Institutional Animal Care and Use Committee (IACUC) of Sichuan University. The HT29, c-Myc-KD, p300-KD, CARM1-KD and Max-KD cells (5×106 each) in 100 μL PBS were mixed with Matrigel (1:1 ratio, v/v) (BD Biosciences, San Jose, CA, USA, #354234). Cells were subcutaneously injected into mice to establish tumor xenografts and tumors were measured with fine calipers at 5-day intervals. In addition, mice injected with HT29 cells were randomly assigned to four groups (n = 5 in each group), followed by injecting with PBS, sAJM, CARM-IN-1 or C646 every five days. Tumor volumes were determined with a formula: Volume =(Length×Width2)/2.
Statistical analysis
The mean ± standard deviation (SD) in each experiment represented three independent replicates. Data were analyzed using a two-sided Student's t test. Significance was set at P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***).
Results
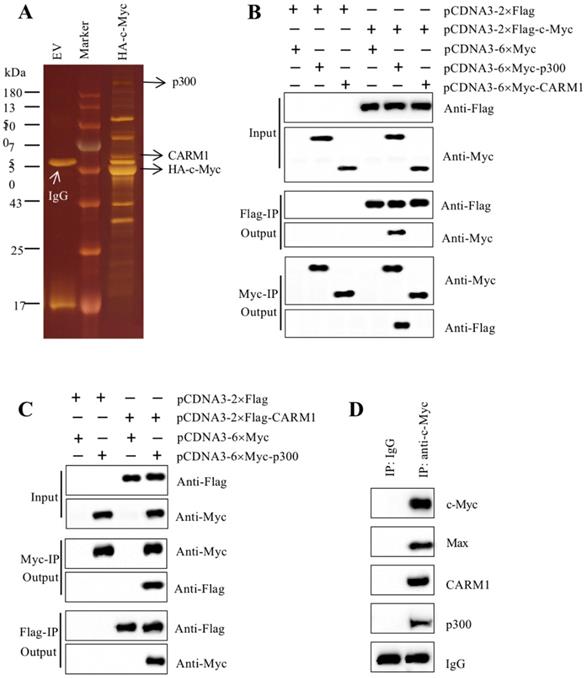
c-Myc associated with Max, p300, and CARM1 to assemble the CPCM complex in vitro and in vivo
As mentioned earlier, our recent findings reported that the amplified c-Myc in CRC cells specifically bound to the promoters of CUL4A/4B and to activate their expression [27]. The induced CUL4A and CUL4B separately formed a CRL4 E3 ligase to ubiquitinate ST7, resulting in tumorigenesis [27]. To reveal how c-Myc cooperates with other proteins to assemble a transcriptional complex in this process, we constructed a c-Myc overexpression vector pCDNA3-2×Flag-3×HA-c-Myc. After transfecting it into HT29 cells, we performed a two-step purification using anti-Flag agarose and anti-HA agarose to enrich c-Myc-associated proteins (Figure 1A). We then applied this complex to mass spectrometry assay to identify proteins. The results identified a total of 33 proteins in this complex (Supplementary Table 3). After analyzing the results, we found Max was a well-known partner of c-Myc and they could form a heterodimer, which could directly bind to DNA [41]. Based on this notion, we thought it was not necessary to determine the interaction between c-Myc and Max in our study. Moreover, we also found two other proteins including p300 and CARM1 have been previously reported to form a transcriptional complex with other transcription factors such as NF-κB (Nuclear Factor Kappa B) [42], Runx2 (Runt-related Transcription Factor 2) and bHLH [43, 44]. Given the conserved assembly mechanism of transcription factors with coactivators and corepressors, we speculated that c-Myc might interact with p300 and CARM1. To verify this hypothesis, we performed Co-IP assays to determine the direct interactions of c-Myc-p300, c-Myc-CARM1, and CARM1-p300. Accordingly, we transfected the Myc-tag and Flag-tag vectors of these three proteins into HT29 cells. After immunoprecipitation using both anti-Flag agarose and anti-Myc agarose in each combination of plasmids, the output proteins were subjected to immunoblots to determine protein interactions. Our results indicated that c-Myc could directly interact with p300 instead of CARM1 (Figure 1B), and p300 could directly interact with CARM1 (Figure 1C). These in vitro results suggested that p300 functioned as an adaptor protein to connect c-Myc-Max heterodimer and CARM1, forming the CPCM complex. We next aimed to determine if these three proteins formed a complex in vivo. For this purpose, we performed in vivo immunoprecipitation using anti-c-Myc antibody in the cancerous tissue from an advanced colitis-associated cancer (CAC, a subtype of CRC) patient under stage IV. Immunoblot detection results using the purified protein complex indicated that c-Myc could pull down Max, CARM1, and p300 (Figure 1D). These in vitro and in vivo results demonstrated that c-Myc-Max heterodimer recruited p300 and CARM1 to assemble the CPCM complex.
c-Myc associated with Max, p300 and CARM1 to assemble the CPCM complex in vitro and in vivo. (A) The c-Myc-associated complex. The HT29 cells expressing pCDNA3-2×Flag-3×HA (empty vector, EV) or pCDNA3-2×Flag-3×HA-c-Myc were lysed and immunoprecipitated sequentially with the anti-Flag and anti-HA resins. After two-step purification, the resulting protein complexes were loaded onto an SDS-PAGE gel for separation. The protein gel was stained with a sliver-staining kit. The positions of IgG, CARM1, and HA-c-Myc were shown. (B) c-Myc interacted directly with p300 but not CARM1. The HT29 cells cotransfected with different combinations of plasmids including pCDNA3-2×Flag + pCDNA3-6×Myc, pCDNA3-2×Flag + pCDNA3-6×Myc-p300, pCDNA3-2×Flag + pCDNA3-6×Myc-CARM1, pCDNA3-2×Flag-c-Myc + pCDNA3-6×Myc, pCDNA3-2×Flag-c-Myc + pCDNA3-6×Myc-p300, and pCDNA3-2×Flag-c-Myc + pCDNA3-6×Myc-CARM1. The resulting cells were applied for Co-IP analyses with anti-Flag and anti-Myc resins. The input and out proteins were detected using anti-Flag and anti-Myc antibodies, respectively. (C) p300 interacted directly with CARM1. The HT29 cells cotransfected with different combinations of plasmids including pCDNA3-2×Flag + pCDNA3-6×Myc, pCDNA3-2×Flag + pCDNA3-6×Myc-p300, pCDNA3-2×Flag-CARM1 + pCDNA3-6×Myc, and pCDNA3-2×Flag-CARM1 + pCDNA3-6×Myc-p300. The resulting cells were applied for Co-IP analyses with anti-Flag and anti-Myc resins. The input and out proteins were detected using anti-Flag and anti-Myc antibodies, respectively. (D) c-Myc associated with Max, p300 and CARM1 in vivo. One cancerous tissue from a CAC patient under stage IV was applied to immunoprecipitation analysis using anti-c-Myc and anti-IgG antibodies, respectively. The purified protein complexes were applied to western blotting assays to examine c-Myc, Max, p300 and CARM1.
