Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Material and Methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(11):1861-1875. doi:10.7150/ijbs.45112 This issue Cite
Research Paper
Osblr8 orchestrates intrachromosomal loop structure required for maintaining stem cell pluripotency
Yanbo Zhu1, Zi Yan2,3, Zhonghua Du1, Shilin Zhang1, Changhao Fu3, Ying Meng1,3, Xue Wen1, Yizhuo Wang1, Andrew R. Hoffman3, Ji-Fan Hu1,3 ![]() , Jiuwei Cui1
, Jiuwei Cui1 ![]() , Wei Li1
, Wei Li1 ![]()
1. Key Laboratory of Organ Regeneration and Transplantation of Ministry of Education, Stem Cell and Cancer Center, The First Hospital of Jilin University, Changchun, Jilin 130021, China
2. Division of Endocrinology and Metabolism, The First Hospital of Jilin University, Changchun, Jilin 130021, China
3. Stanford University Medical School, VA Palo Alto Health Care System, Palo Alto, CA 94304, USA
Received 2020-2-19; Accepted 2020-2-22; Published 2020-4-6
Abstract

Induced pluripotent stem cells (iPSCs), derived from reprogramming of somatic cells by a cocktail of transcription factors, have the capacity for unlimited self-renewal and the ability to differentiate into all of cell types present in the body. iPSCs may have therapeutic potential in regenerative medicine, replacing injured tissues or even whole organs. In this study, we examine epigenetic factors embedded in the specific 3-dimensional intrachromosomal architecture required for the activation of endogenous pluripotency genes. Using chromatin RNA in situ reverse transcription sequencing (CRIST-seq), we identified an Oct4-Sox2 binding long noncoding RNA, referred as to Osblr8, that is present in association with pluripotency status. Osblr8 was highly expressed in iPSCs and E14 embryonic stem cells, but it was silenced in fibroblasts. By using shRNA to knock down Osblr8, we found that this lncRNA was required for the maintenance of pluripotency. Overexpression of Osblr8 activated endogenous stem cell core factor genes. Mechanistically, Osblr8 participated in the formation of an intrachromosomal looping structure that is required to activate stem cell core factors during reprogramming. In summary, we have demonstrated that lncRNA Osblr8 is a chromatin architecture modulator of pluripotency-associated master gene promoters, highlighting its critical epigenetic role in reprogramming.
Keywords: lncRNA, pluripotency, stem cell, intrachromosomal interaction, epigenetics
Introduction
Induced expression of four defined factors that are important for maintaining the pluripotent properties of embryonic stem cells can epigenetically reprogram somatic cells to a pluripotent state, resulting in the generation of induced pluripotent stem cells (iPSCs) [1-3]. Unfortunately, the induction of iPSCs from somatic cells using defined factors is an extremely inefficient process [4, 5]. In reprogramming, epigenetic factors play a very important role in activating the transcriptional network associated with pluripotency [6, 7]. A clear understanding of these cellular factors is very important for improving the efficiency of iPSC induction, allowing for the successful therapeutic application of these pluripotent cells.
In a previous study characterizing epigenetic barriers in reprogramming, we compared the promoter DNA binding and chromatin architecture between iPSCs that completed reprogramming and non-iPSCs that expressed the four defined factors (Oct4-Sox2-klf4-c-Myc, OSKM) but failed to complete reprogramming [8]. The virally expressed OSKM factors bound to their target genes to an equal degree in both groups of cells. However, in non-iPSCs, the endogenous core stem cell genes were not activated, partially due to the lack of a promoter-enhancer intrachromosomal loop architecture [8, 9]. We further demonstrate that the downstream enhancer and promoter of iPSC stemness genes are spatially connected through the formation of a chromatin inner loop structure [8], which is necessary for inducing the transformation to produce iPSCs [10, 11]. In addition, maintaining pluripotency also requires the genome of pluripotent stem cells to be organized in the form of higher-order architecture [12-14]. However, the molecular factors that orchestrate this pluripotency- specific intrachromosomal network are still poorly understood.
Long noncoding RNAs (lncRNAs) have recently been recognized as functional molecules taking part in epigenetic, transcriptional, and post-transcriptional control of gene expression [15-19]. Some lncRNAs are differentially expressed and may control pluripotency and stemness, or they may promote differentiation of pluripotent cells [20-23]. We have used an RNA reverse transcription-associated capture sequencing (RAT-seq) approach to characterize functional lncRNAs that interact with the Oct4 promoter [24], a key transcription factor required for reprogramming somatic cells into iPSCs. The data from conventional RNA-seq were combined with RAT-seq to identify differentially-expressed lncRNAs that may be associated with intrachromosomal looping [25]. Using this strategy, we identified a series of functional lncRNA candidates that are associated with pluripotency [24, 25]. We characterized Peblr20, an Oct4 enhancer binding lncRNA, as an essential chromatin factor for the maintenance of stem cell pluripotency. Notably, Peblr20 controls stem cell pluripotency in trans by recruiting TET2 to the Oct4 enhancer locus, thereby activating the enhancer for the initiation of reprogramming [26].
In the present study, we focused on the role of Osblr8, an Oct4-Sox2 promoter-interacting lncRNA, in pluripotent reprogramming. We show that Osblr8 is a pluripotency-associated lncRNA and is required for the maintenance of the stem cell pluripotent state. By interacting with multiple pluripotency-associated transcriptional factor genes, Osblr8 epigenetically regulates their activity by coordinating pluripotency- specific intrachromosomal looping. This study highlights the role of Osblr8 as a chromatin architecture modulator in the enhancement of reprogramming.
Results
CRIST-seq identifies Osblr8 as an essential lncRNA for pluripotency
In order to explore the epigenetic mechanism in chromatin remodeling, we focused on the lncRNAs that interact with the promotor of core pluripotency maintenance factors Oct4 or Sox2, two core pluripotency regulators in reprogramming. We proposed that lncRNAs embedded in or interacting with these loci were involved in the regulation of pluripotency. For this purpose, we used a CRIST-Seq approach [24] to profile lncRNAs that interact with the promoters of Oct4 and Sox2 (Fig.1A). This assay combines the simplicity of nuclear in situ RNA biotin labeling with the specificity of CRISPR Cas9 gene targeting. Cas9 gRNAs and control gRNA (gCT) were transfected into iPSCs, and chromatin-associated RNAs were in situ labelled by reverse transcription with biotin. After Cas9-FLAG immunoprecipitation and purification of the promoter-associated cDNAs from genomic DNAs by streptavidin beads, we constructed a cDNA library for Illumina sequencing.
Mapping pluripotency-associated lncRNAs by RNA-seq and CRIST-seq. A) Chromatin-lncRNA in situ reverse transcription trap sequencing (CRIST-Seq) assay. dCas9: Catalytically inactive CRISPR Cas9; FLAG: a tag octapeptide at the N-terminal of Cas9; gRNA: Cas9 gRNAs that target the Oct4/Sox2 promoter. After fixation, the Oct4/Sox2 promoter-interacting RNAs were reverse transcribed into cDNAs in the isolated nuclei with biotin-dCTP. The Cas9 Oct4/Sox2 promoter biotin-cDNA complex was immunoprecipitated by a Cas9-FLAG antibody, and biotin-cDNAs were further purified from genomic DNAs by biotin-streptavidin beads. The CRIST-captured cDNAs were used for Illumina library sequencing to identify the RNA components in the Oct4 and Sox2 promoters. B) Profiling pluripotency-associated lncRNAs by CRIST-seq and RNA-Seq. The Oct4/Sox2-interacting lncRNAs identified by CRIST-seq were integrated with RNA-seq data. The combination of these two datasets helps identify lncRNAs that interact with the Oct4 and Sox2 promoters and are also expressed differentially in reprogramming. C) Pluripotency-associated RNA candidates identified by RNA-Seq and CRIST-Seq. The RNA candidates are ranked on the basis of the RNA expression-fold between fibroblasts (FIBs) and iPSCs from the high (red) to the low (blue). Gm43558 (Osblr8) was chosen for further studies.
