Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(11):1774-1784. doi:10.7150/ijbs.42197 This issue Cite
Research Paper
Histone Acetyltransferase (HAT) P300/CBP Inhibitors Induce Synthetic Lethality in PTEN-Deficient Colorectal Cancer Cells through Destabilizing AKT
Yifan Liu, Eun Ju Yang, Changxiang Shi, Pui Kei Mou, Baoyuan Zhang, Changjie Wu, Junfang Lyu, Joong Sup Shim ![]()
Cancer Centre, Faculty of Health Sciences, University of Macau, Taipa, 999078, Macau
Received 2019-11-18; Accepted 2020-2-24; Published 2020-3-25
Abstract

PTEN, a tumor suppressor, is found loss of function in many cancers, including colorectal cancer. To identify the synthetic lethal compounds working with PTEN deficiency, we performed a synthetic lethality drug screening with PTEN-isogenic colorectal cancer cells. From the screening, we found that PTEN-/- colorectal cancer cells were sensitive to anacardic acid, a p300/CBP histone acetyltransferase (HAT) inhibitor. Anacardic acid significantly reduced the viability of PTEN-/- cells not in PTEN+/+ cells via inducing apoptosis. Inhibition of HAT activity of p300/CBP by anacardic acid reduced the acetylation of histones at the promoter region and inhibited the transcription of Hsp70 family of proteins. The down-regulation of Hsp70 family proteins led to the reduction of AKT-Hsp70 complex formation, AKT destabilization and decreased the level of phosphorylated AKT at Ser473, all of which are vital for the survival of PTEN-/- colorectal cells. The synthetic lethality effect of anacardic acid was further validated in tumor xenograft mice models, where PTEN-/- colorectal tumors showed greater sensitivity to anacardic acid treatment than PTEN+/+ tumors. These data suggest that anacardic acid induced synthetic lethality by inhibiting HAT activity of p300/CBP, thereby reducing Hsp70 transcription and destabilizing AKT in PTEN deficient colorectal cancer cells.
Keywords: PTEN, Synthetic lethality, Histone acetyltransferases, Anacardic acid, AKT, Hsp70
Introduction
Phosphatase and tensin homolog (PTEN), a negative regulator of AKT, is a key tumor suppressor gene, which are found mutated or deleted in many cancers. Tumor suppressor genes are the safeguards of regulations of cell cycle, cell adhesion, cell proliferation, cell migration, cell death and DNA damage repairs [1]. Mutation or deletion of tumor suppressor genes is considered as loss of function and lead to functional disruption of these genes by losing inhibition to tumor development [1]. Loss-of-function of PTEN can also be gained from epigenetic silencing. Hypermethylation of CpG islands of PTEN promoter which leads to PTEN transcription silence is found in colorectal, breast, prostate cancer and other cancers [2-4]. The importance of PTEN makes it become a vital target for cancer therapy.
Based on the strategy of synthetic lethality, taken mutation or deletion of tumor suppressor genes as one part in synthetic lethality system, it is prospective to screen partners working in combination with these genes to kill specific tumor suppressor deficient cancer cells. As PTEN is often found mutated or deleted in colorectal cancer CRC [2, 5, 6], synthetic lethality approach targeting PTEN-/- cancer may provide new treatment opportunities for such cancer types [7]. To identify drugs that are selective for PTEN-/- CRC cells, a synthetic lethality drug screening with PTEN-isogenic CRC cell line pair was carried out. Anacardic acid (AA) was identified as a synthetic lethality partner of PTEN deficiency in the screening. AA, a salicylic acid derivative, was a natural HAT inhibitor extracted from Cashew nut shell liquid [8]. AA was reported to inhibit the HAT activity of p300 and p300/CBP-associated factor (PCAF) [9]. It also has been suggested to have anti-microbial, anti- inflammatory and anti-tumor activities in previous research [10-13]. Recent studies suggested that AA could sensitize prostate cancer to ion-radiation (IR) in the androgen receptor dependent pathway [14, 15]. AA induced cell apoptosis through autophagy by ER stress and suppressing DAPK3/ AKT/m-TOR signaling pathway in prostate cancer [16]. However, the anti-cancer activities of AA and its relationship with different PTEN status are unclear in CRC. We here report that AA induces cell apoptosis in a synthetic lethal way in PTEN-/- CRC cells. Mechanistic exploration shows the involvement of HAT-Hsp70- AKT pathway in the AA-induced synthetic lethality in PTEN-/- CRC cells.
Materials and Methods
Cell culture and reagents
The colorectal cancer cell line: HCT116 cells and the prostate cancer cell lines: PC3 and 22RV1 cells were purchased from American Type Tissue Collection (ATCC, Manassas, VA, USA). They were cultured with RPMI-1640 supplemented with 10% Fetal Bovine Serum (#26140079, Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin and streptomycin (#15140163, Thermo Fisher Scientific) at 37° of CO2 incubator. The kinase inhibitor drug library (#L1200, Selleck), Anacardic acid, C646, cycloheximide (CHX), MG132 and MKT077 were purchased from Selleck Chemicals (Houston, TX, USA).
Synthetic lethality screening
The synthetic lethality screening was carried out with PTEN+/+ and PTEN-/- HCT116 isogenic cells in 384-well plate format following the established procedure [17]. A kinase inhibitor library containing 430 small molecules were used for synthetic lethality screening.
Immunoblotting and antibodies
Cells were treated with RIPA buffer (20 mM Tris pH 7.6, 150 mM NaCl, 1% NP40) combined with cOmplete protease inhibitor cocktail (#06538282001, Roche, West Sussex, UK) and lysed for immunoblots. Immunoblots were carried out with 15% and 6% SDS-PAGE gels based on the molecular weight of proteins. The primary antibodies used were listed in Supplementary Table S1. The anti-rabbit and anti-mouse secondary antibodies conjugated with HRP (horse radish peroxidase) were purchased from Santa Cruz and life technology. Western blot signals were detected using Clarity™ Western ECL Substrate (Bio-Rad, Hercules, CA) under a Bio-Rad ChemiDoc MP imaging system.