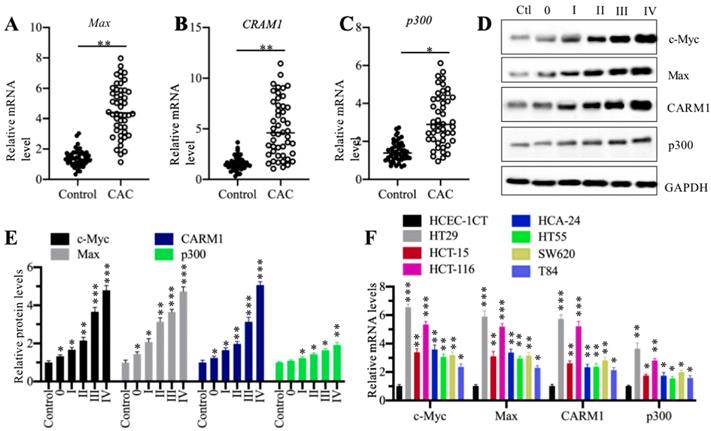
The components of CPCM complex were upregulated in cancerous tissues of CAC patients and cultured CRC cells
Our previous publication has reported that c-Myc is overexpressed in 48 cancerous tissues from CAC patients in comparison to their adjacent noncancerous tissues [27]. To determine the expression levels of other CPCM components in the same RNA samples of CAC cancerous tissues, we performed qRT-PCR analyses to measure mRNA levels of Max, p300 and CARM1. Our results showed that all of these three CPCM components were upregulated in 48 cancerous tissues compared to their adjacent noncancerous tissues (Figures 2A-2C). Meanwhile, we also detected their protein levels in five CAC cancerous tissues (n=1 in each TNM grade). Consistent with their mRNA levels, the CPCM member protein levels were gradually increased in the CAC tumor tissues with the severity of TNM stages (Figures 2D and 2E). To determine if the overexpression of CPCM components happens in CRC cells, we measured their mRNA levels in seven human CRC cell lines including HT29, HT55, HCT-15, HCT-116, HCA-24, SW620 and T84. The qRT-PCR results indicated that these seven cell lines exhibited varying mRNA levels of CPCM components (Figure 2F). Of these cell lines, HT29 exhibited the highest mRNA levels of c-Myc (~6.5-fold), Max (~5.8-fold), p300 (~3.5-fold), and CARM1 (~5.6-fold), followed by HCT-116, HCT-15, HCA-24, HT55, SW620 and T84 (Figure 2F). These results suggested that the overexpression of CPCM components in CAC cancerous tissues and cultured CRC cells was a universal phenomenon. Based on the higher expression levels of the CPCM components in HT29 and HCT-116 cells, we carried out the following experiments in these two cell lines unless otherwise specified.
The CPCM components were amplified in CRC patients. (A-C) The relative mRNA levels of Max (A), CARM1 (B) and p300 (C) in 48-paired cancerous tissues (CAC) and their adjacent noncancerous tissues (Control) were determined by qRT-PCR. *P < 0.05 and **P < 0.01. (D) The protein levels CPCM components in tumor samples. Five tumor samples from CAC patients at different TNM stages (0-IV) and one noncancerous volunteer donor (HC) were applied to western blotting assay to determine protein levels of c-Myc, Max, CARM1, and p300. GAPDH was set as a loading control. (E) The relative protein levels of CPCM components. The protein band signals in (D) were quantified using the Image J software and normalized to GAPDH. *P < 0.05, **P < 0.01 and ***P < 0.001. (F) The mRNA levels of CPCM components in CRC cell lines. Seven CRC cell lines including HT29, HT55, HCT-15, HCT-116, HCA-24, SW620 and T84, and one normal HCEC-1CT cell line were used to examine mRNA levels of c-Myc, Max, CARM1, and p300. *P < 0.05, **P < 0.01 and ***P < 0.001.
Knockdown of CPCM components resulted in downregulation of CUL4A/4B
Both CUL4A and CUL4B are direct targets of c-Myc and knockdown of c-Myc leads to the downregulation of CUL4A/4B [27]. Thus, we next sought to determine the effects of other CPCM components on CUL4A/4B expression. For this purpose, we transfected CPCM component siRNAs or their overexpression vectors into HT29 and HCT-116 cells to specifically knock down or overexpress CPCM members. After determining their successful downregulation or overexpression (Figures 3A-3D), we determined the expression of CUL4A/4B in these cells. As expected, our qRT-PCR results showed that both CUL4A and CUL4B were significantly downregulated in all CPCM knockdown cells compared to controls (Figures 3E). Conversely, the expression of CUL4A/4B was markedly upregulated in all CPCM overexpression cells compared to control cells (Figure 3F). In addition, we also examined the protein levels of CUL4A/4B and ST7 in these knockdown and overexpression cells. Consistent with their mRNA levels, we also observed a significant decrease or increase in CUL4A/4B protein levels in CPCM knockdown or overexpression cells, respectively (Supplementary Figure 1). In contrast, the protein level of ST7 was accumulated or decreased in CPCM knockdown or overexpression cells, respectively (Supplementary Figure 1). These results clearly supported that the CPCM complex was responsible for the regulation of CUL4A/4B expression. Since CARM1, p300, c-Myc and Max could assemble a complex, knocking down or overexpressing any two of them simultaneously should cause a similar effect on CUL4A/4B expression as in cells only knocking down or overexpressing a single member. To verify this hypothesis, we simultaneously knocked down or overexpressed c-Myc and CARM1 in HT29 cells, and then examined the mRNA and protein levels of CUL4A/4B. As expected, our results showed that the mRNA and protein levels of CUL4A/4B in cells knocking down or overexpressing c-Myc+CARM1 were similar to cells knocking down or overexpressing c-Myc or CARM1 alone (Supplementary Figure 2).