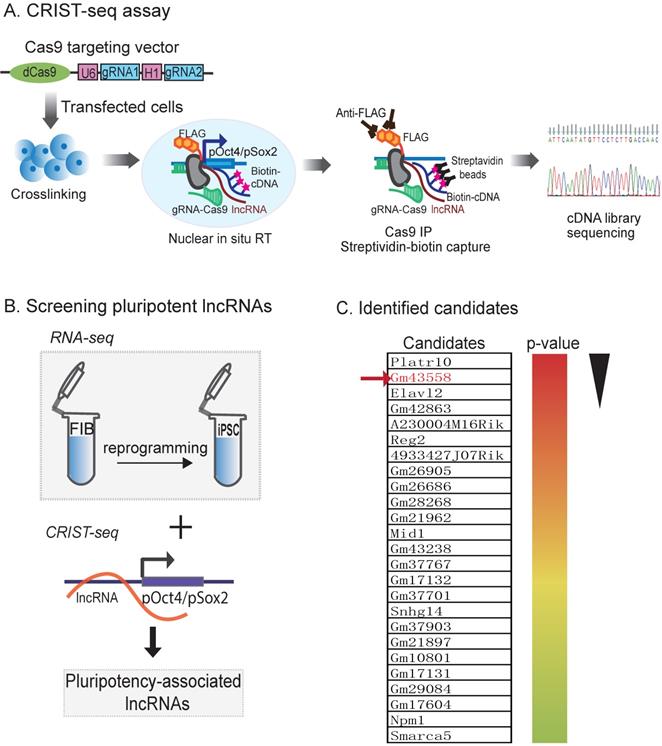
We reasoned that an ideal pluripotent lncRNA candidate should also become activated in reprogramming. Therefore, we collected cells at different stages of reprogramming [27, 28], and RNA-Seq was performed to identify RNAs that were differentially expressed in association with reprogramming [25]. To identify the pluripotency-associated lncRNAs candidates, we integrated the Oct4 and Sox2 CRIST lncRNA data with the RNA-Seq data (Fig.1B). By combining these three datasets, we identified 25 RNA candidates that not only interacted with the Oct4 and Sox2 promoters but were also differentially activated during reprogramming (Fig.1C) [24].
Using this approach, we identified an Oct4/Sox2 promoter-binding lncRNA ENSMUSG00000106628 as a pluripotency-associated lncRNA candidate. We referred it to as Osblr8 (Oct4-Sox2 binding long noncoding RNA 8) to better reflect its function in stem cells. Osblr8 is a 210 bp long lncRNA located in chromosome 3 (Fig.S1A); there was a large-fold increase in Osblr8 abundance when fibroblasts were reprogrammed into iPSCs.
LncRNA Osblr8 is highly expressed in pluripotent stem cells
To determine the role of Osblr8 in regulating pluripotency, we first verified its expression in different reprogramming stages, including fibroblasts, iPSCs, E14, and in non-iPSCs that expressed the lentiviral OSKM factors but failed to complete reprogramming. We confirmed that Osblr8 was highly expressed in fully reprogrammed iPSCs and E14, whereas Osblr8 was nearly undetectable in fibroblasts and non-iPSCs (Fig.2A).
Osblr8 is a pluripotency-associated lncRNA. A) Differential expression of Osblr8 in reprogramming. Fib: fibroblasts; non-iPSC: unreprogrammed cells that express four OSKM cocktail factors but failed to complete reprogramming; iPSC: induced pluripotent stem cells; E14: mouse pluripotent stem cell line used as a positive control. Gene expression was measured by Q-PCR and normalized to β-Actin. For comparison, E14 embryonic pluripotent stem cells were used as the positive control. B) The expression of Osblr8 is associated with Oct4, Sox2, and Nanog expression in embryoid body (EB) differentiation. iPSCs were collected at different stages of EB differentiation for quantitative PCR. C) Subcellular localization of Osblr8 lncRNA. RNA extracted from each fraction was analyzed by Q-PCR. Data shown are mean ± SD (n = 3). β-Actin was used as the cytoplasmic control and U6 was used as the nuclear control. D) RNA FISH of Osblr8. LncRNA probes were synthesized using DIG-11-dUTP and detected by anti-digoxigenin-fluorescein (green). DAPI was used to stain the nucleus of iPSCs (blue). Osblr8 lncRNA was predominantly located in the nucleus.
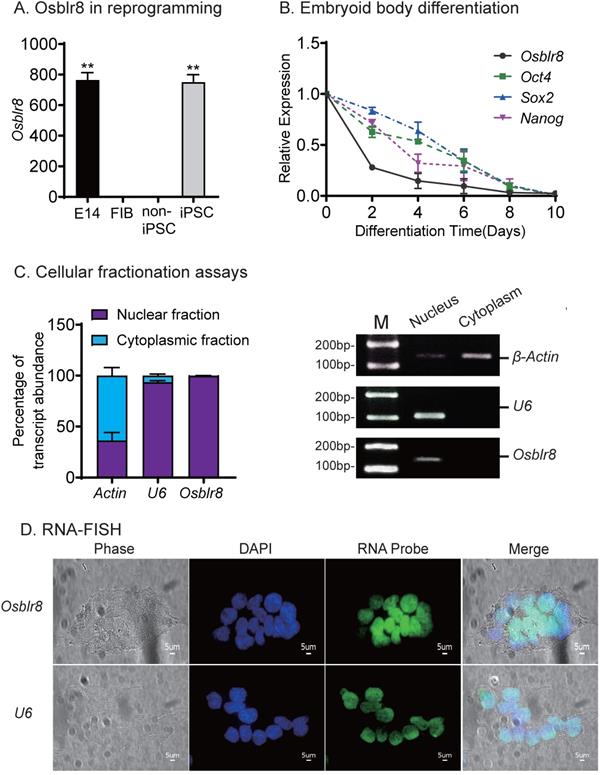
We also collected cells during the process of embryoid body differentiation and examined the expression of Osblr8. Using quantitative PCR, we found that Osblr8 was significantly downregulated during embryoid body differentiation, showing a similar expression pattern with core stem cell factors Oct4, Sox2, and Nanog (Fig.2B). Using cellular fractionation PCR, we found that Osblr8 was predominantly located in the nucleus (Fig.2C), and the nuclear localization was also confirmed by RNA FISH (Fig.2D).
LncRNA Osblr8 Is required for pluripotency maintenance of stem cells
To characterize its role in pluripotency maintenance, we silenced Osblr8 in iPSCs using two pairs of lentivirus-mediated short hairpin RNAs (shRNAs)(Fig.S2A). After lentiviral transfection, iPSCs were selected by puromycin. Single colonies emitting the copGFP green signal were selected, expanded, and collected for Q-PCR. We found that both shRNAs achieved high knockdown efficiency (Fig.3A). Notably, the depletion of Osblr8 lncRNA significantly reduced the expression of pluripotency core factors Oct4, Sox2, and Nanog compared with scrambled control (shCT) cells and untreated iPSCs (Fig.3B). Using an MTT assay, we examined the proliferation capacity of these cells and found that knockdown of Osblr8 altered the proliferation capacity of iPSCs compared to iPSCs and the cells transfected with shCT (Fig.3C).
Osblr8 is required for the maintenance of pluripotency. A) Osblr8 knockdown in iPSCs. After lentiviral shRNA transfection and puromycin selection, iPSCs colonies were selected and expanded for Q-PCR. shCT: random shRNA control; shOsblr8-1, -2: Lenti Osblr8 shRNA-1 and Osblr8 shRNA-2 vectors. For comparison, the abundance of Osblr8 in iPSCs was set as 1. ** p<0.001 as compared with iPSCs and random shCT controls. B) Osblr8 knockdown downregulates stem cell core factors in iPSCs. * p<0.05 as compared with iPSC and random shCT controls. C) Effects of Osblr8 on iPSCs proliferation. MTT assay was performed to determine the viability of iPSCs transfected with shOsblr8, shCT and untreated iPSCs, respectively. D) Osblr8 knockdown induces iPSC differentiation. The Osblr8 shRNA and random shRNA lentiviral vectors carry the copGFP reporter gene (green). After lentiviral transfection, cells were fixed and immunostained by an antibody against the stem cell pluripotent marker OCT4 (red). Compared to the control group, shOsblr8 transfected iPSCs were negative for OCT4 immunostaining and were differentiated morphologically (yellow dotted line).