RNA interferences (RNAi)
The shRNAs of p300 and CBP (#TG313197) were purchased from Origene (Rockville, MD). Reverse transient transfections of shRNAs were performed by using Lipofectamine 3000 transfection reagent (#L3000015, Thermo Fisher Scientific). Cells were seeded in 24-well plates (1 × 105 cells/well) and transfected with shRNAs or siRNAs for 48 h before measuring the cell viability with AlamarBlue. Knockdown efficacy of the shRNAs or siRNAs was assessed by quantitative PCR (q-PCR, RT-PCR) and immunoblotting separately.
Drug combination treatments in vitro
PTEN+/+ and PTEN-/- HCT116 isogenic cells were seeded in 96-well plates for 24 h before adding DMSO control, Anacardic acid, C646, Cycloheximide (CHX), MG132 and MKT077. After incubating with the compounds for 72 h, the cell viability was measured by AlamarBlue assay and were double checked with cell images taken by IncuCyte™ZOOM ((Essen BioScience, Inc., Ann Arbor, Michigan, USA).
Immunoprecipitation
PTEN-/- HCT116 cells was treated with 100 μM Anacardic acid for 0, 3, 6 and 9 hours firstly. The cells were then washed with cold PBS once and lysed with the cell lysis buffer containing 20 mM Tris HCl pH 8, 137 mM NaCl, 10% glycerol, 1% Nonidet P-40 (NP-40), 2 mM EDTA and protease inhibitors (1 mM PMSF and a cOmplete protease inhibitor cocktail) on ice for 5 mins. Cell lysates were centrifuged at 12,000 rpm for 5 mins and the supernatant were used for co-immunoprecipitation with an anti-AKT1 antibody, the mouse IgG as the negative control and Dynabeads™ Protein G Immunoprecipitation Kit (Invitrogen#10007D). The immunoprecipitates were subjected to western blotting with the Hsp70, Hsp90 and AKT1 antibodies separately.
Chromatin Immunoprecipitation
PTEN-/- HCT116 cells were treated with 100μM Anacardic acid for 9hrs after being seeded. 2.0 × 105 cells were used for each single strip well following the protocol with Imprint Chromatin Immunoprecipitation kit from Sigma-Aldrich (Sigma #CHP1-96RXN). The Ac-H4 antibody (sc-8662-R) was used to immunoprecipitate DNA-protein cross-linked complex while the mouse IgG was used as the negative control. The immunoprecipitated promoter sequences of Hsp70s and Hsp90 were detected with real-time PCR.
Real-time quantitative PCR (RT-qPCR)
Cells were washed with cold PBS once and lysed in the Real-time cell lysis buffer containing 10mM Tris pH 7.4, 0.25% Igepal CA-630, and 150mM NaCl for 5 mins [18]. Cell lysates were directly used for real-time as the amplification templates. All the primers used in RT-qPCR are listed in Supplementary Table S2.
Overexpression of Hsp70 (HSPA1A) and Hsc70 (HSPA8)
For overexpression of Hsp70 and Hsc70, HCT116-PTEN-/- cells (2.0 × 105 cells/well in a 24-well plate) were transfected with pcDNA5/FRT/TO GFP HSPA1A (Addgene plasmid, #19483) or pcDNA5/ FRT/TO GFP HSPA8 (Addgene plasmid, #19487) plasmid mixed in Lipofectamine 3000. The transfected cells were co-treated with or without 100 μM AA for 24 h and the cell viability was measured with AlamarBlue assay.
In vivo drug combination studies on mice xenograft models
Drug effects were checked in vivo efficacy on the mice where HCT116- PTEN+/+ and HCT116-PTEN-/- cells (1 × 106 cells/injection) were injected subcutaneously in each flank (bilaterally) of female Nude mice. After 4 days of tumor cell inoculation, both tumors became palpable. The mice were then randomized into 3 groups (n=5/group) of equal tumor volume for treatments with vehicle alone (DMSO) and Anacardic acid (2.5 mg/kg and 5 mg/kg, solved in DMSO) by subcutaneous injection around the tumor [19]. Treatment was done daily for 4 weeks. Tumor size was measured periodically with a vernier caliper and tumor volume was calculated based on the modified ellipsoid formula (long axis × short axis2 × π/6). All in vivo modelling was carried out in accordance with the approved animal protocol (UMARE-010-2018) by Animal Research Ethics Committee of the University Macau.
Statistical analysis
The data are represented as the mean ± SD. Statistical analyses were performed using analysis of variance (ANOVA) and Student's t-test with Graphpad Prism. A P-value less than 0.05 was considered statistically significant.