Knockdown or overexpression of the CPCM components changed the expression of CUL4A/4B. (A-D) The mRNA levels of c-Myc (A), Max (B), p300 (C), and CARM1 (D) in their corresponding knockdown and overexpression cells. The HT29 and HCT-116 cells were transfected with sic-Myc, pCDNA3-2×Flag-c-Myc, siMax, pCDNA3-2×Flag-Max, sip300, pCDNA3-2×Flag-p300, siCARM1, or pCDNA3-2×Flag-CARM1 to decrease or increase the expression of individual CPCM components. The expression of c-Myc, Max, p300, and CARM1 was determined by qRT-PCR analyses. **P < 0.01 and ***P < 0.001. (E and F) The mRNA levels of CUL4A/4B in the CPCM knockdown and overexpression cells. The CPCM knockdown (E) and overexpression (F) cells used in (A-D) were applied to determine the mRNA levels of CUL4A/4B by qRT-PCR analyses. **P < 0.01 and ***P < 0.001.

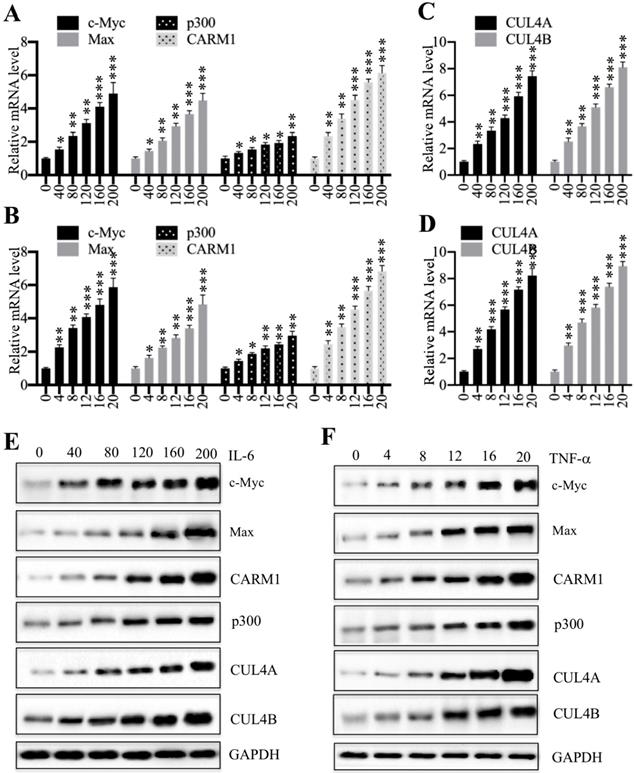
Proinflammatory cytokines activated the expression of CPCM components
We previously showed that proinflammatory cytokines including interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) could induce the expression of c-Myc, CUL4A and CUL4B [27]. Thus, we next aimed to determine if treatments with proinflammatory cytokines also affect the expression of other CPCM components. Accordingly, we treated HCEC-1CT cells with a series of concentrations of IL-6 (0, 40, 80, 120, 160 and 200 ng/mL) and TNF-α (0, 4, 8, 12, 16 and 20 ng/mL), followed by examining mRNA levels of CPCM components. The qRT-PCR results showed that the expression of CPCM components was gradually induced with the increase of both IL-6 and TNF-α concentrations (Figures 4A and 4B). Specifically, treatments with 200 ng/mL IL-6 and 20 ng/mL TNF-α resulted in the induction of c-Myc (~4.7-fold and ~5.5-fold, respectively), Max (~4.3-fold and ~4.8-fold, respectively), p300 (~2.7-fold and ~2.9-fold, respectively), and CARM1 (~6.1-fold and ~6.7-fold, respectively) (Figures 4A and 4B). Consistent with previous results, we also examined CUL4A/4B mRNA levels in these treatments and found that they were gradually induced with the increase of IL-6 and TNF-α concentrations (Figures 4C and 4D). In addition, we also examined the protein levels of CPCM components and CUL4A/4B. The immunoblot results indicated that these proteins shared similar patterns to their corresponding mRNA levels (Figures 4E and 4F). These results consistently supported that intracellular inflammatory status was responsible for the upregulation of CPCM components and their target genes CUL4A and CUL4B.
Treatments with recombinant IL-6 or TNF-α increased the mRNA and protein levels of CPCM components in HCEC-1CT cells. (A and B) The mRNA levels of CPCM components in cells treated with IL-6 (A) or TNF-α (B). The HCEC-1CT cells were treated with different concentrations of IL-6 (0, 40, 80, 120, 160, or 200 ng/mL), or TNF-α (0, 4, 8, 12, 16, or 20 ng/mL), followed by measurement of mRNA levels of c-Myc, Max, CARM1, and p300 by qRT-PCR. *P < 0.05, **P < 0.01 and ***P < 0.001. (C and D) The mRNA levels of CUL4A/4B in cells treated with IL-6 (C) or TNF-α (D). The same RNA samples used in (A and B) were applied to measure mRNA levels of CUL4A/4B by qRT-PCR. **P < 0.01 and ***P < 0.001. (E and F) The protein levels of CPCM components and CUL4A/4B in cells treated with IL-6 (E) or TNF-α (F). The same cells used in (A and B) were applied to measure protein levels of CPCM components and CUL4A/4B. GAPDH was set as a loading control.