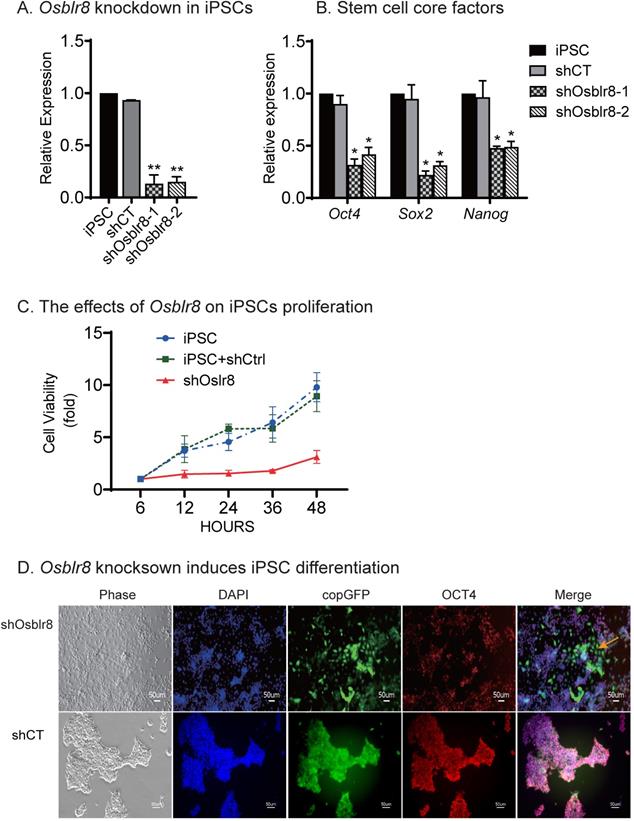
We next examined if Osblr8 knockdown would affect pluripotency of iPSCs (Fig.3D). In the random shRNA control group (shCT), the copGFP-positive cells maintained the same cell morphology as pluripotent stem cells. However, knockdown of Osblr8 dramatically altered cell morphology (top panel5, yellow arrow, shOsblr8). The Osblr8-knockdown cells became spindle-shaped and flat, appearing like fibroblasts. The pluripotency of treated iPSCs was examined by immunohistochemical staining of the pluripotency-associated marker protein OCT4. As expected, the shCT control group showed extensive expression of OCT4 (Fig.3D, bottom panel 4). After Osblr8 shRNA knockdown, however, iPSCs became differentiated and lost the OCT4 expression (top panel 5, green area without red, yellow arrow). In the shOsblr8 group, there were some cells that escaped lentiviral infection and did not express the copGFP tack marker, and they maintained the original compact shape of iPSCs and expressed OCT4. Thus, lncRNA Osblr8 knockdown causes stem cells to exit from the pluripotency state.
LncRNA Osblr8 enhances the activation of stem cell core factors
We then conducted a series of studies to explore the molecular mechanisms by which Osblr8 regulates reprogramming. First, we examined if Osblr8 was able to activate the expression of core stem cell factor genes in fibroblasts. We synthesized a lentipCMV-DsRed/ Puro-Osblr8 plasmid (Fig.S2B) and packaged the lentivirus in 293T cells. Fibroblasts were transfected with Osblr8 lentiviruses. After puromycin selection, ~60% of fibroblasts overexpressed Osblr8 as assessed using DsRed as a tracking marker. Using Q-PCR, we confirmed a >10 fold overexpression of Osblr8 as compared with the vector control (Fig.4A). The endogenous Oct4, Sox2 and Nanog genes were also upregulated in cells in which Osblr8 was overexpressed (Fig.4B).
Osblr8 activates stem cell core factor genes. A) Lentiviral Osblr8 overexpression in fibroblasts. Vector: empty lentiviral vector; lncCT: lncRNA random control; Osblr8: Lentiviral Osblr8 overexpression. ** p<0.001 as compared with the Vector and lncRNA controls. B) Lentiviral overexpression of Osblr8 activates the endogenous stem cell core factor genes. *** p<0.0001, ** p<0.001 as compared with the Vector and lncRNA controls. C) Osblr8 activates promoter activities of core pluripotent factors. 293T cells were co-transfected by reporter plasmids and Osblr8 plasmid. Forty-eight hours after transfection, cells were collected for luciferase activity measurements. Reporter plasmid empty vector and random lncRNA (lncCT) vectors were used as the controls. For comparison, the untreated fibroblasts were set as 1. * P < 0.05, and ** P < 0.001.
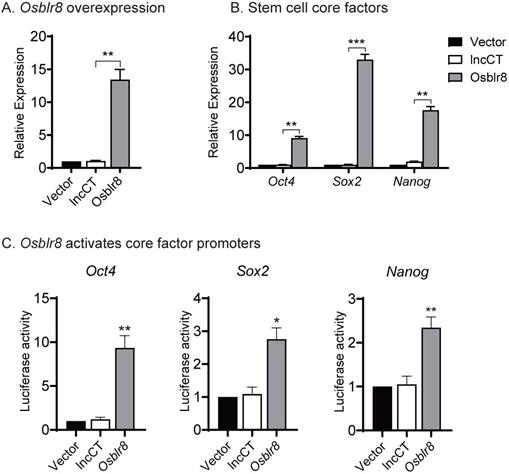
Next, we explored whether Osblr8 could activate the Oct4, Sox2 and Nanog promoters. We synthesized three luciferase plasmids containing the promoters of these three master pluripotency transcript factors, respectively, which were then co-transfected with the Osblr8 overexpression plasmid in 293T. Forty-eight hours after transfection, cells were harvested, and the luciferase activity was measured using the Dual-Luciferase reporter assay system. As shown in Figure 4C, the overexpression of Osblr8 increased the luciferase activity of these promoters in 293T by 9, 2, and 2-fold greater, respectively.
LncRNA Osblr8 interacts with TET Family enzymes
We then focused on the epigenetic mechanisms by which Osblr8 regulates Oct4. In order to initiate reprogramming, the methylated CpGs in the Oct4 promoter must be demethylated to initiate transcription in somatic cells. The ten-eleven translocation (TET) family enzymes catalyze the stepwise oxidation of 5-methylcytosine in DNA to 5-hydroxymethylcytosine and play important biological functions in embryonic stem cells, development, aging, and disease [29]. Thus, we examined if Osblr8 regulates the Oct4 gene through the TET family enzymes.
We compared the expression of the TET family genes in iPSCs and Osblr8-knockdown iPSCs and showed that the expression of Tet1, Tet2 and Tet3 was downregulated in parallel with Osblr8 knockdown (Fig.5A and 5B), suggesting that Osblr8 affects the TET family genes at the transcriptional level.
Osblr8 interacts with TET protein family. A) Knockdown of Osblr8 in iPSCs. B) TET family expression in Osblr8 knockdown iPSCs. cDNAs were extracted from iPSCs loss-of-function experiment. The TET1/2/3 primers were used for Q-PCR. C) Interaction of Osblr8 with TET enzymes by RNA-chromatin immunoprecipitation (RIP). The TET-RNA chromatin complex was immunoprecipitated with an antibody against TET1, TET2 and TET3. After de-crosslinking, the immunoprecipitated RNAs were reverse transcribed. The TET-interacting Osblr8 was measured by Q-PCR. IgG was use as the antibody control. Input: aliquot DNAs collected during the RIP assay. * P < 0.05, ** P < 0.01, and *** P < 0.001. NS: not significant.
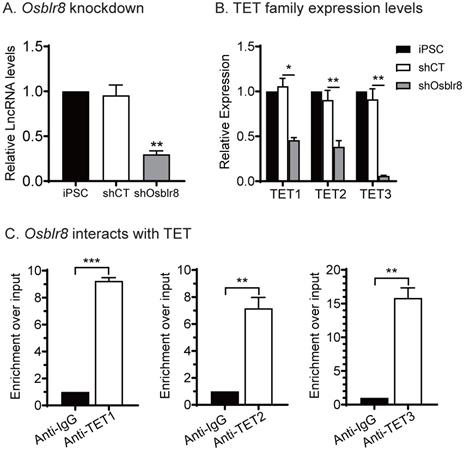
We then performed an RNA-binding protein immunoprecipitation (RIP) assay in iPSCs to examine if Osblr8 interacts directly with TET1, TET2, and TET3 enzymes. The TET 1/2/3-RNA chromatin complex was immunoprecipitated by anti-TET1/2/3 antibodies, respectively. A mouse IgG was used as the negative control. The RNA pull-downs were reverse transcribed and detected by real-time Q-PCR using primers from Osblr8. As compared with the IgG control, we detected a marked enrichment of Osblr8 in the TET antibody-precipitated complexes (Fig.5C). These data suggest that Osblr8 may activate the endogenous Oct4 gene via TET-induced DNA demethylation.
Osblr8 orchestrates pluripotency-specific intrachromosomal looping
Chromatin remodeling is a major epigenetic barrier that determines the initiation of the pluripotency status in reprogramming [30-32]. By comparing local chromatin structure of the Oct4 locus, we previously revealed that there was a pluripotency-associated intrachromosomal loop in iPSCs that juxtaposes a downstream enhancer to the gene's promoter, enabling activation of endogenous stemness genes to achieve reprogramming [8]. We asked whether Osblr8 lncRNA participates in the orchestration of intrachromosomal looping for reprogramming.