Results
Small Molecule Screening Identifies Anacardic Acid as Synthetic Lethality Compound in PTEN-Deficient CRC Cells
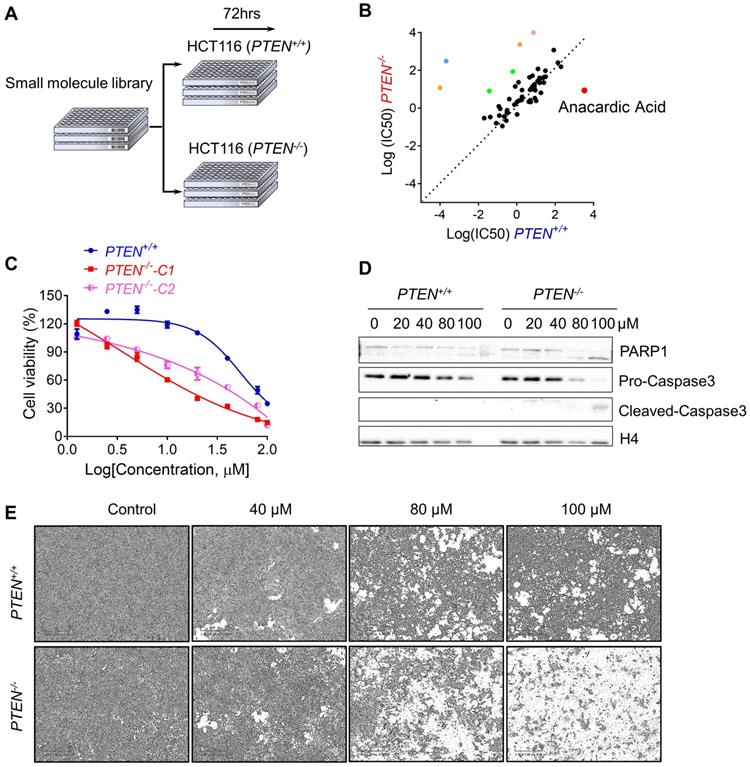
To perform the synthetic lethality drug screening based on the differences of PTEN status, PTEN isogenic HCT116 pair: PTEN+/+ and PTEN-/- was used to screen for synthetic lethality compounds[17]. Total 430 small molecules from kinase inhibitor library that targets most of drug-targetable human kinases were screened (Fig. 1A). From a pair-wise, 8-dose titration screening of 430 small molecules, 7 compounds that showed different selectivity in PTEN+/+ cells over the PTEN-/- part were identified (Fig. 1B). Among the compounds, 2 MEK inhibitors (Trametinib and TAK- 733), 2 VEGFR inhibitors (Apatinib and Nintedanib), a BTK inhibitor (AVL-290) and an SRC inhibitor (PP1) showed the greater selectivity toward PTEN+/+ CRC cells. Anacardic acid (AA), a p300/CBP HAT inhibitor, showed the greater selectivity toward PTEN-/- CRC cells (Fig. 1B). It is known that PTEN deficiency contributed to the drug resistance to trametinib in M14 melanoma cells [20]. Other MEK inhibitors also showed drug resistance with PTEN deletion in acute myeloid leukemia [21]. These data demonstrated the feasibility of our synthetic lethality screening system. Since AA was the only compound that showed the selectivity toward PTEN-/- CRC cells, we studied the effect of AA in greater detail with regard to the PTEN deficiency in CRC cells. AA significantly inhibited the viability of PTEN-/- cells, while they marginally affected PTEN+/+ cell viability (Fig. 1C and E). Similar results were observed in another PTEN-/- HCT116 cell line (PTEN-/--C2), suggesting that the differential sensitivity of the compound was likely due to the difference in the PTEN status (Fig. 1C). It was further observed that AA significantly induced caspase-3 and PARP1 cleavage in PTEN-/- cells but not in PTEN+/+, suggesting that the synthetic lethality effect of AA was via the induction of apoptosis in PTEN-/- cells (Fig. 1D).
Synthetic lethality screening with small compound library identified anacardic acid as a synthetic lethality compound in PTEN-/- colorectal cancer cells. (A) Schematic representation describing workflow for synthetic lethality screening with small compound library in PTEN+/+ and PTEN-/- HCT116 cell pairs. (B) The log10 plot of the IC50 values of each compound against PTEN+/+ and PTEN-/- HCT116 cells. Two MEK inhibitors (Trametinib and TAK-733, Green colored), 2 VEGFR inhibitors (Apatinib and Nintedanib, Orange colored), a BTK inhibitor (AVL-290, Blue colored), an SRC inhibitor (PP1, Pink colored) and a p300/CBP inhibitor (Anacardic acid, AA, red colored) are marked as selective inhibitors toward PTEN+/+ or PTEN-/- HCT116 cells. (C) Survival curves of PTEN+/+ and PTEN-/- HCT116 cells treated with anacardic acid for 72 h. PTEN-/--C1 and PTEN-/--C2 represent PTEN-/- clone 1 and 2, respectively. (D) Western blots of cleaved PARP1 and cleaved caspase 3 in PTEN+/+ and PTEN-/- HCT116 cells treated with anacardic acid for 72 h. (E) Representative cell images from PTEN+/+ and PTEN-/- HCT116 cells treated with anacardic acid for 72 h.
Inhibition of p300/CBP HAT Activity by shRNA Silencing or Another Small Molecule p300/CBP Inhibitor C646 Recapitulated the Phenotype of Anacardic Acid
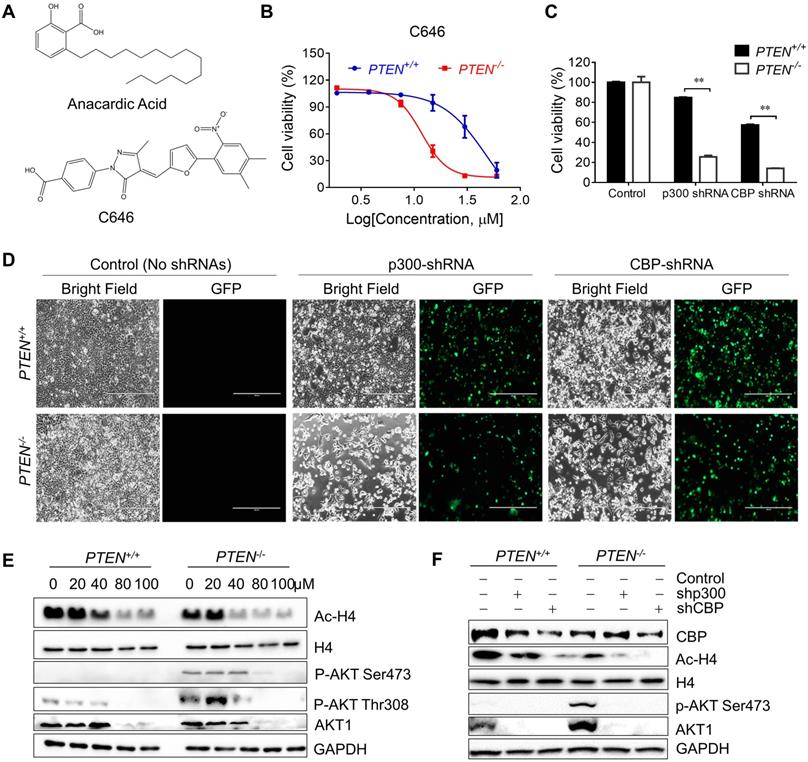
Since AA was reported as a HAT inhibitor, we first checked whether the synthetic lethality effect of AA in the PTEN-/- cells was based on inhibition of HAT activity of p300/CBP or not. The isogenic CRC cells were treated with another specific p300/CBP inhibitor C646 and it was found that PTEN-/- cells were more sensitive to C646 (Fig. 2B). Then, further exploration was made into the effects of inhibition of p300 and CBP by silencing with specific p300/CBP- EGFP tagged shRNAs separately. Silencing p300 or CBP selectively inhibited the viability of PTEN-/- HCT116 cells, but not PTEN+/+ ones, indicating that the inhibition of p300 and CBP was likely to mediate the synthetic lethality phenotype induced by AA (Fig. 2C and D; Supplementary Fig. S1). Further analysis of the downstream pathways that might be affected by p300/CBP inhibition or silencing showed that the acetylation of histone H4 decreased while total H4 showed no difference, which was as expected (Fig. 2E and F). These data indicated that AA indeed inhibited the HAT activity of p300 and CBP.