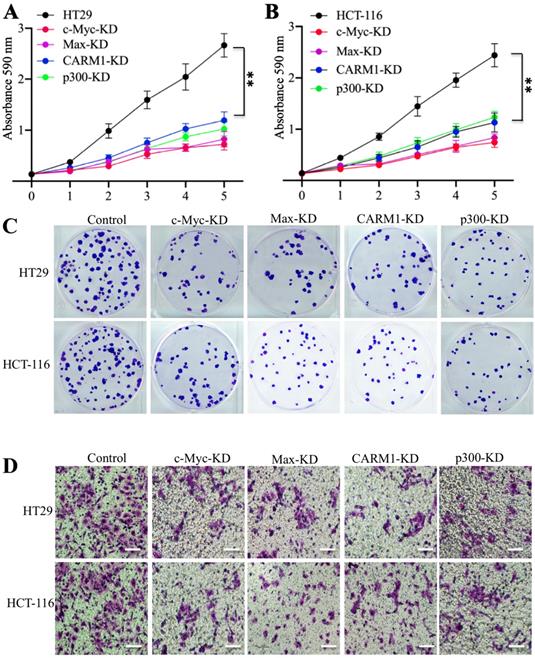
Knockdown or inhibition of the CPCM components caused oncogenic phenotype defects
Our previous results have shown that knockdown of c-Myc can inhibit oncogenic phenotypes of colorectal cells [27]. We next aimed to determine if knockdown of the other CPCM components also had similar effects. For this purpose, we subjected CPCM knockdown cells to evaluate their oncogenic phenotypes. The cell proliferation results indicated knockdown either Max, CARM1 or p300 significantly inhibited the growth of colorectal cancer cells to a comparable level in c-Myc-knockdown cells (Figures 5A and 5B). The colony formation results also indicated that the downregulation of CPCM components caused decreased colonies (Figure 5C and Supplementary Figure 3A). Moreover, we also observed a significant decrease in invading cells in these knockdown cells compared to controls (Figure 5D and Supplementary Figure 3B). Since knockdown of CPCM components inhibited cancer cell growth, we speculated that inhibition of this complex by the inhibitors that specifically targeted CPCM components should also cause similar effects. To verify this hypothesis, we treated colorectal cells with a c-Myc inhibitor (sAJM-589), a CARM1 inhibitor (CARM1-IN-1) and a p300 inhibitor (C646), respectively. After these treatments, we primarily measured mRNA and protein levels of CPCM components. The results indicated that all these treatments could not change mRNA and protein levels of these CPCM components (Supplementary Figures 4A and 4B). However, these treatments caused the downregulation of CUL4A/4B and the accumulation of ST7 (Supplementary Figure 4B), which suggested that oncogenic phenotypes might be inhibited following these inhibitor treatments. To verify this hypothesis, we also evaluated cell proliferation, colony formation and cell invasion abilities under CPCM inhibitor treatments. Similar to the results in their knockdown cells, we also observed the significant repression of cell proliferation (70% deduction at the 5-day point) (Supplementary Figures 5A-5C), colony numbers (75% deduction at the 5-day point) (Supplementary Figures 5D-5F, and Supplementary Figure 6A) and invading cell numbers (70% deduction at the 5-day point) (Supplementary Figures 5G-5I, and Supplementary Figure 6B) in comparison to non-treatment cells.
Knockdown of the CPCM components caused oncogenic phenotype defects. (A and B) The knockdown of CPCM components decreased cell proliferation. The CPCM component knockdown cells in HT29 (A) and HCT-116 (B) backgrounds were subjected to determine cell proliferation using an MTT assay. **P <0.01. (C) The knockdown of CPCM components decreased colony formation. The same cells as used in (A and B) were seeded into six-well plates with a density of 103 cells per well, followed by continuously growing for two weeks. Colonies were stained with 0.2% crystal violet. (D) The knockdown of CPCM components inhibited cell invasion. The same cells as used in (A and B) were seeded into the upper chamber of Boyden chambers and incubated in 37°C for 24 h. Cells in the lower chambers were fixed in methanol and stained with 0.2% crystal violet. Bars=50 μm.
Inhibition of the CPCM components impaired their bindings on the promoters of CUL4A/4B
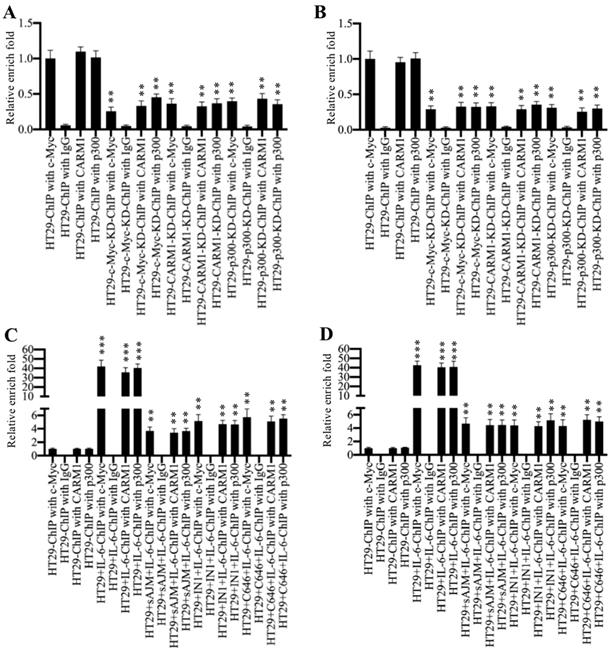
To determine if the CPCM complex regulated CUL4A/4B expression through binding to their promoters, we carried out ChIP assays using antibodies that recognized CPCM components. Firstly, we performed ChIP assays in HT29, c-Myc-KD, CARM1-KD and p300-KD cells without any treatment. The qRT-PCR results showed that the occupancies of CPCM components on the promoters of CUL4A/4B were significantly decreased (~60-70% deduction) in c-Myc-KD, CARM1-KD and p300-KD cells compared to HT19 cells (Figures 6A and 6B). Besides, we also evaluated the occupancies of CPCM components in cells treated with IL-6 (200 ng/mL) alone and in cells treated with both CPCM component inhibitors and IL-6. The results showed that IL-6 treatment significantly increased (~40-fold) the occupancies of CPCM components on the promoters of CUL4A/4B compared to controls (Figures 6C and 6D). The combined treatments of CPCM component inhibitors and IL-6 significantly decreased their occupancies from ~40-fold to ~4-fold (Figures 6C and 6D). At the same time, we also examined mRNA levels of CUL4A/4B in cells treated with CPCM component inhibitors and IL-6. The results showed that the expression of CUL4A/4B was significantly induced (~8.3-fold) in cells treated with IL-6 alone compared to HT29 control cells, while their expression was only slightly induced (~1.5-fold) by IL-6 after CPCM component inhibitor treatments (Supplementary Figure 7). These data suggested that the CPCM complex bound explicitly to the CUL4A/4B promoters and activated their expression under IL-6 treatment.