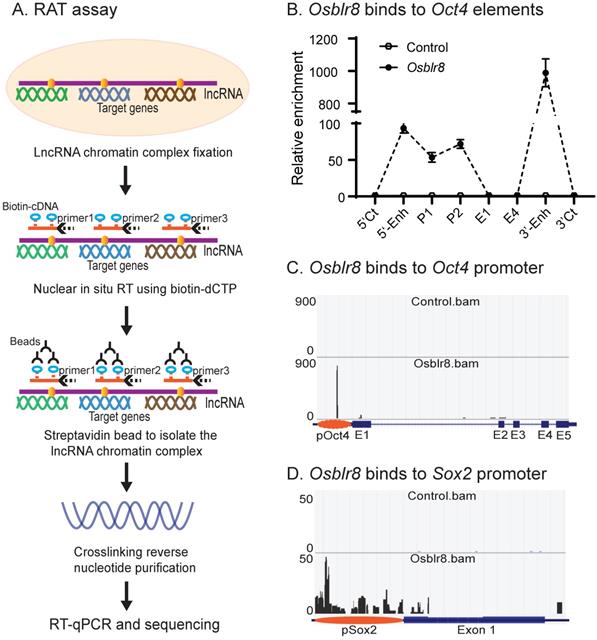
We first examined the binding of Osblr8 in the pluripotency-associated network. A detailed IGV analysis of the CRIST-seq data indicated that the signal of Osblr8 binding to the Oct4 and Sox2 promoter was enriched only in the Cas9 Oct4 and Sox2 gRNA cells, but not in the Cas9 gCT control and the IgG immunoprecipitation control (Fig.S3A). The CRIST-seq IGV analysis revealed that Osblr8 bound to the Oct4 and Sox2 promoter using two 50 bp fragments (5'-CCCCCTTCCTTCATAACTAGTGTCGCAACAATAAAATTTGAGCCTTGATC-3' and 5'-CCAGCCTGAAACCTGCTTGCTCGGGGTGGAGCTTCCTGCTCATTCGTTCT-3'). Using RAT-seq (Fig. 6A), we found that Osblr8 bound to the Oct4 promoter and to 5'- and 3'-enhancer elements (Fig. 6B and 6C). The RAT-seq IGV analysis also showed that Osblr8 binds to the Sox2 promoter. No such interaction signals were detected in the RAT random control library products (CTL).
Mapping the Osblr8 interaction network by RAT-seq. A) RNA reverse transcription-associated trap (RAT) assay. After fixation, Osblr8 lncRNA was reverse transcribed into cDNAs in the isolated nuclei with biotin-dCTP using three gene specific reverse primers. The DNA-protein-lncRNA-biotin-cDNA complex was further purified from genomic DNAs by biotin-streptavidin beads. After the crosslinking reversal, the captured DNAs were used for Illumina library sequencing or analyzed by Q-PCR to identify the Osblr8 interaction network. B) Osblr8 lncRNA-Oct4 DNA interactions by Q-PCR. RAT library samples were used to perform Q-PCR to quantitate binding intensity. The results were normalized to the value of the streptavidin bead pulldown control. Control: the RAT-seq library was constructed using random oligo primers; Obelr20: the RAT-seq library was constructed using Osblr8-speccific primers; 5'-CT, 3'-CT: the RAT 5'- and 3'- control sites; P: promoter; E1 and E4: Oc4 exons 1 and 5. C) Interaction of Osblr8 at the Oct4 locus. Control: the RAT library was constructed with random oligo primers. Osblr8: the RAT library was constructed using Osblr8 complementary primers; 5'-Enh: 5'-enhancer; E1-E5: Oct4 exons 1-5; 3'-Enh: 3'-enhancer. Note the enriched binding of Osblr8 lncRNA at the Oct4 promoter. D) Interaction of Osblr8 at the Sox2 locus. Osblr8 binds to the Sox2 promoter area.
We then used chromatin conformation capture (3C) [33, 34] to compare the intrachromosomal looping between iPSCs, shRNA control cells, and Osblr8 knockdown cells. Cells were fixed with 1% formaldehyde, digested with restriction enzymes BamH1/BglII, and then connected with T4 DNA ligase. After crosslinking reversal and DNA purification, chromatin interactions were detected by specific 3C primers located in the promoter and the enhancers of Oct4 (Fig.7A). As previously reported [8], we detected reprogramming-associated intrachromosomal interaction products in iPSCs: the P780/ P783 and P780/P785 loops between the 5'-upstream enhancer and the promoter, and the P790/P785 loop between the 3'-enhancer and the promoter (Fig.7B). In the control iPSCs transfected with shRNA control (shCT), these intrachromosomal loops were intact. However, shRNA knockdown of Osblr8 abolished these intrachromosomal interaction signals and caused the iPSCs to exit from pluripotency.
Osblr8 orchestrates pluripotency-specific intrachromosomal looping. A) Location of 3C primers used to detect the interaction between the Oct4 promoter and enhancer. Enh: enhancers; pOct4: Oct4 promoter; E1-E5: exons; Arrows: intrachromosomal interactions. B) Knockdown of Osblr8 abolishes the intrachromosomal interaction loop. shCT: negative control shRNA, shOsblr8: shRNA that targets Osblr8 lncRNA. iPSC: induced pluripotent stem cells. Primer sets that detect the presence of looping are marked in Fig.7A. The 3C interaction was quantitated by qPCR and was standardized over the 3C control Ercc3 gene. For comparison, the relative 3C interaction was calculated by setting the 5' or 3' control as 1. *P<0.05, ** P< 0.01 as compared with the shOsblr8 treatment and shRNA control. C) Overexpression of Osblr8 induces de novo formation of intrachromosomal looping. FIB: fibroblasts. Fib+Vector: fibroblasts transfected with vector control; Fib+Osblr8: fibroblasts overexpressed Osblr8. D) Sequencing of the Oct4 intrachromosomal loop products. Blue line on the top of the sequence: the 3C ligation product between the BamH1 and Bgl2 sites. Red line on the top of the sequence: the 3C ligation product between the BamH1 and BamH1 sites.
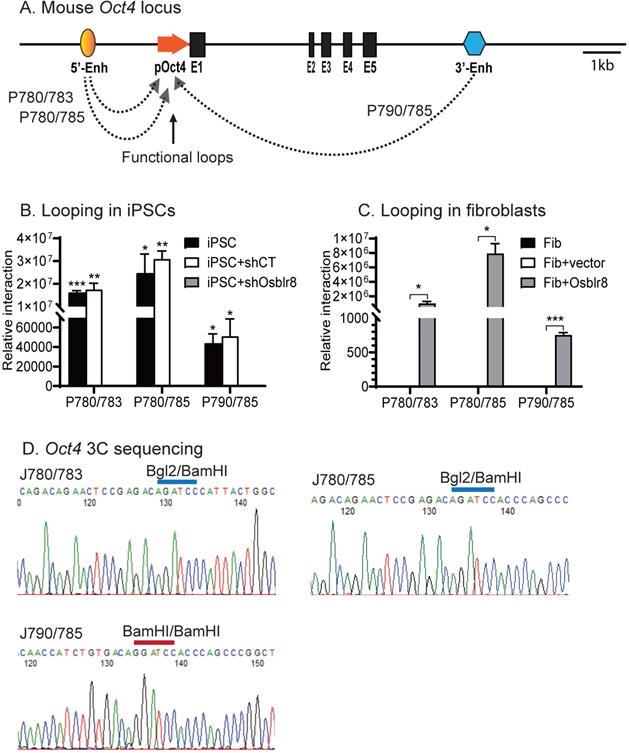
We also observed de novo formation of intrachromosomal interactions in fibroblasts when Osblr8 was overexpressed in these cells (Fig. 7C). We sequenced the 3C products and verified the presence of the ligated BamHI or BamHI/BglII sites, which were flanked by the sequences from the promoter and enhancers of Oct4, respectively (Fig.7D). These data suggest that Osblr8 is critical in the maintenance of intrachromosomal interactions that are known to associate with reprogramming and maintenance of pluripotency [8].