Inhibiting p300/CBP induced synthetic lethality in PTEN-/- colorectal cancer cells. (A) The structures of AA and C646. (B) Survival curves of PTEN+/+ and PTEN-/- HCT116 cells treated with C646. Cells were treated with C646 for 96 h. (C) Bar charts of cell viability in PTEN+/+/PTEN-/- cells after transfecting with p300 and CBP shRNAs for 72 h. (D) Phase-contrast and fluorescence images of PTEN+/+/PTEN-/- cells transfected with p300 or CBP shRNAs with GFP for 72 h. (E) Western blots of Ac-H4, H4, p-AKT Ser473, p-AKT Thr308, AKT1 and GAPDH in PTEN isogenic HCT116 pairs after treated with 0, 20,40, 80, 100 μM anacardic acid for 24 h. (F) Western blots of CBP, Ac-H4, p-AKT Ser473, AKT1 and GAPDH after knock-downing p300/CBP with shRNAs for 72 h in PTEN isogeneic HCT116 cell pairs. **P-values ≤ 0.01 in Student's t-test.
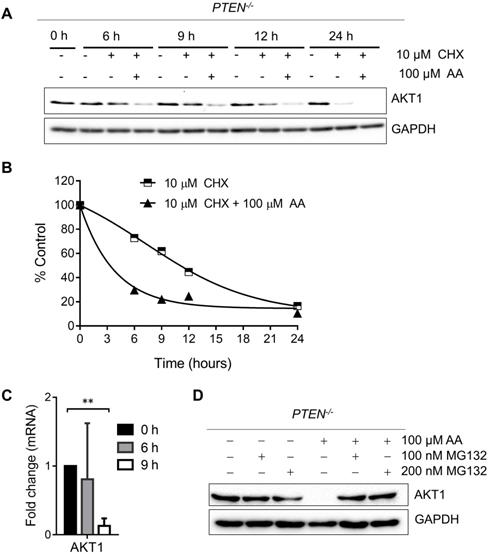
As PI3K-AKT pathway was well known to be significantly affected by PTEN status, a check on downstream proteins of PTEN was made. The levels of p-AKT-Ser473 and total AKT1 were reduced by AA treatment or the silencing of p300 and CBP (Fig. 2E and F). The reduction of the level of p-AKT-Ser473 was likely to be resulted from the reduction of total AKT level as their reduction dynamics was similar. To test the mechanism how AA reduced the AKT protein level, we treated the PTEN-/- HCT116 cells with AA alone or combined with the protein translation inhibitor cycloheximide (CHX) for 24 h. Western blotting of AKT1 at 0, 3, 6, 9, 12, 24 h showed that the half-life of AKT1 was around 12 hours after CHX treatment and it was shortened to 3.5 hours when combined with AA (Fig. 3A and B), suggesting that AA significantly reduced the AKT1 protein stability. As acetylation of histones regulates transcription activation and repression [22], we also tested whether AA affect AKT1 transcription. RT-qPCR analysis showed that AA treatment also reduced AKT1 mRNA level (Fig. 3C). In addition, co-treatment of MG132, a proteasome inhibitor, can reverse the effect of AA on the reduction of AKT1 level (Fig. 3D), suggesting that AA reduces AKT1 level at both the transcription and the post-translational levels after inhibition of p300/CBP HAT activity.
Anacardic acid reduces the level of AKT at both transcription and post-translational levels. (A) Western blots of AKT1 in PTEN-/- cells treated with 10 μM cycloheximide (CHX) combined with or without 100 μM AA for indicated time points. (B) Quantification curve of AKT1 protein level based on the western blot of (A). (C) RT-qPCR analysis of AKT1 mRNA in PTEN-/- cells treated with 100 μM anacardic acid for 9 h. (D) Western blots of AKT1 and GAPDH in PTEN-/- cells treated with anacardic acid, MG132 and the combination of both for 24 h. **P-values ≤ 0.01 in Student's t-test.
Anacardic Acid Reduces the Transcription of Hsp70 Family by Inhibiting p300/CBP HAT Activity
The heat shock protein 70 (Hsp70s) family works in protein folding, helps misfolded client proteins to refold and degrades the abnormal proteins [23-25]. Heat shock protein 90 (Hsp90s) family stabilizes AKT by forming HSP-client complex [24, 26, 27]. To explore the mechanisms that underlie the destabilization of AKT and the related decrease of AKT phosphorylation at Ser473, firstly, a check was made on the heat shock proteins which were reported to bind with AKT and form the functional complex [24, 26]. Surprisingly, it was found that the protein level of Hsp70, but not Hsp90, was dose-dependently reduced by AA (Fig. 4A). Immunoprecipitation analysis of AKT1/Hsp complex showed that while Hsp70 and Hsp90 proteins co-immunoprecipitated with AKT1, AA significantly reduced the Hsp70 level from the complex (Fig. 4B; Supplementary Fig. S2). These data gave clues that the reduction of Hsp70 level might contribute to the AKT1 destabilization after AA treatment. There are two major cytosolic members of Hsp70 family: the constitutive Hsc70, and the inducible Hsp70 (Hsp70A1A and Hsp70A1B) which are induced under stress and are important for cancer cell survival [28, 29]. We thus used MK077, an allosteric inhibitor of Hsp70, which binds to ADP-bound state of Hsp70 and inhibits the release of its substrates [30], to test the effect of Hsp70 inhibition on AKT1 stability and synthetic lethality. Either silencing of the Hsp70 protein family or small molecule inhibitor recapitulated the synthetic lethality phenotype in PTEN-/- HCT116 cells (Fig. 4C, and D). In order to assess the causal relationship between Hsp70 and AA-induced synthetic lethality, we analyzed the rescue effect of Hsp70 overexpression on the synthetic lethality using two Hsp70 overexpression vectors, including HSPA1A (Hsp70)-GFP and HSPA8 (Hsc70)- GFP (Supplementary Fig. S3). Overexpression Hsp70 or Hsc70 significantly rescued the synthetic lethality effects of AA in PTEN-/- HCT116 cells (Fig. 4H). These data suggested that the reduction of Hsp70 level was likely the key factor mediating the synthetic lethality effect of AA.