Inhibition of CPCM components decreased their occupancies on the promoters of CUL4A/4B. (A and B) The knockdown of CPCM components decreased their occupancies on the promoters of CUL4A/4B. The HT29, c-Myc-KD, CARM1-KD and p300-KD cells were subjected to ChIP assays using anti-c-Myc, anti-CARM1, anti-p300 or IgG for immunoprecipitation. The purified DNA was used to examine the enrichment of CPCM components on the promoters of CUL4A (A) and CUL4B (B). **P < 0.01. (C and D) Inhibition of CPCM components impaired their enrichment on the promoters of CUL4A/4B. The HT29 cells were treated with 5 μM sAJM, 20 μM CARM-IN-1 or 50 nM C646 for 6 h, followed by treatment with 200 ng/mL IL-6 for another 6 h. The resulting cells were subjected to ChIP assays using anti-c-Myc, anti-CARM1, anti-p300 or IgG for immunoprecipitation. The purified DNA was used to examine the enrichment of CPCM components on the promoters of CUL4A (C) and CUL4B (D). **P < 0.01 and ***P < 0.001.
Inhibition of the CPCM components repressed the ubiquitination of ST7
One possibility for the reason that caused the defects of oncogenic phenotypes in cells with CPCM component knockdown or inhibition was that CRL4 E3 ligase activities were repressed. To very this possibility, we measured protein levels of ST7 and its ubiquitination under the conditions of inhibitor treatments. As expected, the results indicated that the ST7 protein level was significantly accumulated (Figure 7A). Consistent with the results in CPCM knockdown cells, we also observed inhibition of the CPCM components caused the decrease of CUL4A and CUL4B protein levels, while these inhibitors could not change the protein levels of CPCM components (Figure 7A). To evaluate the ubiquitination level of ST7 in the treatments of CPCM component inhibitors, we primarily cotransfected pCDNA3-2×Flag-ST7 with pCDNA3-2×HA-Ubiquitin into HCEC-1CT cells, followed by treated with sAJM-589, CARM1-IN-1 or C646. After immunoprecipitation with anti-Flag resin, we detected ST7 ubiquitination level. The results indicated that CPCM component inhibitors significantly decreased the ST7 ubiquitination level to a similar pattern (Figure 7B). These results suggested that the inhibition of the CPCM components repressed the ubiquitination of ST7.
Inhibition of CPCM components repressed the ubiquitination of ST7. (A) Inhibition of CPCM components increased the ST7 protein level. The HT29 cells were treated with 5 μM sAJM, 20 μM CARM-IN-1 or 50 nM C646 for 6 h, followed by subjecting cells to examine protein levels of c-Myc, Max, CARM1, p300, CUL4A, CUL4B, and ST7. GADPH was used as a loading control. (B) Inhibition of CPCM components repressed the ubiquitination of ST7. The HT29 cells coexpressing pCDNA3-3×HA-ubiquitin and pCDNA3-2×Flag-ST7 were treated with 5 μM sAJM, 20 μM CARM-IN-1 or 50 nM C646 for 6 h. Cells were immunoprecipitated with an anti-Flag resin. Equal amounts of Flag-ST7 from different treatments were loaded to an SDS-PAGE gel to detect the ubiquitination of ST7 with an anti-HA antibody. The membrane was probed with anti-Flag to indicate the equal loading of ST7 (bottom panel).

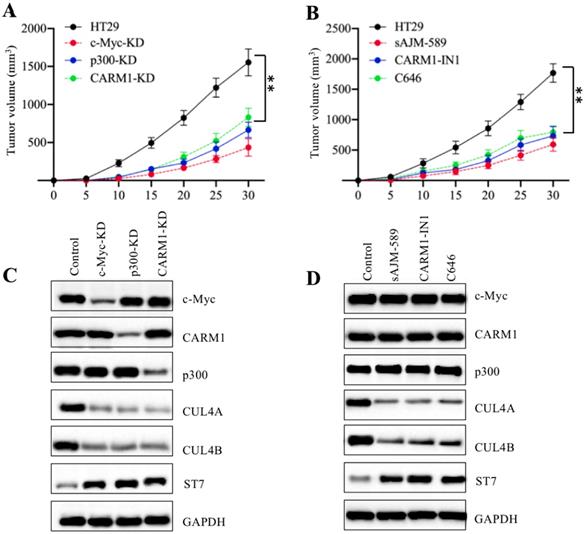
Knockdown or inhibition of CPCM components decreased the tumor formation in vivo
Our above in vitro results showed that knockdown or inhibition of CPCM components decreased the growth of CRC cells. To evaluate their in vivo effects, we injected HT29, c-Myc-KD, p300-KD, and CARM1-KD cells into nude mice to establish tumor xenografts. Our results indicated that the tumors volumes in mice injected with c-Myc-KD, p300-KD, and CARM1-KD cells were similar and they were much smaller than tumors from mice injected with HT29 cells (Figure 8A). To determine the in vivo effects of CPCM component inhibitors, we primarily injected mice with HT-29 cells and then these mice were randomly assigned to four groups (n = 5 in each group). These four-group mice were subsequently injected with PBS, sAJM, CARM-IN-1 or C646 every five days, respectively. The measurement of tumor volumes indicated that these inhibitors significantly inhibited tumor growth in vivo, and there was no significant difference in mice injected different CPCM component inhibitors (Figure 8B). In addition, we also examined in vivo protein level changes in tumors derived from different group mice. As shown in Figure 8C, knockdown of CPCM components caused the decrease of CUL4A/4B protein levels but the increase of ST7 (Figure 8C). Similarly, we also observed CPCM inhibitor treatments resulted in the deduction of CUL4A/4B protein levels but the increase of ST7 (Figure 8D). These results suggested that the impaired CPCM complex decreased CUL4A/4B protein levels, which led to the deduction of ST7 ubiquitination and resulted in its accumulation. The accumulated ST7 functioned as a tumor suppressor to inhibit tumor growth.