Discussion
Formation of a specific promoter-enhancer intrachromosomal architecture constitutes a critical epigenetic barrier in pluripotent reprogramming [8, 35]. In this study, we focused on the chromatin factors that interact with the Oct4 and Sox2 promoters. Using CRIST-seq, we identified Osblr8 as a crucial regulator that helps maintain the pluripotency of iPSCs. Osblr8 was differentially expressed during the reprogramming progress, silenced in fibroblasts, but highly expressed in iPSCs and E14 cells. The loss of Osblr8 results in the exit of E14 cells from pluripotency, while an increased abundance of Osblr8 activates the stem cell core factors in fibroblasts. Most importantly, Osblr8 participates in the formation of a loop structure that is required for the activation of stemness gene expression. Loss of Osblr8 abolishes the long-range interaction and leads to cell differentiation. Thus, Osblr8 plays a crucial role in coordinating a topological architecture network that is necessary for maintenance of pluripotency.
Subcellular localization predicts the function of lncRNAs [36, 37]. Nuclear lncRNAs usually act as guiders and tethers to enhance or activate the expression of specific genes. They can help chromatin modification complexes bind to specific genomic loci or act as scaffolds to tether together distinct related functional complexes [38, 39]. Nuclear lncRNAs can interact with proteins and RNAs by base-pairing with other nucleic acids to activate or repress the transcription of defined genes [39-41]. LncRNAs localized in the cytoplasm, on the other hand, usually regulate gene expression by base-pairing complementary regions on target RNAs [42]. Osblr8 is highly expressed in the nucleus and functions by binding to the Oct4 promoter and interacting with TET proteins. Osblr8 coordinates the formation of intrachromosomal loops, thereby enhancing the expression of stemness-related genes. Through this complex network, Osblr8 can control the pluripotent state of iPSCs.
DNA methylation plays an important role during reprogramming. Inhibition of DNA methyltransferase DNMT1 can enhance the efficiency of reprogramming [43]. The CpG methylation status of the promoter is critical in the regulation of the Oct4 gene, as demonstrated by the different expression patterns seen in somatic and pluripotent cells [44-46]. TET proteins can successfully induce DNA demethylation by the oxidation of C-5 position of cytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). 5fC and 5caC are selectively recognized and excised by thymine DNA glycosylase (TDG), leading to DNA demethylation [47, 48] These pathways could allow turnover or resetting of DNA methylation to promote and maintain pluripotent states. TET1 and TET2 are highly expressed in mESCs, which is consistent with the relatively high levels of 5hmC and detectable levels of 5fC and 5caC in these cells [49-51]. In the absence of TET1 and TET2, ESCs are intrinsically blocked from reaching complete global hypomethylation [52]. Numerous studies show that the TET protein family plays an important role in maintaining pluripotency status. Through the CRIST-seq and RIP-PCR, we demonstrate that Osblr8 may help recruit the TET protein family to the Oct4 promoter, where DNA demethylation promotes the expression of the Oct4 gene.
The 3D chromatin architecture is very important for transcriptional regulation. Looping between core promoter elements and distal enhancer or insulator elements controls the transcriptional activation or repression of genes, respectively [53-55]. Some studies reported that Mediator and Cohesin physically and functionally connect the enhancers and core promoters of active genes in embryonic stem cells. Mediator, a transcriptional coactivator, forms a complex with cohesin, which can form rings that connect two DNA segments [56, 57]. At the Oct4 gene locus, for example, the intrachromosomal loops between the gene promoter and enhancer regulatory regions are specific for pluripotent stem cells. This topological structure helps bring distal regulatory elements, like enhancers, into physical proximity with gene target promoters, thereby activating them to initiate cellular reprogramming [8]. In this study, we showed that Osblr8 was enriched in the promoter and 5'- and 3'-enhancers of Oct4. A similar mechanism has been observed for a well-known lncRNA Xist, which initiates X-chromosome inactivation (XCI) in females by coating the inactive X-chromosome in cis [58-62]. We demonstrated that Osblr8 was essential for the maintenance of this intrachromosomal looping. Knockdown of Osblr8 abolishes this intrachromosomal loop structure, and causes the exit of iPSCs from pluripotency. Thus, during reprogramming, Osblr8 is actively transcribed and acts in concert with other chromatin factors to coordinate a topological architecture network that is necessary to initiate pluripotency. In addition to Oct4, our RAT-seq data show that Osblr8 also binds to other pluripotency- associated factor genes, like Sox2. It will be interesting to explore if Osblr8 also utilizes a similar mechanism to regulate the expression of Sox2 in reprogramming.
Overall, our study identifies Osblr8 as a novel pluripotency-associated lncRNA. Osblr8 becomes activated in reprogramming. Depletion of Osblr8 caused E14 cells to differentiate and lose pluripotency. Osblr8 maintains stem cell pluripotency through multiple epigenetic mechanisms, including activation of endogenous stem cell core factor genes and coordination of intrachromosomal looping. Osblr8 also regulates TET protein family genes, with which to control the expression of stemness genes. Thus, lncRNA Osblr8 exerts its function as a chromatin epigenetic modulator in the regulatory network of stem cell pluripotency and reprogramming.
Material and Methods
CRIST-Seq to map the Oct4/Sox2-interacting lncRNAs
The CRIST-seq method used to identify lncRNAs that bind to the Oct4 and Sox2 promoters was described by Zhang et al [24]. After CRIST-seq, the called peaks that overlapped with the IgG control enriched regions were removed, and the CRIST-Seq signal intensities were further normalized over that of the non-targeting Cas9 gCT control using parameters of fold-change difference ≥2 and p-value < 0.05, with false discovery rate (FDR) <0.1. The adjusted CRIST-Seq data were then used for mapping the Oct4 and Sox2 RNA interactions [24, 25].
Identification of lncRNAs by RNA-seq in reprogramming
Mouse fibroblasts were reprogrammed with Oct4-Sox2-Klf4-c-Myc (OSKM) lentiviruses [8, 63]. The isolated iPSC colonies were characterized by immunostaining stem cell markers, alkaline phosphatase staining, karyotype analysis, and teratoma formation. The fibroblast-like cells that expressed OSKM but were not reprogrammed were termed “non-iPSCs” and used in parallel with iPSCs in the study [8, 25]. The lncRNAs that are differentially expressed in reprogramming were identified by RNA-seq. RNAs that are differentially expressed in reprogramming were identified using the fold-change > 2 and p < 0.05 with an unpaired two-sided t-test [25].
RNA extraction and cDNA synthesis
Total RNA was extracted by Trizol reagent (Invitrogen). The concentration and quality of all RNA samples were evaluated by Nanodrop 1000 (ThermoScientific, CA), and the 260/280 and 260/230 values of all samples were more than 1.8 and 1.9, respectively. The extracted RNA samples were stored at -80°C, and cDNAs were synthesized using M-MLV reverse transcriptase (Invitrogen). Briefly, 400 - 800 ng total RNA was added to 12 µl liquid wax per reaction; we then used DNase I (Millipore Sigma, MA) to remove genomic DNA contamination. The reverse transcription reaction was performed with M-MLV reverse transcriptase at 37°C for 1 h, followed by 95°C for 10 min. After 10-fold dilution, cDNA was stored at -20°C or used for PCR and RT-qPCR.
Quantitation of gene expression by Q-PCR
Real-time PCR was carried out using 3 X Klen-Taq I Mix with a Bio-Rad Thermol Cycler. PCR amplification was performed by PCR of 1 cycle at 98°C for 5 min, 32 cycles at 95°C for 20 s, 62°C for 15s and 72°C for 15 s, and 1 cycle at 72°C for 10 min. β-Actin was used as PCR input. Quantitative real-time PCR (Q-PCR) was performed using the FastStart Universal SYBR Green Master mix (Millipore Sigma, MA) with a StepOnePlus real-time PCR system (ABI Prism 7900HT; Applied Biosystems, USA). For quantitative real-time PCR, the threshold cycle (Ct) values of target genes were normalized over the Ct of the β-Actin control. Primers used for real-time PCR and qPCR are listed in Table S1.
Preparation of Cytoplasmic and Nuclear Fractions
Cells were briefly digested by Trypsin-EDTA and gently resuspended in DMEM. After completely aspirating the PBS, 800 μl hypotonic buffer (10 mM Hepes, pH 7.9, 1.5 mM MgCl2, 10 mM KCl) were added and placed on ice for 2 min. 10% Nonidet P-40 was added to a final concentration of 0.4% (35 μl). Samples were inverted a few times and spun at 3,000 g for 7 min. Supernatants (cytoplasmic fractions) were collected for processing, and the pellet (nuclear fraction) was gently resuspended in 500 μl hypotonic buffer and spun at 3,000 g for 2 min. This washing step was repeated three to four times. Both the cytoplasmic and nuclear fractions were processed for RNA extraction, cDNA synthesis and real-time PCR or qPCR. To verify that the cytoplasmic and nuclear fractions were completely separated, we used U6 as a nuclear control and β-Actin as cytoplasmic control. The primers for PCR are listed in Table S1.