Anacardic acid decreased the transcription of Hsp70 family by inhibiting P300/CBP HAT activity on promoters of Hsp70 family and induced destabilization of Hsp70/AKT complex. (A) Western blots of heat shock proteins in PTEN isogenic HCT116 pairs after treated with 0, 20, 40, 80, 100 μM anacardic acid for 24 h. (B) Western blots of co-immunoprecipitation experiment of heat shock protein members with AKT1 in PTEN-/- cells treated with 100 μM AA for 3, 6 and 9 h. (C) Bar charts of cell viability after incubating PTEN+/+ and PTEN-/- HCT116 cell lines with the Hsp70 inhibitor MKT077. (D) Western blots of p-AKT Ser473, AKT1, cleaved-caspase3, PARP1, Hsc70, Hsp70, Hsp90 and beta-actin in PTEN isogenic HCT116 pairs after treating with 0, 2.5, 5, 10, 20 μM MKT077 for 24 h. (E) Western blots of AKT1, Hsp70, Hsp90, Ac-H4 and GAPDH in PTEN-/- cells treated with anacardic acid, MG132 and the combination of both for 24 h. (F) Real-time qPCR showing the changes in the expression level of Hsp70 and Hsp90 family at 0, 6, 9 h with 100 μM anacardic acid in PTEN-/- cells. (G) Bar charts of immunoprecipitated promoter regions of heat shock protein members and AKT1 with acetylation H4 antibody in ChIP experiments after treating with anacardic acid for 9 h in PTEN-/- cells. (H) The effect of the overexpression of Hsp70/Hsc70 on anti-proliferative effect on AA in PTEN-/- cancer cells. PTEN-/- HCT116 cells were transfected with Hsp70 or Hsc70 plasmid and treated with 100 μM AA for 24 h. The cell viability was measured with AlamarBlue assay. *P-values ≤ 0.05; **P-values ≤ 0.01 in Student's t-test.
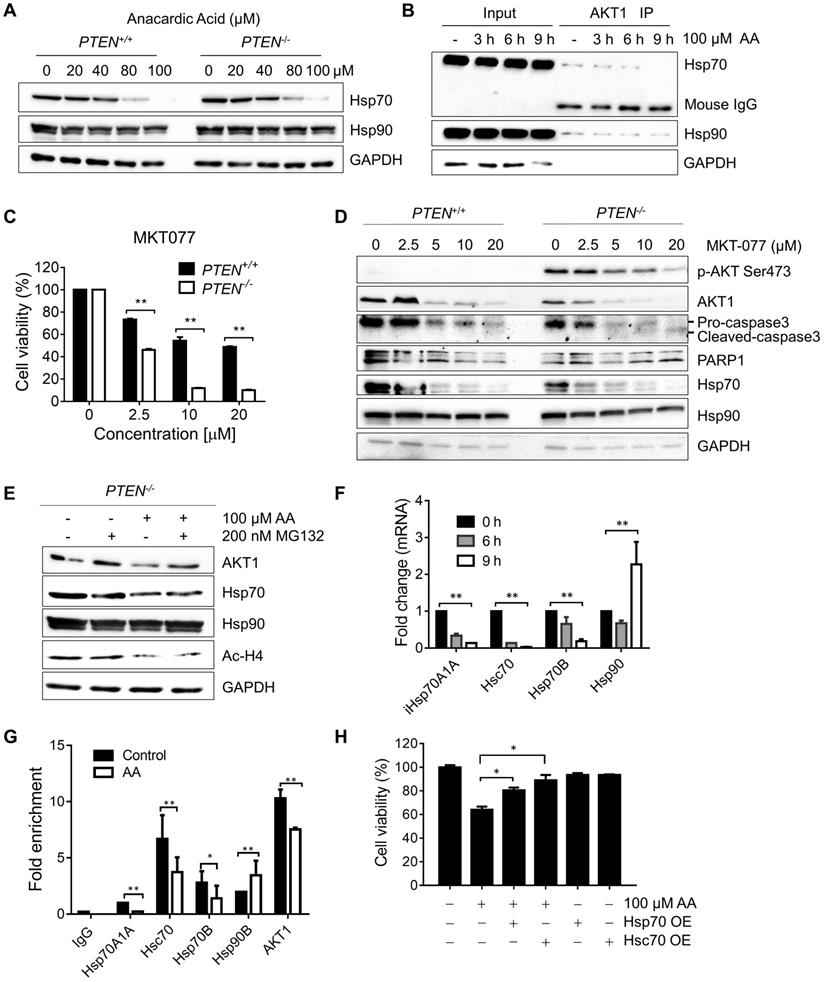
To test whether the reduction of Hsp70 by AA is due to the inhibition at the transcription or post-translational level, the PTEN-/- HCT116 cells were treated with AA with or without MG132 and Hsp70, Hsp90, AKT and acetyl-H4 levels were analyzed. As a result, MG132 treatment did not reverse the effect of AA on decreasing Hsp70 or acetyl-H4, while it reversed the AKT1 level, indicating that the decrease in Hsp70 level might be due to the inhibition at the transcription level (Fig. 4E). RT-qPCR verified that the mRNAs of Hsp70 family members decreased significantly by AA treatment, while no inhibition on Hsp90 mRNA was observed (Fig.4F). As the acetylation of histones regulates transcription activation and repression [22], further exploration was made in the relationship between the down-regulation of Hsp70 transcription and histone acetylation changes induced by AA. For this, we conducted chromatin immunoprecipitation (ChIP) analysis of the promoter regions of Hsp70 family and Hsp90 genes using anti-acetyl-H4 antibody in PTEN-/- CRC cells treated with AA. It was found that AA treatment significantly reduced the level of local H4 acetylation on the promoter regions of Hsp70 (Fig. 4G). AA did not affect the H4 acetylation on the promoter region of Hsp90 gene, while it partially reduced the H4 acetylation on the promoter region of AKT1. These data further suggested that AA reduced Hsp70 transcription through the inhibition of p300/CBP HAT activity, and the reduced Hsp70 protein in turn destabilized AKT1 protein level. AA also reduced AKT1 transcription level through the inhibition of HAT activity. The decrease in AKT1 level through transcription and post-translational regulations was likely to make a significant impact on the viability of PTEN-/- CRC cells.