Knockdown or inhibition of the CPCM components repressed tumor cell growth in vivo. (A) The knockdown of the CPCM components reduced tumor volumes in vivo. The HT29, c-Myc-KD, CARM1-KD and p300-KD cells were injected into nude mice (n=5 for each cell line) to generate tumors. Tumor volumes were measured in a five-day interval. **P < 0.01. (B) Inhibition of the CPCM components reduced tumor volumes in vivo. The HT29 cells were injected into nude mice and then mice were randomly assigned to four groups (n = 5 in each group). Mice were injected with PBS, 5 μM sAJM, 20 μM CARM-IN-1 or 50 nM C646 every five days. Tumor volumes were measured in a five-day interval. **P < 0.01. (C and D) Knockdown or inhibition of the CPCM components caused the decrease of CUL4A/4B but increase of ST7 in vivo. Tumors from (A) and (B) were subjected to examine protein levels of c-Myc, Max, CARM1, p300, CUL4A, CUL4B, and ST7. GADPH was used as a loading control.
Discussion
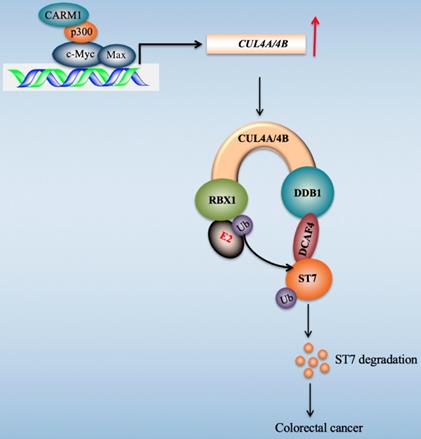
Transcription factors regulate gene expression through coordinating with other proteins such as coactivators and corepressors, and recruiting RNA polymerase II [45]. Our recent publication found that c-Myc induced the expression of CUL4A/4B in CRC cells [27]. To explore the transcriptionally regulatory mechanism of c-Myc in the regulation of CUL4A/4B expression, we purified c-Myc-coupled complex and identified several interesting partners including Max, p300, and CARM1 in this study. We then determined the interactions of these proteins and revealed how these four proteins assembled to a CPCM complex. We also evaluated the effects of knockdown and inhibition of CPCM components on CUL4A/4B expression, ST7 ubiquitination, oncogenic phenotypes, and in vivo tumor growth. Our results supported a model in which the CPCM complex specifically bound to the promoters of CUL4A/4B and activated their expression. The amplified CUL4A/4B assembled two separate CRL4 E3 ligases with DDB1, RBX1 and DCAF4, thereby promoting the ubiquitination of ST7 and leading to its degradation. The degraded ST7 lost its role in preventing tumor cell growth and resulted in the occurrence of CRC (Figure 9).
A schematic diagram of CPCM transcriptional complex and its downstream signaling. In CRC cells, c-Myc is amplified and it dimerizes with Max. The c-Myc-Max heterodimer recruits p300 and CARM1 to assemble the CPCM complex, which specifically binds to the promoters of CUL4A/4B to activate their expression. The overexpressed CUL4A/4B recruit RBX1, DDB1 and DCAF4 to assemble two independent CRL4A/4BDCAF4 E3 ligases, which ubiquitinate a tumor suppressor ST7 and cause its degradation, leading to tumorigenesis.
c-Myc is amplified in multiple human cancers and its overexpression can affect a variety of tumorigenic processes such as cell proliferation, invasion and migration, metastasis, apoptosis, and cell cycle progression [46, 47]. However, it is still not fully understood how c-Myc assembles transcriptional complexes with coactivators and corepressors to regulate downstream target genes in these processes [46, 47]. Biochemically, c-Myc binds gene promoter DNA through a consensus sequence CACGTG, where c-Myc dimerizes with its partner Max [48, 49]. After binding to DNA, c-Myc recruits cofactors through a transcription activation domain (TAD) located in its N-terminus [48, 49]. Like many transcription factors, c-Myc also can directly interact with HATs such as p300, lysine acetyltransferase 2A (KAT2A, also known as GCN5) and KAT5 [50, 51]. Some studies reveal that p300 and KAT2A can acetylate the c-Myc-Max complex, and p300 also can stabilize c-Myc through a mechanism independent on acetylation [50, 52]. p300 can further recruit other proteins such as corepressors CtBP1/2 (C-Terminal binding protein 1 and 2) [53], and coactivator CARM1 [54]. CtBP1 is able to directly bind to the PXDLS motif localized in the bromodomain of p300, and this interaction causes the repression of p300-mediated transactivation [53]. CtBP2 can associate with p300 and transcription factor RUNX2 to form a complex, which specifically binds to the promoters of several bone differentiation and development genes and repress their expression [43]. CARM1 also can be recruited by p300 and it functions as a coregulator of multiple transcription factors such as p53 [55], YY1 (Yin and Yang 1 protein) [56], NF-κB [42], PPARγ (Peroxisome proliferator-activated receptor gamma) [57], RUNX1 and E2F1 (E2F transcription factor 1) [58, 59]. However, no studies have reported so far that c-Myc can recruit p300-CARM1complex to regulate the expression of its downstream target genes. Therefore, our research not only clearly explains how CUL4A/4B is activated, but also enriches the role of the c-Myc-associated transcriptional complex. Our study will provide a reference for elucidating how c-Myc coordinates with other coactivators to activate the expression of downstream genes in c-Myc-overexpressing cancer cells.