RNA FISH
RNA FISH was performed by a modification of the method published previously [64-66]. The RNA FISH probe was prepared as an antisense single strand DNA (ssDNA) by asymmetric PCR [67]. Briefly, the ssDNA probe was synthesized by 3x Klen-Taq I DNA polymerase mix, the iPSC cDNA was used as the template, PCR primers were JH6195 and JH6196 as listed in Table S1. The DNA probe was purified by electrophoresis on 2% agarose gel and eluted in 20 μl TE buffer. For hybridization, 0.1 μg ssDNA probe and 10 μg salmon sperm DNA (Boehringer, Meylan, France) were precipitated with ethanol and suspended in 10 μl RNA hybridization buffer (2xSSC, 10% dextran sulfate, 0.2mg/mL BSA (Invitrogen, CA), 2mM VCR, 10% formamide). After sequential RNA FISH, slides were counterstained with DAPI, and FISH signals were detected using the Keyence BZ-X710 fluorescence microscope with GFP filter (EX 470/40 nm, DM 495nm, BA 525/50nm), Cy5 filter (EX 620/60 nm, DM 660nm, BA 700/75nm) and DAPI filter (EX 360/40 nm, DM 400nm, BA 460/50nm), respectively. Images were captured and merged to confirm the subcellular localization.
Lentiviral overexpression of Osblr8 lncRNA in fibroblasts
Full-length Osblr8 lncRNA was amplified with PCR primers containing the EcoRI and EcoRV restriction sites. The PCR products were gel-purified, cut by restriction enzymes, and ligated into the pCMV-DsRed/Puro vector constructed in our lab. The Osblr8 lncRNA clone was confirmed by sequencing and then packaged in 293T packing cells [68] using the method described in our lab [48]. After transfecting fibroblasts, cells were selected by puromycin. The DsRed reporter in the vector was used to track lentivirus transfection efficiency. After 14 days, cells were collected for next experiments.
MTT assay
Cell proliferation was quantitated using the Cell Growth determination KIT MTT Bases (Sigma, USA, Stock No. CGD-1) following the manufacturer's instructions. Cells are cultured in 96-well plates. When cells were adherent to the plate, medium was removed and MTT SOLUTION was added in an amount equal to 10% of the culture. Cells were incubated in a 37°C incubator with 5% CO2 for 4 h. Then the MTT SOLUTION was removed and the MTT SOLVENT was added in an amount equal to the original culture volume. After gentle shaking, the absorbance was spectrophotometrically measured at a wave length of 570 nm. We measured cell number at 6, 12, 24, 36, and 48 h. Cells were cultured in three wells and the study was repeated for three replicates.
Knockdown of Osblr8 lncRNA in iPSCs
LncRNA Osblr8 was knocked down by two shRNA lentiviruses. The shRNA vector was constructed by cloning two shRNAs into pGreenPuro vector (#SI505A-1, SBI, CA). shRNAs were designed online (http://katahdin.cshl.edu/homepage/siRNA/RNAi). For cloning, two pairs of shRNAs (5'- CTGGAACCTGAGGAGCCACACACGT-3' and 5'- TGCACCTTTCTACTGGACCAGAGAT-3') combined with loop were linked to the H1 and U6 promoter using PCR and were ligated into the EcoR1/BamH1 site in pGreenPuro vector. The copGFP reporter in the vector was used to track lentivirus transfection in iPSCs. A random shRNA (GCAGCAACTGGACACGTGATCTTAA) was cloned in the same vector as the assay control (shCT). After lentiviral transfection, iPSCs were selected by puromycin. Single colonies were selected and cultured for expansion. Cells were collected for RNA quantitation of Osblr8 lncRNA and related genes using RT-qPCR.
Immunofluorescent staining of stem cell markers
Immunofluorescent staining was used to examine stem cell markers in iPSC colonies [69]. Briefly, cells were fixed by freshly made 4% paraformaldehyde for 10 min at room temperature, permeabilized with freshly made 0.5% v/v Triton X-100/PBS on ice for 5min, then blocked in 1% w/v BSA for 30 min at room temperature. After incubation with primary antibodies diluted in 1% BSA (2 μg/ml 1:500) for 1-3 h at room temperature, samples were washed three times in PBS for 5 min each, and then incubated with the secondary antibodies for 1 h at room temperature. The following antibodies were used in the immunostaining: rabbit anti-OCT4 (1:100 dilution, Santa Cruz). The cell samples were subsequently incubated with Cy3 or Alexa Fluor 647 labeled secondary antibodies for 1 h. After washing three times with PBS, samples were counterstained with Hoechst 33258 (Invitrogen). Alternatively, the pluripotency of stem cells was examined by Fluorescent Mouse ES/iPS Cell Characterization kit (Cat.#SCR077, Millipore, MA) following the protocol provided by the manufacturer. Fluorescence images were acquired with a Zeiss AxioCam Camera.
Embryoid body differentiation
E14 cells were cultured by the hanging drop method in a 10-cm culture dish without LIF. After 3 days, EBs were transferred to 10-cm ultra-low-adherence plates for suspension culture with slow shaking for up to 12 days. The media were replenished by sedimentation every other day. Embryoid bodies were collected on D0, D2, D4, D6, D8, D10 and D12 for quantitation of Osblr8 lncRNA and targeted genes using RT-qPCR.
Luciferase assay
The function of Osblr8 in activating the promoters was first examined in 293T cells by using a dual-luciferase reporter assay. A 3.9 kb genomic DNA fragment covering the Oct4, Sox2 and Nanog promoters respectively. The promoter DNA fragment was cloned into pGL3 vector by Kpn1/Xho1.
For the luciferase assay, cells were seeded at a density of 5 × 104 cells/well in 24-well plates. The lentiviral Osblr8 overexpression vector was co-transfected with an Oct4-luciferase plasmid and Renilla luciferase control plasmid (Promega) using Lipofectamine 3000 (Invitrogen, CA). The empty lentiviral vector and random lncRNA vector were used as controls. Forty-eight hours after transfection, firefly and Renilla luciferase activities were measured with the dual-luciferase reporter system (Promega) using a luminometer (Turner Biosytem, CA). The relative activity of the promoter was calculated by setting the untreated control cells as 1. All luciferase assays were repeated three times with three culture replicates each.
Profiling the Osblr8 genome-wide gene targets by RAT-seq
The RAT-seq approach was used to map the genome wide interacting target genes for lncRNA candidates[25, 70, 71]. Briefly, 1.0 × 107 cells were cross-linked with 2% formaldehyde and lysed with cell lysis buffer (10 mM Tris [pH 8.0], 10 mM NaCl, 0.2% NP-40, 1X protease inhibitors). Nuclei were collected, suspended in 1X reverse transcription buffer in the presence of gene-specific primer, biotin-14-dCTP, RNase inhibitor and Maxima Reverse Transcriptase (Thermo Fisher Scientific, CA). After 30 min of reverse transcription of Osblr8 lncRNA labeled by biotin-14-dCTP with Maxima Reverse Transcriptase at 65◦C. After nuclear lysis, the complex was sonicated for 180s (10s on and 10s off) on ice by 2-mm microtip at 40% output control and 90% duty cycle settings. The biotin-cDNA/chromatin DNA complex was pulled down with biotin-streptavidin magic beads (Invitrogen, CA). After reversing the cross-links and washing with 10 mg/ml proteinase K at 65°C overnight and treatment with 0.4 μg/ml RNase A for 30 min at 37°C, the genomic DNA that interacts with the lncRNA was extracted and digested by MboI, and ligated with the NEBNext adaptors (NEBNext® ChIP-Seq Library Prep Master Mix Set for Illumina) to construct the library. The library DNAs were used to Illumina sequencing (Shanghai Biotechnology, Shanghai) and binding PCR with primers shown in Table S1. We performed a RAT assay with random primers and constructed a control library for sequencing using the same protocol.