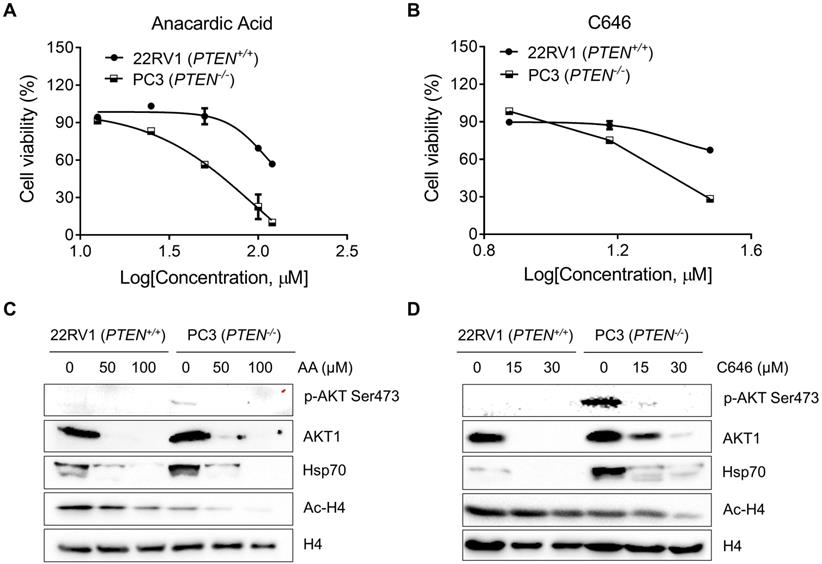
Anacardic Acid and C646 Selectively Inhibit PTEN-Deficient Prostate Cancer Cells
To further validate that PTEN-mediated synthetic lethality to AA could apply to other cancers; two p300/CBP inhibitors, AA and C646 in a pair of prostate cancer cell lines with different PTEN status: 22RV1 (PTEN wild-type, PTEN+/+) and PC3 (PTEN null, PTEN-/-). AA and C646 induced significant cell death in PC3 cells, while they showed marginal effects in 22RV1 (Fig. 5A and B). Hsp70, p-AKT Ser473, AKT1, and Ac-H4 protein levels showed an identical changing trend as what CRC isogenic cell line pair did (Fig. 5C and D). These data further demonstrated that PTEN deficiency resulted in synthetic lethality with the inhibition of HAT activity of p300/CBP in prostate cancer cells.
Anacardic acid and C646 sensitize PTEN deficient prostate cancer cells to die. (A and B) Survival curves of 22RV1 and PC3 cells treated with AA (A) and C646 (B) for 96 h. (C and D) Western blots of p-AKT Ser473, AKT1, Hsp70s, Ac-H4 and H4 in prostate cancer cells treated with two p300/CBP inhibitors, AA (C) and C646 (D) for 24 h.
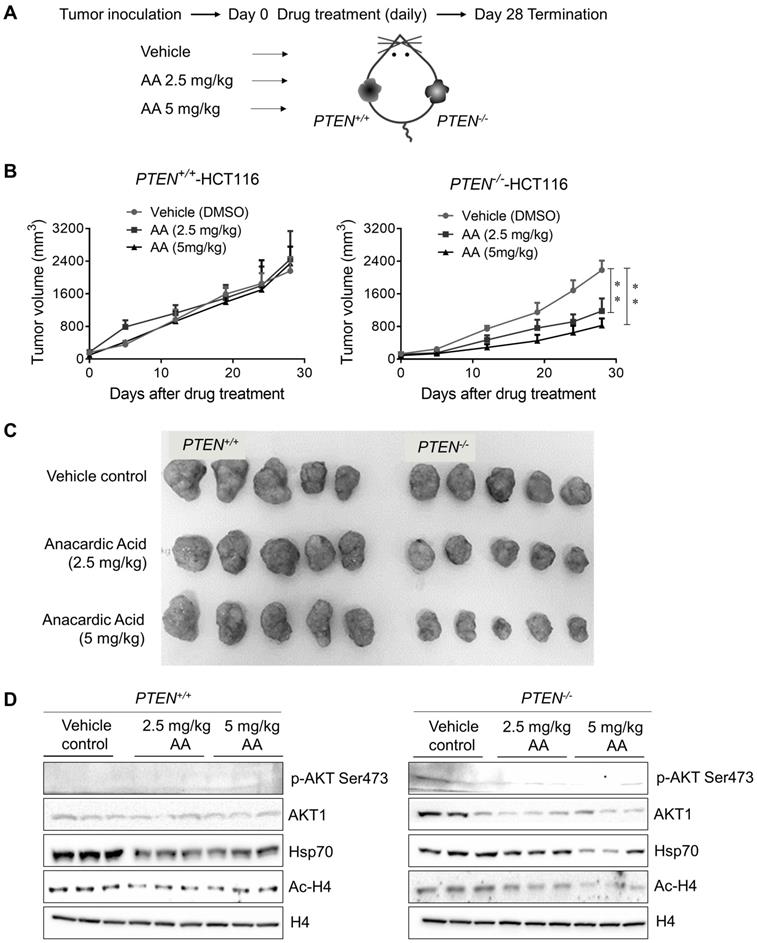
Anacardic Acid Showed Synthetic Lethality with PTEN Loss in CRC Mice Xenograft Model
To further validate PTEN-mediated synthetic lethality to AA, a PTEN-isogenic tumor xenograft mouse model experiment was employed as shown in Fig. 6A. The growth rate of PTEN isogenic CRC cell line pair was identical in 72 h in vitro (Supplementary Fig. S4A), which precluded the affection of different growth rates of two cell lines. Mice bearing PTEN- isogenic tumors in both flanks were given AA for 28 days, and tumor volume and mouse body weight were measured periodically. These measurements indicated that all the drug treatment conditions in this study did not show any apparent toxicity in mice (Supplementary Fig. S4B). The treatment with AA significantly delayed the tumor growth of PTEN-/- HCT116 xenografts (Fig. 6B and C). Whereas the growth of PTEN+/+ HCT116 xenografts was not affected by the same dosage of AA. Similar to that observed in cells, the level of phosphorylated AKT at Ser473 was much higher in PTEN-/- tumors than in PTEN+/+ ones and was significantly reduced by AA treatment (Fig. 6D). AA treatment also showed the inhibition of histone acetylation at H4 and the reduction of Hsp70 and AKT1 levels in both PTEN+/+ and PTEN-/- tumors, recapitulating in vitro data. These data further demonstrated that the p300/CBP HAT inhibitor AA induced synthetic lethality in PTEN- deficient CRC tumor in vivo.