Due to its central regulatory role in the oncogenic process, c-Myc is considered as an attractive target in developing anti-cancer medicines [60, 61]. However, it is a challenge to directly targeting c-Myc in the developing c-Myc inhibitors owing to its undruggable protein structure [60, 61]. In the present study, we identified c-Myc could assemble a complex with Max, p300, and CACM1. Importantly, knockdown or inhibition of CPCM components could repress tumor cell growth in vitro and in vivo. Thus, alternatively targeting the CPCM components and the interactions of c-Myc-Max, c-MYc-p300 and p300-CARM1 may be options for developing medicines to inhibit c-Myc downstream targets in CRC.
In summary, our studies identify a CPCM complex, which binds to the promoters of CUL4A/4B and induces their expression, and also activates CUL4A/4B-associated E3 ligases-CRL4. The CRL4 E3 ligases ubiquitinate ST7 and cause its degradation, leading to the tumorigenesis of CRC.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study is supported by four grants including Technology Support Program of Sichuan Province (2015SZ0145, 2017SZ0151, 2018SZ0113) and Technology Support Program of Sichuan Traditional Chinese Medicine Administration (2014D002).
Author Contributions
HL designed the research, analysed the data, coordinated the figures and wrote the manuscript. WL performed most of the experiments including protein purification, RT-PCR, ChIP assay, oncogenic phenotype assays in vitro and in vivo. CY and HH performed some of the experiments including mouse maintenance, cell culture and transfection, and western blotting. All authors revised the manuscript.
Competing Interests
The authors declare no competing financial, professional or personal interests that might have influenced the performance or presentation of the work described in this manuscript.
References
1. Rafiemanesh H, Mohammadian-Hafshejani A, Ghoncheh M, Sepehri Z, Shamlou R, Salehiniya H. et al. Incidence and Mortality of Colorectal Cancer and Relationships with the Human Development Index across the World. Asian Pac J Cancer Prev. 2016;17:2465-73
2. Vuik FE, Nieuwenburg SA, Bardou M, Lansdorp-Vogelaar I, Dinis-Ribeiro M, Bento MJ. et al. Increasing incidence of colorectal cancer in young adults in Europe over the last 25 years. Gut. 2019;68:1820-6
3. Ewing I, Hurley JJ, Josephides E, Millar A. The molecular genetics of colorectal cancer. Frontline Gastroenterol. 2014;5:26-30
4. Grady WM, Markowitz SD. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig Dis Sci. 2015;60:762-72
5. Kriegl L. [In situ analyses of molecular mechanisms of colorectal carcinogenesis]. Pathologe. 2013;34(Suppl 2):269-73
6. Nguyen HT, Duong HQ. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol Lett. 2018;16:9-18
7. Tariq K, Ghias K. Colorectal cancer carcinogenesis: a review of mechanisms. Cancer Biol Med. 2016;13:120-35
8. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073-87 e3
9. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059-72
10. Kastrinos F, Syngal S. Inherited colorectal cancer syndromes. Cancer J. 2011;17:405-15
11. Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138:2044-58
12. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423-37
13. Lee EY, Muller WJ. Oncogenes and tumor suppressor genes. Cold Spring Harb Perspect Biol. 2010;2:a003236
14. Druliner BR, Ruan X, Sicotte H, O'Brien D, Liu H, Kocher JA. et al. Early genetic aberrations in patients with sporadic colorectal cancer. Mol Carcinog. 2018;57:114-24
15. Testa U, Pelosi E, Castelli G. Colorectal cancer: genetic abnormalities, tumor progression, tumor heterogeneity, clonal evolution and tumor-initiating cells. Med Sci (Basel). 2018:6
16. Ding L, Lan Z, Xiong X, Ao H, Feng Y, Gu H. et al. The Dual Role of MicroRNAs in Colorectal Cancer Progression. Int J Mol Sci. 2018:19
17. Masuda T, Hayashi N, Kuroda Y, Ito S, Eguchi H, Mimori K. MicroRNAs as Biomarkers in Colorectal Cancer. Cancers (Basel). 2017:9
18. Cai J, Zuo X, Chen Z, Zhang Y, Wang J, Wang J. et al. Long Noncoding RNAs Serve as Potential Diagnostic Biomarkers for Colorectal Cancer. J Cancer. 2019;10:611-9
19. Han ZJ, Feng YH, Gu BH, Li YM, Chen H. The post-translational modification, SUMOylation, and cancer (Review). Int J Oncol. 2018;52:1081-94
20. Chen Z, Sui J, Zhang F, Zhang C. Cullin family proteins and tumorigenesis: genetic association and molecular mechanisms. J Cancer. 2015;6:233-42
21. Lee J, Zhou P. Cullins and cancer. Genes Cancer. 2010;1:690-9
22. Li Y, Wang X. The role of cullin4B in human cancers. Exp Hematol Oncol. 2017;6:17
23. Sharma P, Nag A. CUL4A ubiquitin ligase: a promising drug target for cancer and other human diseases. Open Biol. 2014;4:130217
24. Chen Z, Wang K, Hou C, Jiang K, Chen B, Chen J. et al. CRL4B(DCAF11) E3 ligase targets p21 for degradation to control cell cycle progression in human osteosarcoma cells. Sci Rep. 2017;7:1175
25. Li B, Jia N, Kapur R, Chun KT. Cul4A targets p27 for degradation and regulates proliferation, cell cycle exit, and differentiation during erythropoiesis. Blood. 2006;107:4291-9
26. Chen Z, Zhang W, Jiang K, Chen B, Wang K, Lao L. et al. MicroRNA-300 Regulates the Ubiquitination of PTEN through the CRL4B(DCAF13) E3 Ligase in Osteosarcoma Cells. Mol Ther Nucleic Acids. 2018;10:254-68