After RAT sequencing, the low quality reads were filtered using Fastx (version:0.0.13) software (http://hannonlab.cshl.edu/fastx_toolkit/index.html). Clean reads were mapped to the mouse genome (genome version: mm10) using the Bowtie (version:0.12.8) software with default parameters[72]. Enriched regions of the genome were identified by comparing the RAT-Seq peaks to input samples using MACS2 (version:2.1.1) and q-value of 0.05 was used as the initial cutoff threshold to minimize peak caller bias[73]. The upstream 2 k of the transcription start sites and the downstream 5k of the transcription termination region were defined as the gene regions. The significant GO terms of biological processes with a p-value < 0.05 were selected. We also used the MEME suite [74] for the discovery and analysis of the peaks' sequence motifs. The resulting coverage tracks (bedgraph file) were visualized in the UCSC genome browser. To reduce the background, the RAT-Seq data were further normalized over the peaks of the control RAT-Seq data that were generated by using random oligonucleotide primers in the RAT assay. Differential binding analysis was performed with the DiffBind package using parameters of fold change difference ≥2 and p-value < 0.05, with false discovery rate (FDR) <0.1. The adjusted RAT-Seq data were used for mapping the lncRNA target gene interaction network.
Chromosome conformation capture (3C)
We used a 3C assay to determine intrachromosomal interactions [75, 76]. Briefly, fibroblasts and iPSCs were cross-linked with 2% formaldehyde and lysed with cell lysis buffer. An aliquot of nuclei (2×106) was digested with 800 U BamHI/BglII at 37°C overnight. Chromatin DNA was diluted with NEB ligation buffer and ligated with 4,000 U of T4 DNA ligase. After reversing the crosslinks, DNA was purified and used for PCR amplification using primers that are derived from different regions of the Oct4 locus. The 3C PCR products were sequenced to validate the intrachromosomal interaction by affirming the presence of the BamHI/BglII or BamHI ligation site.
RNA binding protein immunoprecipitation (RIP) assay
A lncRNA-affinity binding precipitation assay (RIP) [77] was performed to examine the binding of TET1/2/3 protein with Osblr8 lncRNA. RIP was performed using the Magna RIPTMRNA-Binding Protein Immunoprecipitation Kit (Millipore, Germany) according to the manufacturer's instructions. Osblr8-overexpressed fibroblasts were collected and lysed using RIP lysis buffer. Then 100 μl cell extract was incubated with RIP buffer containing magnetic beads conjugated with anti-TET1/2/3 antibody (Abcam, MA). Mouse IgG was used as the negative control. The samples were incubated with proteinase K to digest protein, and then the immunoprecipitated RNA was isolated. The purified RNAs were sequenced and detected by reverse transcription qPCR. The primers for Q-PCR are listed in Supplementary Table S1. CHIP-qPCR was performed with three replicates. The Ct values were normalized over the input and compared with the IgG control.
Statistical analysis
All experiments were performed in triplicate. The data were expressed as mean ± standard error of mean (SEM) and were analyzed using SPSS software (version16.0, IL). The data were analyzed with Student's t-test or by one-way analysis of variance, and statistically significant differences by Student's t test.
Data availability
The RNA-Seq data generated in this study have been deposited in NIH GEO databases with accession number GSE116605, including 1). PSC RNA-seq.fq.gz (GSM3243668, iPSC RNA sequencing fastq data); 2). FIB RNA-seq.fq.gz (GSM3243669, Fibroblast RNA sequencing fastqdata) [25]. The CRIST-Seq data generated in this study have been deposited in NIH GEO databaseswith accession number GSE107945. The folder contains four raw data files, including1). Cas9 Sox2-gRNA.fq.gz (Sox2 promoter CRIST lncRNA sequencing fastq data); 2).Cas9 Oct4-gRNA.fq.gz (Oct4 promoter CRIST lncRNA sequencing fastq data); 3).Cas9-gCT2.fq.gz (Oct4 promoter CRIST Cas9-gCT random control library sequencingfastq data); 4). Cas9-IgG.fq.gz (CRIST IgG control library sequencing fastq data) [24].
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFA0106902), National Natural Science Foundation of China (31430021, 81874052, 81672275, 31871297, 81670143), the Key Project of Chinese Ministry of Education grant (311015), the National Basic Research Program of China (973 Program)(2015CB943303), Nation Key Research and Development Program of China grant (2016YFC13038000), Research on Chronic Noncommunicable Diseases Prevention and Control of National Ministry of Science and Technology (2016YFC1303804), National Health Development Planning Commission Major Disease Prevention and Control of Science and Technology Plan of Action, Cancer Prevention and Control (ZX-07-C2016004), Natural Science Foundation of Jilin Province (20150101176JC, 20180101117JC, 20130413010GH), Provincial Science Fund of Jilin Province Development and Reform Commission (2014N147 and 2017C022), and California Institute of Regenerative Medicine (CIRM) grant (RT2-01942). This work was supported in part by a Merit Review from the United States (U.S.) Department of Veterans Affairs, Biomedical Laboratory Research and Development Service (BX002905).
Author Contributions
Y.Z. performed majority of the assays and analyzed data. Z.Y., Z.D., S.Z., C.F., Y.M., X.W., and Y.W. performed partial experiments and data collection. A.R.H., J.C., W.L., and J.F.H. designed the study and supervised the project. Y.Z. and J.F.H. wrote the manuscript. A.R.H. edited the manuscript. All authors approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Warren L, Lin C. mRNA-Based Genetic Reprogramming. Mol Ther. 2019;27:729-34
2. Watanabe A, Yamada Y, Yamanaka S. Epigenetic regulation in pluripotent stem cells: a key to breaking the epigenetic barrier. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120292
3. Lin YC, Murayama Y, Hashimoto K, Nakamura Y, Lin CS, Yokoyama KK. et al. Role of tumor suppressor genes in the cancer-associated reprogramming of human induced pluripotent stem cells. Stem Cell Res Ther. 2014;5:58
4. Haridhasapavalan KK, Raina K, Dey C, Adhikari P, Thummer RP. An Insight into Reprogramming Barriers to iPSC Generation. Stem Cell Rev Rep. 2019
5. Yoshida Y, Yamanaka S. Induced Pluripotent Stem Cells 10 Years Later: For Cardiac Applications. Circ Res. 2017;120:1958-68
6. Nashun B, Hill PW, Hajkova P. Reprogramming of cell fate: epigenetic memory and the erasure of memories past. EMBO J. 2015;34:1296-308
7. Abdelmohsen K, Panda A, Kang MJ, Xu J, Selimyan R, Yoon JH. et al. Senescence-associated lncRNAs: senescence-associated long noncoding RNAs. Aging cell. 2013;12:890-900
8. Zhang H, Jiao W, Sun L, Fan J, Chen M, Wang H. et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell. 2013;13:30-5
9. Hu JF, Hoffman AR. Chromatin looping is needed for iPSC induction. Cell Cycle. 2014;13:1-2
10. Dean A. In the loop: long range chromatin interactions and gene regulation. Brief Funct Genomics. 2011;10:3-10
11. Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S. et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341-7
12. Denholtz M, Bonora G, Chronis C, Splinter E, de Laat W, Ernst J. et al. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 2013;13:602-16
13. Ji X, Dadon DB, Powell BE, Fan ZP, Borges-Rivera D, Shachar S. et al. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell. 2016;18:262-75
14. Sexton T, Cavalli G. The 3D Genome Shapes Up For Pluripotency. Cell Stem Cell. 2013;13:3-4
15. Fernandes JCR, Acuna SM, Aoki JI, Floeter-Winter LM, Muxel SM. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Noncoding RNA. 2019 5
16. Dinescu S, Ignat S, Lazar AD, Constantin C, Neagu M, Costache M. Epitranscriptomic Signatures in lncRNAs and Their Possible Roles in Cancer. Genes (Basel). 2019 10
17. Huang H, Sun J, Sun Y, Wang C, Gao S, Li W. et al. Long noncoding RNAs and their epigenetic function in hematological diseases. Hematological oncology. 2019;37:15-21
18. Berabez N, Durand S, Gabut M. Post-transcriptional regulations of cancer stem cell homeostasis. Curr Opin Oncol. 2019;31:100-7
19. Zhao Y, Sun L, Wang RR, Hu JF, Cui J. The effects of mitochondria-associated long noncoding RNAs in cancer mitochondria: New players in an old arena. Critical reviews in oncology/hematology. 2018;131:76-82
20. Smith KN, Miller SC, Varani G, Calabrese JM, Magnuson T. Multimodal Long Noncoding RNA Interaction Networks: Control Panels for Cell Fate Specification. Genetics. 2019;213:1093-110
21. Yan P, Luo S, Lu JY, Shen X. Cis- and trans-acting lncRNAs in pluripotency and reprogramming. Curr Opin Genet Dev. 2017;46:170-8
22. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW. et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353-60