Xenograft mice models showed anacardic acid reduced tumor growth significantly in PTEN-/- cancer cells in vivo. (A) Schematic representation of AA drug experiment in mice bearing established PTEN+/+ / PTEN-/- xenografts. (B) Mice were randomized into treatment cohorts of AA (2.5 mg/kg and 5 mg/kg, daily by subcutaneous injection), and vehicle treatments. Mice were treated for a subsequent 28 days' period. Tumor volume was monitored twice weekly. **P-values ≤ 0.01 in ANOVA analysis. (C) Whole tumor tissues isolated from mice xenograft cohorts. (D) Western blots p-AKT Ser473, total AKT1, Hsp70, Ac-H4 and H4 levels in mice tumor samples.
Discussion
Epigenetic regulation of PTEN-AKT axis occurs commonly in a variety cancers. In Alveolar rhabdomyosarcoma (ARMS) cells, PTEN transcription was repressed by lysine methyltransferase G9a, which activates AKT and promotes the viability of ARMS cells [31]. Epigenetic silencing of PTEN was supposed to be related with the inactivation of the tumor suppressor function and the de-repression of AKT pathway in melanoma development [32, 33]. Long term treatment of imatinib caused drug resistance by down-regulating PTEN expression with hypermethylation on the promoter of PTEN and activating AKT with the help of highly-enriched DNMT3A and EZH2 in leukemia cells [34, 35]. There are also evidences of pathway crosstalk between PTEN and histone acetyltransferases (HATs) in regulating gene transcription. PCAF, a HAT working with p300/CBP in regulating gene transcription, was known to interact and acetylate PTEN, causing down-regulation of PI3K/AKT signaling pathway [36]. Loss of PTEN inhibited the activity of Forkhead transcription factors (FOXO) [37]. Inactivation of FOXO3a activated p300-dependent hypoxia-inducible factor-1 (HIF-1) transcriptional activity and promoted tumorigenesis subsequently [37]. PTEN deficiency facilitated p300 to bind and acetylate androgen receptor (AR) by increasing the phosphorylation of AR in prostate cancer cells. The acetylation of AR by p300 precluded the poly-ubiquitination and hence inhibited proteasome-dependent degradation of AR [38]. Knocking down p300 with siRNAs inhibited the proliferation and progression of PTEN-deficient prostate cancer cells and tumors in mice [38]. These data suggested the existence of the functional crosstalk between PTEN and HAT via direct interaction or indirectly through regulating epigenetics machinery in cancer.
In this study, from a small molecule screening, we identified the small molecule p300/CBP inhibitor AA as a synthetic lethality compound in PTEN- deficient CRC cells. Mechanistically, AA inhibition of the HAT activity of p300/CBP led to the reduced transcription of Hsp70 family proteins, which in turn caused reduced AKT1-Hsp70 complex formation, inducing AKT1 destabilization. Inhibition of p300/ CBP by AA also reduced the transcription of AKT1, contributing partially to the depletion of AKT1 protein level in AA-treated CRC cells. PTEN-/- CRC cells have very high level of phosphorylated AKT1 at Ser473 due to the lack of its phosphatase activity, and this high level of phospho-AKT1 makes the cells highly dependent on AKT1 signaling for their survival, a phenomenon called oncogene addiction. Cancer cells with an oncogene addiction are generally hyper-vulnerable to the inhibitors of the oncogene signaling that the cells are highly relying on. Our previous study showed that PTEN-/- CRC cells were sensitive to AKT inhibition or inhibition of upstream pathways that phosphorylate Ser473 of AKT in CRC [17]. PTEN deletion or mutation was also reported to increase the sensitivity to AKT inhibitors, PI3K inhibitors, mTOR inhibitors and Hsp90 chaperone inhibitors in breast and other types of cancers [17, 39, 40]. Indeed, a number of PI3K/AKT inhibitors are currently under Phase 1 or 2 clinical trials for cancer patients with PTEN mutations (https://clinicaltrials.gov/). Therefore, our study suggests that AA-induced depletion of AKT1 protein could be the main factor mediating the synthetic lethality effect in PTEN-/- CRC cells.