27. Liu H, Lu W, He H, Wu J, Zhang C, Gong H. et al. Inflammation-dependent overexpression of c-Myc enhances CRL4(DCAF4) E3 ligase activity and promotes ubiquitination of ST7 in colitis-associated cancer. J Pathol. 2019;248:464-75
28. Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18:5546-53
29. Rughooputh S, Manraj S, Eddoo R, Greenwell P. Expression of the c-myc oncogene and the presence of HPV 18: possible surrogate markers for cervical cancer? Br J Biomed Sci. 2009;66:74-8
30. Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7:143-64
31. Smith DR, Myint T, Goh HS. Over-expression of the c-myc proto-oncogene in colorectal carcinoma. Br J Cancer. 1993;68:407-13
32. Gamberi G, Benassi MS, Bohling T, Ragazzini P, Molendini L, Sollazzo MR. et al. C-myc and c-fos in human osteosarcoma: prognostic value of mRNA and protein expression. Oncology. 1998;55:556-63
33. Little CD, Nau MM, Carney DN, Gazdar AF, Minna JD. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature. 1983;306:194-6
34. Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429-40
35. Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM. et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003;4:575-80
36. Zhang K, Faiola F, Martinez E. Six lysine residues on c-Myc are direct substrates for acetylation by p300. Biochem Biophys Res Commun. 2005;336:274-80
37. Elliott K, Sakamuro D, Basu A, Du W, Wunner W, Staller P. et al. Bin1 functionally interacts with Myc and inhibits cell proliferation via multiple mechanisms. Oncogene. 1999;18:3564-73
38. Wiese KE, Walz S, von Eyss B, Wolf E, Athineos D, Sansom O. et al. The role of MIZ-1 in MYC-dependent tumorigenesis. Cold Spring Harb Perspect Med. 2013;3:a014290
39. Santos TM, Han S, Bowser M, Sazani K, Beauchamp RL, Murthy V. et al. Alternative splicing in protein associated with Myc (Pam) influences its binding to c-Myc. J Neurosci Res. 2006;83:222-32
40. McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20:556-62
41. Kato GJ, Lee WM, Chen LL, Dang CV. Max: functional domains and interaction with c-Myc. Genes Dev. 1992;6:81-92
42. Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C. et al. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J. 2005;24:85-96
43. Zhang W, Duan N, Zhang Q, Song T, Li Z, Chen X. et al. The intracellular NADH level regulates atrophic nonunion pathogenesis through the CtBP2-p300-Runx2 transcriptional complex. Int J Biol Sci. 2018;14:2023-36
44. Eckner R, Yao TP, Oldread E, Livingston DM. Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes Dev. 1996;10:2478-90
45. Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121-41
46. Chen Y, Olopade OI. MYC in breast tumor progression. Expert Rev Anticancer Ther. 2008;8:1689-98
47. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov. 2015;5:1024-39
48. Sabo A, Amati B. Genome recognition by MYC. Cold Spring Harb Perspect Med. 2014:4
49. Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J. et al. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115-29
50. Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, White RJ. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci U S A. 2007;104:14917-22
51. Wei X, Cai S, Boohaker RJ, Fried J, Li Y, Hu L. et al. KAT5 promotes invasion and metastasis through C-MYC stabilization in ATC. Endocr Relat Cancer. 2019;26:141-51
52. Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ. et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol. 2004;24:10826-34
53. Kim JH, Cho EJ, Kim ST, Youn HD. CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat Struct Mol Biol. 2005;12:423-8
54. Bao J, Rousseaux S, Shen J, Lin K, Lu Y, Bedford MT. The arginine methyltransferase CARM1 represses p300*ACT*CREMtau activity and is required for spermiogenesis. Nucleic Acids Res. 2018;46:4327-43
55. Behera AK, Bhattacharya A, Vasudevan M, Kundu TK. p53 mediated regulation of coactivator associated arginine methyltransferase 1 (CARM1) expression is critical for suppression of adipogenesis. FEBS J. 2018;285:1730-44
56. Behera AK, Kumar M, Shanmugam MK, Bhattacharya A, Rao VJ, Bhat A. et al. Functional interplay between YY1 and CARM1 promotes oral carcinogenesis. Oncotarget. 2019;10:3709-24
57. Yadav N, Cheng D, Richard S, Morel M, Iyer VR, Aldaz CM. et al. CARM1 promotes adipocyte differentiation by coactivating PPARgamma. EMBO Rep. 2008;9:193-8
58. Vu LP, Perna F, Wang L, Voza F, Figueroa ME, Tempst P. et al. PRMT4 blocks myeloid differentiation by assembling a methyl-RUNX1-dependent repressor complex. Cell Rep. 2013;5:1625-38
59. El Messaoudi S, Fabbrizio E, Rodriguez C, Chuchana P, Fauquier L, Cheng D. et al. Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc Natl Acad Sci U S A. 2006;103:13351-6
60. Carabet LA, Rennie PS, Cherkasov A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int J Mol Sci. 2018:20
61. McKeown MR, Bradner JE. Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med. 2014:4
Author contact
![]() Corresponding author: Dr. Hong Liu, Department of Integrated Traditional and Western Medicine, West China Hospital, NO. 37 Guoxue Xiang, Wuhou District, Chengdu, Sichuan Province, China, 610041. Email: liuhong1980edu.cn
Corresponding author: Dr. Hong Liu, Department of Integrated Traditional and Western Medicine, West China Hospital, NO. 37 Guoxue Xiang, Wuhou District, Chengdu, Sichuan Province, China, 610041. Email: liuhong1980edu.cn