23. Ghosal S, Das S, Chakrabarti J. Long noncoding RNAs: new players in the molecular mechanism for maintenance and differentiation of pluripotent stem cells. Stem Cells Dev. 2013;22:2240-53
24. Zhang S, Wang Y, Jia L, Du Z, Wen X, Wang C. et al. Profiling the long noncoding RNA interaction network in the regulatory elements of target genes by chromatin in situ reverse transcription sequencing. Genome Res. 2019;29:1521-32
25. Du Z, Jia L, Wang Y, Wang C, Wen X, Chen J. et al. Combined RNA-seq and RAT-seq mapping of long noncoding RNAs in pluripotent reprogramming. Sci Data. 2018;5:180255
26. Wang C, Jia L, Wang Y, Du Z, Zhou L, Wen X. et al. Genome-wide interaction target profiling reveals a novel Peblr20-eRNA activation pathway to control stem cell pluripotency. Theranostics. 2020;10:353-70
27. Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41:7429-37
28. Zhai Y, Chen X, Yu D, Li T, Cui J, Wang G. et al. Histone deacetylase inhibitor valproic acid promotes the induction of pluripotency in mouse fibroblasts by suppressing reprogramming-induced senescence stress. Experimental cell research. 2015;337:61-7
29. Best J, Schotten C, Theysohn JM, Wetter A, Muller S, Radunz S. et al. Novel implications in the treatment of hepatocellular carcinoma. Ann Gastroenterol. 2017;30:23-32
30. Abrahao Kde S, Bergmann A, Aguiar SS, Thuler LC. Determinants of advanced stage presentation of breast cancer in 87,969 Brazilian women. Maturitas. 2015;82:365-70
31. Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H. et al. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151-9
32. Chedin F. Nascent Connections: R-Loops and Chromatin Patterning. Trends Genet. 2016;32:828-38
33. Pian L, Wen X, Kang L, Li Z, Nie Y, Du Z. et al. Targeting the IGF1R Pathway in Breast Cancer Using Antisense lncRNA-Mediated Promoter cis Competition. Mol Ther Nucleic Acids. 2018;12:105-17
34. Zhao X, Liu X, Wang G, Wen X, Zhang X, Hoffman AR. et al. Loss of insulin-like growth factor II imprinting is a hallmark associated with enhanced chemo/radiotherapy resistance in cancer stem cells. Oncotarget. 2016;7:51349-64
35. de Wit E, Bouwman BA, Zhu Y, Klous P, Splinter E, Verstegen MJ. et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 2013;501:227-31
36. Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7-21
37. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298-307
38. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F. et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689-93
39. Morlando M, Ballarino M, Fatica A, Bozzoni I. The role of long noncoding RNAs in the epigenetic control of gene expression. ChemMedChem. 2014;9:505-10
40. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA. et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311-123
41. Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y. et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120-4
42. Facciorusso A, Villani R, Bellanti F, Mitarotonda D, Vendemiale G, Serviddio G. Mitochondrial Signaling and Hepatocellular Carcinoma: Molecular Mechanisms and Therapeutic Implications. Curr Pharm Des. 2016;22:2689-96
43. Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P. et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49-55
44. Imamura M, Miura K, Iwabuchi K, Ichisaka T, Nakagawa M, Lee J. et al. Transcriptional repression and DNA hypermethylation of a small set of ES cell marker genes in male germline stem cells. BMC Dev Biol. 2006;6:34
45. Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313-7
46. Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042-7
47. Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341-56
48. Bao X, Wu H, Zhu X, Guo X, Hutchins AP, Luo Z. et al. The p53-induced lincRNA-p21 derails somatic cell reprogramming by sustaining H3K9me3 and CpG methylation at pluripotency gene promoters. Cell Res. 2015;25:80-92
49. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129-33
50. Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA. et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398-402
51. Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J. et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200-13
52. Hackett JA, Dietmann S, Murakami K, Down TA, Leitch HG, Surani MA. Synergistic mechanisms of DNA demethylation during transition to ground-state pluripotency. Stem Cell Reports. 2013;1:518-31
53. Engel JD, Tanimoto K. Looping, linking, and chromatin activity: new insights into beta-globin locus regulation. Cell. 2000;100:499-502
54. Abe S, Funato T, Takahashi S, Yokoyama H, Yamamoto J, Tomiya Y. et al. Increased expression of insulin-like growth factor i is associated with Ara-C resistance in leukemia. Tohoku J Exp Med. 2006;209:217-28
55. Huang SL, Wu Y, Yu H, Zhang P, Zhang XQ, Ying L. et al. Inhibition of Bcl-2 expression by a novel tumor-specific RNA interference system increases chemosensitivity to 5-fluorouracil in Hela cells. Acta Pharmacol Sin. 2006;27:242-8
56. Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL. et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430-5
57. Ohlsson R. Gene expression: The coherent Mediator. Nature. 2010;467:406-7
58. Goodrich L, Panning B, Leung KN. Activators and repressors: A balancing act for X-inactivation. Semin Cell Dev Biol. 2016;56:3-8
59. Payer B, Rosenberg M, Yamaji M, Yabuta Y, Koyanagi-Aoi M, Hayashi K. et al. Tsix RNA and the germline factor, PRDM14, link X reactivation and stem cell reprogramming. Mol Cell. 2013;52:805-18
60. Sun BK, Deaton AM, Lee JT. A transient heterochromatic state in Xist preempts X inactivation choice without RNA stabilization. Mol Cell. 2006;21:617-28
61. Vallot C, Rougeulle C. Long non-coding RNAs and human X-chromosome regulation: a coat for the active X chromosome. RNA Biol. 2013;10:1262-5
62. Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750-6
63. Chen M, Zhang H, Wu J, Xu L, Xu D, Sun J. et al. Promotion of the induction of cell pluripotency through metabolic remodeling by thyroid hormone triiodothyronine-activated PI3K/AKT signal pathway. Biomaterials. 2012;33:5514-23
64. Barakat TS, Gribnau J. Combined DNA-RNA fluorescent in situ hybridization (FISH) to study X chromosome inactivation in differentiated female mouse embryonic stem cells. J Vis Exp. 2014;88:e51628
65. Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17:661-78
66. Andriole GL, Crawford ED, Grubb RL 3rd, Buys SS, Chia D, Church TR. et al. Prostate cancer screening in the randomized Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial: mortality results after 13 years of follow-up. J Natl Cancer Inst. 2012;104:125-32
67. Zhao Y, Liu S, Li1 X, Zhou L, Meng Y, Li Y. et al. Aberrant shuttling of long noncoding RNAs during the mitochondria-nuclear crosstalk in hepatocellular carcinoma cells. Am J Cancer Res. 2019;9:999-1008
68. Broussau S, Jabbour N, Lachapelle G, Durocher Y, Tom R, Transfiguracion J. et al. Inducible packaging cells for large-scale production of lentiviral vectors in serum-free suspension culture. Mol Ther. 2008;16:500-7
69. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM. et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405-9
70. Sun J, Li W, Sun Y, Yu D, Wen X, Wang H. et al. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic Acids Res. 2014;42:9588-601
71. Wang H, Li W, Guo R, Sun J, Cui J, Wang G. et al. An intragenic long noncoding RNA interacts epigenetically with the RUNX1 promoter and enhancer chromatin DNA in hematopoietic malignancies. Int J Cancer. 2014;135:2783-94
72. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25
73. Kretlow JD, Jin YQ, Liu W, Zhang WJ, Hong TH, Zhou G. et al. Donor age and cell passage affects differentiation potential of murine bone marrow-derived stem cells. BMC Cell Biol. 2008;9:60
74. Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696-7
75. Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA. et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299-309
76. Zhang H, Niu B, Hu JF, Ge S, Wang H, Li T. et al. Interruption of intrachromosomal looping by CCCTC binding factor decoy proteins abrogates genomic imprinting of human insulin-like growth factor II. J Cell Biol. 2011;193:475-87
77. Grinnell KL, Bickenbach JR. Skin keratinocytes pre-treated with embryonic stem cell-conditioned medium or BMP4 can be directed to an alternative cell lineage. Cell Prolif. 2007;40:685-705
Author contact
![]() Corresponding authors: Wei Li, Tel: +86-0431-8878-2974, e-mail: jdyylwcom; or Jiuwei Cui, +86-431-8878-2178, e-mail: cuijiuweiedu.cn; or Ji-Fan Hu, Tel: +1-650-852-3175, e-mail: jifanedu.
Corresponding authors: Wei Li, Tel: +86-0431-8878-2974, e-mail: jdyylwcom; or Jiuwei Cui, +86-431-8878-2178, e-mail: cuijiuweiedu.cn; or Ji-Fan Hu, Tel: +1-650-852-3175, e-mail: jifanedu.