Considering the critical role of AKT activation in PTEN mutated or deleted cancers, eliminating either AKT total protein level or phospho-AKT at Ser473 level would interrupt PI3K-AKT pathway and induce cell death of those cancers that are dependent on AKT. Our study indicate that targeting upstream pathways that activate AKT phosphorylation, or proteins that help stabilizing AKT protein, such as Hsp70s or Hsp90, would be effective strategies for cancer therapy for patients with PTEN loss.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Prof. Gang LI of the University of Macau to share the shp300 and shCBP plasmids. We also thank to the UM-FHS Animal Facility at the University of Macau for experimental and technical supports in animal experiments. This study was supported by the Multi-Year Research Grant of the University of Macau, MYRG2017-00176-FHS and MYRG2019-00116-FHS.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weinberg RA. Tumor suppressor genes. Science. 1991;254:1138-46
2. Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L. et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res. 2004;64:3014-21
3. Khan S, Kumagai T, Vora J, Bose N, Sehgal I, Koeffler PH. et al. PTEN promoter is methylated in a proportion of invasive breast cancers. Int J Cancer. 2004;112:407-10
4. Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL. et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A. 1998;95:5246-50
5. Behjati S, Tarpey PS, Sheldon H, Martincorena I, Van Loo P, Gundem G. et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet. 2014;46:376-9
6. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330-7
7. Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011;10:351-64
8. Tyman JHP, Johnson RA, Muir M, Rokhgar R. The Extraction of Natural Cashew Nut-Shell Liquid from the Cashew Nut (Anacardium-Occidentale). J Am Oil Chem Soc. 1989;66:553-7
9. Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278:19134-40
10. Muroi H, Kubo I. Bactericidal Activity of Anacardic Acids against Streptococcus-Mutans and Their Potentiation. J Agr Food Chem. 1993;41:1780-3
11. Kubo I, Muroi H, Himejima M, Yamagiwa Y, Mera H, Tokushima K. et al. Structure Antibacterial Activity Relationships of Anacardic Acids. J Agr Food Chem. 1993;41:1016-9
12. Schultz DJ, Wickramasinghe NS, Ivanova MM, Isaacs SM, Dougherty SM, Imbert-Fernandez Y. et al. Anacardic Acid Inhibits Estrogen Receptor alpha-DNA Binding and Reduces Target Gene Transcription and Breast Cancer Cell Proliferation. Mol Cancer Ther. 2010;9:594-605
13. Yasutake T, Wada H, Higaki M, Nakamura M, Honda K, Watanabe M. et al. Anacardic acid, a histone acetyltransferase inhibitor, modulates LPS-induced IL-8 expression in a human alveolar epithelial cell line A549. F1000Res. 2013;2:78
14. Yao K, Jiang XZ, He LY, Tang YX, Yin GM, Zeng Q. et al. Anacardic acid sensitizes prostate cancer cells to radiation therapy by regulating H2AX expression. Int J Clin Exp Patho. 2015;8:15926-32
15. Zhao Q, Zhang XF, Cai HF, Zhang P, Kong D, Ge XS. et al. Anticancer effects of plant derived Anacardic acid on human breast cancer MDA-MB-231 cells. Am J Transl Res. 2018;10:2424 -+
16. Tan J, Jiang XZ, Yin GM, He LY, Liu JY, Long Z. et al. Anacardic acid induces cell apoptosis of prostatic cancer through autophagy by ER stress/DAPK3/Akt signaling pathway. Oncol Rep. 2017;38:1373-82
17. Liu YF, Yang EJ, Zhang BY, Miao ZQ, Wu CJ, Lyu JF. et al. PTEN deficiency confers colorectal cancer cell resistance to dual inhibitors of FLT3 and aurora kinase A. Cancer Lett. 2018;436:28-37
18. Shatzkes K, Teferedegne B, Murata H. A simple, inexpensive method for preparing cell lysates suitable for downstream reverse transcription quantitative PCR. Sci Rep. 2014;4:4659
19. Banerjee S, Kaseb AO, Wang Z, Kong D, Mohammad M, Padhye S. et al. Antitumor activity of gemcitabine and oxaliplatin is augmented by thymoquinone in pancreatic cancer. Cancer Res. 2009;69:5575-83
20. Milella M, Falcone I, Conciatori F, Matteoni S, Sacconi A, De Luca T. et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci Rep. 2017:7
21. Smith AM, Zhang CRC, Cristino AS, Grady JP, Fink JL, Moore AS. PTEN deletion drives acute myeloid leukemia resistance to MEK inhibitors. Oncotarget. 2019;10:5755-67
22. Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin - Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224-9
23. Hartl FU, Hayer-Hartl M. Protein folding - Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852-8
24. Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111-33
25. Duncan EJ, Cheetham ME, Chapple JP, van der Spuy J. The role of HSP70 and its co-chaperones in protein misfolding, aggregation and disease. Subcell Biochem. 2015;78:243-73
26. Liao Y, Hung MC. Physiological regulation of Akt activity and stability. Am J Transl Res. 2010;2:19-42
27. Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277:39858-66
28. Tutar Y, Song Y, Masison D. Primate chaperones Hsc70 (constitutive) and Hsp70 (induced) differ functionally in supporting growth and prion propagation in Saccharomyces cerevisiae. FEBS J. 2006;273:237 -
29. Zorzi E, Bonvini P. Inducible hsp70 in the regulation of cancer cell survival: analysis of chaperone induction, expression and activity. Cancers (Basel). 2011;3:3921-56
30. Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER. Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol. 2011;411:614-32
31. Bhat AV, Kala MP, Rao VK, Pignata L, Lim HJ, Suriyamurthy S. et al. Epigenetic Regulation of the PTEN-AKT-RAC1 Axis by G9a Is Critical for Tumor Growth in Alveolar Rhabdomyosarcoma. Cancer Res. 2019;79:2232-43
32. Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, Hengge UR. Epigenetic silencing of the PTEN gene in melanoma. J Invest Dermatol. 2006;126:86 -
33. Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ, Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000;157:1123-8
34. Nishioka C, Ikezoe T, Yang J, Udaka K, Yokoyama A. Imatinib causes epigenetic alterations of PTEN gene via upregulation of DNA methyltransferases and polycomb group proteins. Blood Cancer J. 2011:1
35. Nishioka C, Ikezoe T, Yang J, Yokoyama A. Long-term exposure of leukemia cells to multi-targeted tyrosine kinase inhibitor induces activations of AKT, ERK and STAT5 signaling via epigenetic silencing of the PTEN gene. Leukemia. 2010;24:1631-40
36. Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. J Biol Chem. 2006;281:26562-8
37. Emerling BM, Weinberg F, Liu JL, Mak TW, Chandel NS. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). P Natl Acad Sci USA. 2008;105:2622-7
38. Zhong J, Ding LY, Bohrer LR, Pan YQ, Liu P, Zhang J. et al. p300 Acetyltransferase Regulates Androgen Receptor Degradation and PTEN-Deficient Prostate Tumorigenesis. Cancer Res. 2014;74:1870-80
39. Dillon LM, Miller TW. Therapeutic targeting of cancers with loss of PTEN function. Curr Drug Targets. 2014;15:65-79
40. Kingston B, Bailleux C, Delaloge S, Schiavon G, Scott V, Lacroix-Triki M. et al. Exceptional Response to AKT Inhibition in Patients With Breast Cancer and Germline PTEN Mutations. JCO Precis Oncol. 2019:1-7
Author contact
![]() Corresponding author: Joong Sup Shim, PhD, Email: jsshimedu.mo; Tel.: +853-8822-4990
Corresponding author: Joong Sup Shim, PhD, Email: jsshimedu.mo; Tel.: +853-8822-4990